
Abstract
A Pd(II) complex of two sterically crowded ligands, specifically an N,S-donor thiosemicarbazone and an N-donor imidazole, has been synthesized and characterized by physicochemical and spectroscopic methods. X-ray single-crystal analysis revealed that the coordination geometry around the palladium center is distorted square planar, and the chloride ligand is involved in intermolecular bifurcated X–H···Y-type (where X = C, N and Y = Cl) hydrogen bonding. This complex proved to be a highly active and retrievable pre-catalyst for additive-free Suzuki–Miyaura cross-coupling reactions of arylboronic acids with aryl bromides or chlorides at room temperature and 60 °C, respectively. The reactions require a low catalyst loading and the complex is converted to ~1.5–2.0 nm-sized Pd nanoparticles (probably the real catalyst). The catalyst can be reused up to seven times without significant loss in activity. Since the reaction proceeds under mild conditions in aqueous medium and the catalyst is recoverable, it provides an environmentally benign alternative to the existing protocols for Suzuki–Miyaura reactions.
Graphical abstract
Biaryls can be synthesized in high yield under greener reaction conditions in the presence of efficient pre-catalyst, thiosemicarbazone–palladium(II)–imidazole.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the first report of the Suzuki–Miyaura reaction [1, 2], this method has become one of the most powerful and versatile procedures for the construction of unsymmetrical biaryls that are used as building blocks in the pharmaceutical, agrochemical and material industries [3, 4]. Thousands of Pd(II) complexes with various ligand systems have been reported to accelerate this coupling reaction [5,6,7,8]. In most cases, the catalytic agent Pd(II) is converted to Pd(0) during catalysis [8]. However, a debate on this issue is still ongoing and it is uncertain with many Pd complexes whether they are the actual catalyst or pre-catalyst. Traditionally, Suzuki–Miyaura coupling relies on toxic, air and/or moisture sensitive and expensive palladium catalysts, usually with phosphine ligands [9,10,11,12]. Moreover, the solvents used in such reactions are mostly not eco-friendly. Since catalysis under phosphine-free conditions is an important challenge, in recent years a plethora of N-based ligands such as amines [13], N-heterocyclic carbenes (NHC) [14, 15], oximes [16], hydrazones [17] and others [18, 19] have been tested as possible alternatives to phosphines in Suzuki–Miyaura cross-coupling reactions. Recently, Schiff bases have been recognized as excellent alternative ligands in Suzuki–Miyaura reactions [5,6,7,8]. There are, however, few reports of Pd catalysts bearing thiosemicarbazone ligands in Suzuki–Miyaura reactions [20–27]. Many of the earlier protocols use severe reaction conditions, such as high temperature (100–155 °C), DMF solvent, long reaction time, high catalyst loading in some cases, and little or no scope for recycling the catalyst, due to its homogeneous nature. Since this reaction is relevant to industry, it is important to design novel catalytic systems with air stable, less expensive and robust ligands which could efficiently catalyze the cross-coupling reactions without employing any additives, especially in green solvents.
Thiosemicarbazones constitute an interesting class of N,S-donor ligands because of their mixed hard–soft character. These ligands are mainly known for their biological activity [28, 29]. They can act as bi- or multidentate ligands [30,31,32], and can occupy two, three or four coordination sites and thereby control the selectivity of the catalyst [30,31,32]. Additional donor atoms in the ligand can act as stabilizing groups during the course of metal-mediated reactions and thereby improve catalytic efficiency [20]. As a part of our continuing efforts to develop efficient Pd catalysts for cross-coupling reactions [33,34,35,36], herein, we report the synthesis of a reusable Pd(II) complex with thiosemicarbazone and imidazole ligands within the same coordination sphere, and its evaluation as a catalyst in Suzuki–Miyaura reactions of aryl halides with aryl boronic acids in aqueous media at room temperature. To the best of our knowledge, this is the first example of a Pd catalyst bearing both thiosemicarbazone and imidazole ligands. We hoped that the presence of two bulky, electron-rich ligands within the same coordination sphere would increase steric congestion around the Pd metal and facilitate the rate of both oxidative addition and reductive elimination steps in the mechanism [37]. However, Pd(0) nanoparticles (~1.5–2.0 nm) were formed while carrying out the reactions, and these appeared to be the real catalyst.
Experimental
Materials and instrumentation
All chemicals were of AnalaR grade and obtained commercially. They were used as received without further drying or purification. Solvents were purchased from Merck. The 4-phenylthiosemicarbazide and N-methylimidazole were purchased from Sigma–Aldrich, and palladium(II) chloride was procured from Arora Matthey Limited. The arylboronic acids were purchased from Spectrochem. FTIR spectra (4000–250 cm−1) were recorded using KBr disks on a Shimadzu Prestige-21 FTIR spectrophotometer. Elemental analyses were obtained on an Elementar Vario EL III Carlo Erba 1108 elemental analyzer. Electrospray ionization (ESI) (+) mass spectra were recorded on a Waters ZQ-4000 liquid chromatograph–mass spectrometer. 1H and 13C (100 MHz) NMR spectra were recorded in DMSO-d6 using TMS as an internal standard on a JEOL JNM ECS NMR spectrometer operating at 400 MHz and an Advance DPX 300 MHz FT-NMR spectrometer operating at 300 MHz. Melting points were determined by using a BUCHI B450 melting point apparatus. SEM images were obtained with a JEOL JSM Model 6390 LV scanning electron microscope, operating at an accelerating voltage of 15 kV. Transmission electron microscopic (TEM) investigations were carried out on a JEM-2100 instrument equipped with a high-resolution CCD camera and an accelerating voltage of 60–100 kV in 50 kV steps. EDX spectra were also recorded on the same instrument attached to the scanning electron microscope. X-ray diffraction analysis (XRD) was performed on a Bruker AXS D8 Advance Diffractometer with Cu-Kα (λ = 1.541Å) radiation. X-ray photoelectron spectra (XPS) were recorded on an XPS-AES Module, Model: PHI 5000 Versa Prob II. The C (1s) electron binding energy corresponding to graphitic carbon was used for calibration of the Pd (3d) core-level binding energy. The amount of Pd leached on the filtrate after the fifth cycle of catalysis was analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) on a Thermo Electron IRIS Intrepid II XSP DUO instrument. The progress of the reactions was monitored by TLC on silica gel plates (E. Merck, silica gel 60F254), using n-hexane–ethyl acetate as eluent. The products of the reactions were confirmed by comparing 1H spectra with those reported in the literature.
Crystallography The X-ray crystallographic analysis was carried out on a Bruker Apex 2 CCD diffractometer using monochromatic Mo-K α radiation (λ = 0.71073Å) at 293 K. The data were corrected for Lorenz and polarization effects. Hydrogen atoms were included in calculated positions and refined in riding mode. The structure was solved using the SHELXL-2014/7 package and refined by full-matrix least squares on F 2. CCDC No. 1497434 contains the supplementary crystallographic data for this paper. This file can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/ structures.
Catalyst preparation
Synthesis of anisaldene-4–phenylthiosemicarbazone (HL1)
HL1 was synthesized by reacting anisaldehyde (1.36 g, 10 mmol) and 4-phenylthiosemicarbazide (1.65 g, 10 mmol) in 1:1 molar ratio in EtOH (25 ml) with a few drops of acetic acid [38]. White crystalline solid; yield: 85%; m.p. 146 °C. Anal. Calcd. for C15H15N3OS (%): C, 63.2; H, 5.3; N, 14.7; S, 11.2. Found: C, 63.6; H, 5.0; N, 14.2; S, 10.8. FW: 285 g mol−1. ESI (+) MS m/z [M]+: 285, [M + H]+; selected IR bands (KBr, cm−1): (N3–H) 3327, (N2–H) 3152, (C=N) 1603, (N–N) 1171, (C=S) 827. 1H NMR [300 MHz, DMSO-d6, δ ppm]: 11.69 (s, 1H, N2–H), 10.04 (s, 1H, N3–H), 8.10 (s, 1H, CH=N), 6.96–7.84 (m, 9H, Ph–H), 3.78 (s, 3H, OCH3). 13C NMR [100 MHz, DMSO-d6, δ ppm]: 175.5 (C=S), 160.8 (Ph–C), 142.9 (CH), 138.9 (Ph–C), 114.1–129.3(Ph–C), 55.2 (CH3).
Synthesis of [PdL1Cl2]
PdCl2 (0.089 g, 0.5 mmol) and HL1 (0.142 g, 0.5 mmol) were added to acetonitrile (20 ml), and the mixture was stirred at room temperature in air for 3 h. An orange precipitate was formed. This was filtered off, washed with acetonitrile and dried over fused CaCl2 in a desiccator. Dark red solid; yield: 75%; d.t. 215–220 °C; selected IR bands (KBr, cm−1): (N3–H) 3242, (N2–H) 3046, (C=N) 1591, (N–N) 1174, (C=S) 825, (Pd–N) 500, (Pd–S) 396, (Pd–Cl) 385. 1H NMR[400 MHz, DMSO-d6, δ ppm]: 10.75 (s, 1H, N2–H), 9.73 (s, 1H, N3-H), 8.29 (s, 1H, CH=N), 8.12 (d, 2H), 7.52 (d, 2H), 7.07 (t, 1H), 7.36 (t, 2H), 7.50 (d, 2H), 3.82 (s, 3H, OCH3); 13C NMR[100 MHz, DMSO-d6, δ ppm]: 176.5 (C=S), 162.8(Ph–C), 143.9 (CH), 138.9 (Ph–C), 116.1–132.3 (Ph–C), 55.0 (CH3).
Synthesis of [PdL1L2Cl] (C1)
The precursor complex, [PdL1Cl2] (0.231 g, 0.5 mmol), was dissolved in acetonitrile (10 ml). To this solution, KPF6 (0.092 g, 0.5 mmol) and N-methylimidazole (0.041 g, 0.5 mmol) were added. The mixture was refluxed with stirring for 1.5 h under air. The resulting yellow precipitate was filtered off, washed with acetonitrile and dried under vacuum. Yellow solid; yield: 60%; d.t. 220–227 °C; FW: 508.3 g mol−1; selected IR bands (KBr, cm−1): (N3–H) 3100, (C=N) 1593, (N–N) 1176, (C–S) 593, (Pd–N) 561,(Pd–S) 387, (Pd–Cl) 382; 1H NMR[400 MHz, DMSO-d6, δ ppm]: 8.47(s, 1H, N3–H), 7.68 (s, 1H, CH=N1), 7.50–7.20 (m, 9H, Ph–H), 6.8 (s, 1H, CH=N4, Im-H), 5.50 (d, 2H, Im-H), 3.82 (s, 3H, OCH3), 3.61 (s, 3H, N5–CH3); 13C NMR[100 MHz, DMSO-d6, δ ppm]: 171.1 (C–S), 160.9(Ph–C), 141.9 (CH), 55.2 (CH3).
Catalytic experiments
The Suzuki–Miyaura cross-coupling reactions were carried out under aerobic conditions at room temperature (28 °C). Progress of the reactions was monitored by TLC. The products were isolated by column chromatography using silica gel (60–120 mesh) and characterized by comparing their 1H NMR, mass spectral data and melting points with authentic samples.
In a typical procedure, a mixture of aryl halide (0.5 mmol), arylboronic acid (0.6 mmol), K2CO3 (3 equiv.) and catalyst (1.18 mol%) was added to solvent (4 ml) and the mixture was stirred at room temperature for the required time. After completion of the reaction, the mixture was centrifuged. The residual solid was filtered off and washed with three portions of the same reaction solvent (4 ml). The residue was extracted from the filtrate using water–ether mixture (1:3) followed by washing with brine and drying over Na2SO4. The products were obtained by column chromatography of the residue using ethyl acetate/hexane (1:9) as eluent. For recycling experiments, the catalyst was washed several times after each cycle with water (3 × 5 ml) followed by diethyl ether (3 × 5 ml). After overnight drying at 120 οC, the recovered catalyst was subjected to subsequent runs under identical conditions.
Results and discussion
Synthesis and spectroscopic characterization
Our study was directed toward developing a cheap and non-toxic Pd-based catalytic system for Suzuki–Miyaura cross-coupling reactions under room temperature, environmentally friendly conditions. To this end, we have synthesized a new complex C 1, [PdL1L2Cl] from [PdL1Cl2] and N-methyl imidazole as per Scheme 1 (L 1 = anisaldene-4-phenylthiosemicarbazone and L 2 = N-methyl imidazole). N-methyl imidazole was chosen as the co-ligand in order to increase the electron density at Pd [39].

Synthesis of precursor complex and C 1
The precursor complex, [PdL1Cl2], and the new complex C 1 are both stable to air and moisture and soluble in dichloromethane and DMSO. They were characterized by ESI mass spectrometry, NMR (1H and 13C) and FTIR. Moreover, the structure of the complex C 1 was determined by single-crystal X-ray diffraction analysis. In the infrared spectra, both complexes exhibit characteristic IR bands due to v(Pd–N) and v(Pd–S) in the ranges of 500–561 cm−1 and 387–396 cm−1, respectively, showing coordination of the ligand to the metal. The ν(C=N) band at 1600 cm−1 for the free ligand is shifted (Δν ≈ 7–9 cm−1) toward lower frequencies on complexation, suggesting coordination to the metal through the azomethine nitrogen [40]. The ν(C=S) band at 828 cm−1 for the free ligand is absent for complex C 1; a new, medium intensity band at 593 cm−1 is attributed to ν(C–S), indicating the presence of only thiolate sulfur in the complex.
Description of the crystal structure of [PdL1L2Cl]
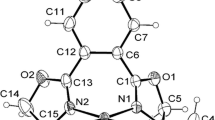
The complex C 1 was crystallized from acetonitrile by slow evaporation over a period of 5 days. The crystal has monoclinic P21/c space group. The molecular structure of C 1 together with the atomic numbering scheme is shown in Fig. 1. Crystallographic data are given in Table S1, and selected bond lengths are listed in Table S2. The single-crystal X-ray diffraction analysis confirmed coordination of the thiosemicarbazone to palladium through thiolate sulfur S1 and azomethine nitrogen N1. The other two coordination sites are occupied by the pyridine nitrogen N4 of the N-methylimidazole ligand and a chloride ligand.

Molecular structure of [PdL1L2Cl], C 1
The coordination geometry around the palladium is distorted square planar, with N(4)–Pd(1)–Cl(1), N(4)–Pd(1)–S(1), N(1)–Pd(1)–S(1) and N(1)–Pd(1)–Cl(1) bond angles at 87.72°(15), 91.21°(15), 84.11°(15) and 97.18°(14), respectively (Table 2). The Pd1–N1, Pd1–N4, Pd1–S1 and Pd1–Cl1 bond distances are 2.015 Å(5), 2.019 Å(5), 2.2260 Å(18) and 2.3407 Å (18), respectively. Interestingly, the chloride ligand is involved in intermolecular bifurcated hydrogen bonding, specifically N3–H···Cl1(d(H···A) = 2.624 Å) and C19–H···Cl1(d(H···A) = 2.927 Å) (Fig. 2) [41, 42]. The second of these hydrogen bonds may be important for the C–H bond activation, since the strength of C–H···Halogen hydrogen bonding can play an important role in determining the affinity and selectivity of catalysts for C–H bond activation [43]. Moreover, the crystal lattice shows a helical structure [44] through the N3–H···Cl1 hydrogen bonds, which run along the b-axis (Fig. 3).

Intermolecular bifurcated hydrogen bonding in the complex

Helical structure of C 1 along the b-axis
To determine the catalytic activity of the precursor complex C 1, a model cross-coupling reaction was investigated at room temperature between 4-bromoanisole and phenyl boronic acid. The results of these experiments are summarized in Table 1. Screening various solvents using K2CO3 as base showed that ethanol was the most effective solvent (Table 1, entries 1, 11 and 22). Increasing the temperature reduced the reaction time, but did not alter the yield substantially (Table 1, entries 3 and 16). Upon trialling different catalyst loadings, we found that 1.18 mol% of catalyst was efficient for the required conversion (Table 1, entry 11). Our investigations also showed that the base plays an essential role, since the reaction did not proceed without base (Table 1, entry 21). We therefore screened different bases, namely K2CO3, Na2CO3, Cs2CO3, NaOH, KOH, NaHCO3 and Na3PO4·12H2O (Table 2); the best results were obtained with K2CO3 (Table 2, entry 7).

Since C 1 is soluble in ethanol, it could not be recovered from this solvent. However, in aqueous medium the catalyst remained as a suspension, allowing easy separation from the organic products. Since reusability is an important concern for a catalyst and also from the green chemistry perspective water is the most suitable solvent, we chose water as solvent to optimize the choice of base (Table 2) and to also examine the scope of the reaction for various aryl bromides and aryl boronic acids having either electron-withdrawing or electron-donating groups as coupling partner (Table 3).
From the substrate study reported in Table 3, it is clear that the rate of cross-coupling depends on the functional group present on both the aryl halide and aryl boronic acid. The coupling between 4-substituted aryl bromides with electron-withdrawing groups and phenyl boronic acid (Table 3, entries 2, 3 and 8) required short reaction times and returned good product yields. 4-Methoxyphenyl boronic acid did not have any appreciable effect on the progress of the reaction with aryl bromides bearing either electron-withdrawing or electron-donating groups, giving good yields in both cases (Table 3, entries 5, 9–12). On the other hand, 3-thienyl and 4-fluoro boronic acids required longer reaction times and gave moderate and trace amounts of product, respectively (Table 3, entries 15 and 16). The primary problem associated with fluoro boronic acid might be a slow rate of transmetalation, associated with the electron deficiency of the aromatic ring. This protocol was not useful for aryl chlorides at room temperature due to the higher bond energy of C–Cl compared to C–Br; however, at elevated temperature (60 °C) moderate isolated yields were obtained (Table 3, entries 19, 20 and 22).
Reusability of the catalyst was checked using phenylboronic acid and 4-bromoanisole as coupling partner. Since C 1 forms a colloidal solution under the reaction conditions, it is easily recoverable by extraction with ethyl acetate and water, washing with brine followed by centrifugal precipitation. The catalyst can then be reused after washing with ethyl acetate and brine and drying. Very interestingly, the second run reaction completed very rapidly (within 1 h) with higher isolated yield (95%) compared to the first run (6 h, 78% yield). However, the reaction time increased slightly in subsequent cycles (Table 4). From the turnover frequency (TOF) determination (Table S3), it was observed that the TOF of the second catalytic cycle (81 h−1) is highest. After the second cycle, the slight gradual decrease in product yield on successive reuse may be due to either physical loss of catalyst or some decomposition of the real catalyst. To investigate for possible catalyst leaching, we performed ICP-AES analysis of the filtrate after the fifth catalytic cycle. Detection of a negligible amount (0.0065 mol%) of palladium in the filtrate suggests no significant leaching [45]. To further confirm the nature of the catalytic system, we carried out a hot filtration test [45], again using 4-bromoanisole and phenylboronic acid. After 15 min, the reaction was stopped and the catalyst was filtered off after centrifugation (20% yield determined by GC–MS). The reaction was then allowed to continue for a further 6 h without the catalyst (Fig. 4). No catalytic activity could be seen in the filtrate (by GC analysis), ruling out the possibility of homogeneous catalysis by leached Pd species. Furthermore, to identify the active catalytic species, we performed an Hg(0) poisoning test. Hg(0) poisons heterogeneous palladium particles by forming an amalgam [46], but does not poison homogeneous complexes of palladium in high oxidation states. Hence, for the mercury poisoning test, Hg(0) (molar ratio to [Pd] ~400) was taken in a reaction flask containing H2O before the addition of reactants and stirred for 0.5 h at room temperature. To this suspension, 4-bromonitrobenzene (0.5 mmol), phenylboronic acid (0.6 mmol), K2CO3 and the palladium catalyst recovered after first run were added, and the mixture was stirred for the required time at room temperature. The reaction was almost stopped (by GC analysis) by the addition of Hg(0). This established that the complex C 1 is actually a pre-catalyst, which gets activated to Pd(0) during the first catalytic cycle and interacts with Hg(0) to stop the reaction. To clarify the catalytic nature of the palladium catalyst recovered after the first run, we have carried out TEM, SEM–EDX and XRD analysis. Also to check the oxidation state of palladium, XPS was performed before and after the first cycle.

Activity of the catalyst for the reaction between phenyl boronic acid and 4-bromoanisole with hot filtration and without filtration
The TEM image (SAED: selected area electron diffraction, in the inset) of the catalyst [Fig. S5] after the first run clearly shows the formation of Pd nanoparticles, with the majority of particles within the size range of 1.5–2.0 nm, as shown in the particle size histogram presented in Fig. 5. We believe that during the course of the reaction, the palladium–ligand bonds dissociate and form Pd(0) nanoparticles which might be stabilized by a surface layer of the thiosemicarbazone. As shown in the figure, the clearly visible lattice fringes and diffraction dots observed in the SAED image indicate the crystalline nature of the PdNPs.

Particle size distribution of C 1 after the first catalytic cycle
Figure S6 shows the SEM–EDX pattern of the catalyst after the first catalytic cycle. The data reveal the palladium content along with the N, S, C and O proportions and suggest that the Pd nanoparticles are stabilized by the thiosemicarbazone ligand. The XRD pattern of C 1 after the first catalytic cycle exhibits diffraction peaks at 2θ values of 40.2, 46.3 and 67.8 which correspond to the (111), (200) and (220) planes of the face centered cubic phase of Pd(0) nanoparticles [JCPDS46-1043]. The peak broadening suggests the presence of small particles as well as a low degree of crystallinity (Fig. S7). This was further confirmed by TEM analysis.
Figure 6a, b shows the characteristic XPS responses of the Pd2+ and Pd0 3d core-level peaks of the catalyst before and after the first catalytic cycle, respectively. The two peaks at 341.2 and 346.1 eV (Fig. 6a) can be assigned to the Pd2+ 3d core-level peaks corresponding to the 5/2 and 3/2 spin–orbit components [47]. The Pd0 nanoparticles 3d core-level spectrum (Fig. 6b) is characterized by a pair of relatively narrow peaks corresponding to the 5/2 and 3/2 spin–orbit components located at 336.2 and 341.4 eV, in agreement with several results previously reported [48, 49].

a 3d core-level XPS spectrum of the catalyst C 1 (i.e., Pd2+ complex before catalysis), b 3d core-level XPS spectrum of the recovered catalyst after the first catalytic cycle
In order to investigate the cause of the decrease in catalytic efficiency on successive cycles, we carried out a TEM analysis of C 1 recovered after the sixth catalytic cycle. This revealed that the Pd nanoparticles of size 1.5–2.0 nm formed after the first run had aggregated to larger particles (Fig. S8). The aggregation appears to be responsible for the decrease in catalytic activity observed for successive runs.
We have compared our catalyst with some previously reported catalysts [21,22,23,24,25,26,27] and found it to have some advantages in terms of greener reaction conditions, recyclability and heterogeneity (Table 5). Most of the previously reported catalysts were homogeneous in nature, so either could not be recycled or could be used for a maximum for three runs [27]. Moreover, some of these reported catalysts contain expensive and toxic phosphine ligands [23, 24].
Conclusion
In summary, the complex C 1 proved to be an efficient, recyclable pre-catalyst for Suzuki–Miyaura cross-coupling reactions of various electronically diverse aryl bromides with arylboronic acids in aqueous media at room temperature. This new method has versatile synthetic utility, providing biaryls in good to excellent yields in water at room temperature. The catalyst can be synthesized very easily from commercially available precursor materials and is stable to air and moisture. The formation of Pd(0) nanoparticles was observed during the course of reaction, which appear to be the real catalyst. Aggregation of Pd(0) nanoparticles appears to decrease the catalytic activity in successive cycles of reuse.
References
Miyaura N, Suzuki A (1979) J Chem Soc Chem Comm 19:866–867
Miyaura N, Yamadnandea K, Suzuki A (1979) Tetrahedron Lett 20:3437–3440
Suzuki A (2011) Angew Chem Int Ed 50:6722–6737
Fihri A, Bouhrara M, Nekoueishahraki B, Basset JM, Polshettiwar V (2011) Chem Soc Rev 40:5181–5203
Miyaura N, Suzuki A (1995) Chem Rev 95:2457–2483
Das P, Linert W (2016) Coord Chem Rev 311:1–23
Paul S, Islam MM, Islam SM (2015) RSC Adv 5:42193–42221
Begum T, Mondal M, Borpuzari MP, Kar R, Kalita G, Gogoi PK, Bora U (2017) Dalton Trans 46:539–546
Marziale AN, Faul SH, Reiner T, Schneider S, Eppinger J (2010) Green Chem 12:35–38
Sabounchei SJ, Ahmadi M, Panahimehr M, Bagherjeri FA, Nasri Z (2014) J Mol Catal A 249:383–384
Schaarschmidt D, Lang H (2011) ACS Catal. 1:411–416
Monnereau L, Mall HE, Semeril D, Matt D, Toupet L (2014) Eur J Inorg Chem 2014:1364–1372
Li JH, Liu WH (2004) J Org Chem 6:2809–2811
Blakemore JD, Chalkley MJ, Farnaby JH, Guard LM, Hazari N, Incarvito CD, Luzik ED, Suh HW (2011) Organometallics 30:1818–1829
Liu T, Zhao X, Shen Q, Lu L (2012) Tetrahedron 68:6535–6547
Susanto W, Chu CY, Ang WJ, Chou TC, Lo LC, Lam Y (2012) Green Chem 14:77–80
Mino T, Shirae Y, Sakamoto M, Fijita T (2005) J Org Chem 70:2191–2194
Xu C, Gong JF, Guo T, Zhang YH, Wu YJ (2008) J Mol Catal A Chem 279:69–76
Hanhan ME, Martinez-Manez R, Ros-Lis RJ (2012) Tetrahedron Lett 53:2388–2391
Kostas ID (2008) Inorg Chim Acta 361:1562–1565
Tenchiu AC, Ventouri IK, Ntasi G, Palles D, Kokotos G, Demertzi DK, Kostas ID (2015) Inorg Chim Acta 435:142–146
Verma PR, Mandal S, Gupta P, Mukhopadhyay P (2013) Tetrahedron Lett 54:4914–4917
Yan H, Chellan P, Li T, Mao J, Chibale K, Smith GS (2013) Tetrahedron Lett 54:154–157
Dutta J, Bhattacharya S (2013) RSC Advances 3:10707–10721
Dutta J, Datta S, Seth DK, Bhattacharya S (2012) RSC Adv 2:11751–11763
Paula P, Datta S, Haldera S, Acharyya R, Basuli F, Butcher RJ, Peng SM, Lee GH, Castineiras A, Drew MGB, Bhattacharya S (2011) J Mol Catal A Chem 344:62–73
Pandiarajan D, Ramesh R, Liu Y, Suresh R (2013) Inorg Chem Commun 33:33–37
Lobana TS, Kumari P, Hundal G, Butcher RJ, Castineiras A, Akitsu T (2013) Inorg Chim Acta 394:605–615
Pelosi G, Bisceglie F, Bignami Ronzi P, Schiavone P, Re MC, Casoli C, Pilotti E (2010) J Med Chem 53:8765–8769
Zhang WH, Chien SW, Hor TSA (2011) Coord Chem Rev 255:1991–2024
Kalinowski J, Fattori V, Cocchi M, Williams JAG (2011) Coord Chem Rev 255:2401–2425
Guerchais V, Fillaut JL (2011) Coord Chem Rev 255:2448–2457
Borah G, Boruah D, Sarmah G, Bharadwaj SK, Bora U (2013) Appl Organomet Chem 27:688–694
Dewan A, Bora U, Borah G (2014) Tetrahedron Lett 55:1689–1692
Gogoi A, Dewan A, Borah G, Bora U (2015) New J Chem 39:3341–3344
Borah G, Sarmah PP, Boruah D (2015) Bull Korean Chem Soc 36:1226–1230
Saikia B, Ali AA, Boruah PR, Sarma D, Barual NC (2015) New J Chem 39:2440–2443
Agarwala BV, Reddy PSN (1988) Trans Met Chem 13:187–189
Tang YQ, Lu M, Shao LX (2011) J Organomet Chem 696:3741–3744
Nakamoto K (1997) Infrared and raman spectra of inorganic and coordination compounds, 5th edn. Wiley, New York
Desiraju GR, Steiner T (1999) The weak hydrogen bond. Oxford University Press, Oxford
Freytag M, Jones PG (2000) Chem Commun 4:277–278
Fiedler D, Leung DH, Bergman RG, Raymond KN (2005) Acc Chem Res 38:351–360
Watson JD, Crick FHC (1953) Nature 171:737–738
Gogoi N, Begum T, Bora U, Gogoi PK (2015) RSC Adv 5:95344
Kitamura Y, Sako S, Tsutsui A, Monguchi Y, Maegawa T, Kitade Y, Sajiki H (2010) Adv Synth Catal 352:718–730
Pathak A, Singh AP (2016) J Porous Mater. doi:10.1007/s10934-016-0266-0
Yang S, Dong J, Yao Z, Shen C, Shi X, Tian Y, Lin S, Zhang X (2014) Sci Rep 4:4501
Jin Y, Zhao J, Li F, Jia W, Liang D, Chen H, Li R, Hu J, Ni J, Wu T, Zhong D (2016) Electrochim Acta 220:83–90
Acknowledgements
The authors acknowledge the analytical services provided by SAIF IISC, Bangalore; SAIF, CIL, Punjab University, Chandigarh; Department of Chemical Sciences, Tezpur University, Assam, India; SAIF, IIT Madras; STIC, Kochi University, Kochi and IIT, Kanpur. The authors are also grateful to UGC, New Delhi, India, for financial support under the SAP-DRS-I program (2016–2021).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Baruah, J., Gogoi, R., Gogoi, N. et al. A thiosemicarbazone–palladium(II)–imidazole complex as an efficient pre-catalyst for Suzuki–Miyaura cross-coupling reactions at room temperature in aqueous media. Transit Met Chem 42, 683–692 (2017). https://doi.org/10.1007/s11243-017-0174-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-017-0174-4