
Abstract
Two new cobalt(II) coordination polymers, [Co(HL)(bbe)] n (1) and [Co(HL)(bbop)] n (2) (H3L = 5-(carboxymethoxy)isophthalic acid, bbe = 1,2-bis(benzimidazol-2-yl)ethane, bbop = 1,3-bis(benzimidazol-2-yl)-2-oxapropane), were synthesized and characterized by elemental analyses, IR spectra, single-crystal X-ray diffraction and thermogravimetric analyses. Both complexes exhibit 1D chain structures, constructed from HL2− anions bridging metal ions. The 1D chains are further connected by hydrogen bonds and π–π stacking interactions to form the 3D supramolecular architectures. The cobalt centers display different coordination environments, with a tetrahedral geometry in 1 and a distorted tetragonal pyramid in 2. The DNA binding properties of both complexes were investigated by UV absorption spectroscopy. The electrochemical properties of both complexes were studied by cyclic voltammetry.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
There is an increasing interest in the design and preparation of metal–organic coordination polymers, owing to their intriguing structures and potential applications in catalysis, magnetism, luminescent materials, gas absorption, etc. [1–6]. In recent years, particular attention has been devoted to the coordination polymers constructed from aromatic polycarboxylic acids and nitrogen heterocycles as mixed organic building blocks [7–13]. These two types of organic ligands were chosen for the following reasons: (1) Organic polycarboxylic acids possess a variety of coordination modes; (2) flexible bis(nitrogen heterocycle) ligands often adopt different configurations to meet the requirements of coordination geometries of metal ions; (3) polydentate aromatic acids and imidazole compounds can all act as both H-bond acceptors and donors; and (4) the extent of deprotonation of polydentate aromatic acids and imidazole-based compounds depends on the reaction pH values. All these properties facilitate the formation of unusual structural and characteristic coordination polymers.
Recently, quite a few mixed-ligand metal–organic coordination polymers with different structures and functions have been reported [14–20]. However, to the best of our knowledge, studies on the mixed-ligand transition metal coordination polymers containing 5-(carboxymethoxy)isophthalic acid (H3L) and bis-benzimidazole ligands are less reported. In this paper, two new Co(II) coordination polymers, [Co(HL)(bbe)] n (1) and [Co(HL)(bbop)] n (2) (bbe = 1,2-bis(benzimidazol-2-yl)ethane, bbop = 1,3-bis(benzimidazol-2-yl)-2-oxapropane), were synthesized and structurally characterized. In addition, the DNA binding and electrochemical properties of both complexes were investigated.
Experimental
Materials and methods
All reagents and chemicals were of analytical grade purity and used without further purification. The organic ligands H3L [21], bbe and bbop [22] were prepared by the procedures reported. Salmon sperm DNA was purchased from Shanghai Huashun Biological Engineering Company. Concentrated stock solutions of dsDNA were prepared in 50 mmol L−1 NaCl–5 mmol L−1 Tris–HCl (pH 7.2). Solutions of dsDNA in 50 mmol L−1 NaCl–5 mmol L−1 Tris–HCl gave the ratio of UV absorbance at 260 and 280 nm, A 260/A 280 = 1.919, indicating that the dsDNA was sufficiently free of protein [23]. The concentration of dsDNA in base pairs was determined by UV absorbance at 260 nm, and the extinction coefficient (ε 260) was taken as 6600 L mol−1 cm−1 [24]. ssDNA was prepared according to an established procedure [25].
The C, H and N contents were determined by using an Elementar Vario EL III analyzer. Infrared spectra were recorded from KBr pellets by a Nicolet 510P FT-IR spectrometer. Thermogravimetric analyses (TGA) were performed on a Perkin-Elmer TG-7 analyzer heated from room temperature to 900 °C under an atmosphere of air at heating rate of 10 °C min−1. Ultraviolet and visible spectra (UV–Vis) were measured using a Cary 500 UV–Vis–NIR spectrophotometer.
Synthesis of complex 1
A mixture of CoCl2·6H2O (0.120 g, 0.5 mmol), bbe (0.131 g, 0.5 mmol), H3L (0.120 g, 0.5 mmol), NaOH (0.060 g, 1.5 mmol) and 10 mL of H2O was placed in a 20-mL Parr Teflon-lined stainless steel vessel. The vessel was sealed and heated to 160 °C for 72 h. Then, the reactant mixture was cooled to room temperature, leading to the formation of violet crystals 1. Yield 62 % (based on CoCl2·6H2O). Anal. Calcd for C26H20N4O7Co (%): C, 55.82; H, 3.60; N, 10.02. Found: C, 55.93; H, 3.51; N, 9.90. IR (KBr, cm−1): 3424 (m), 3169 (m), 3108 (s), 2916 (m), 2874 (m), 2798 (m), 2646 (m), 1695 (s), 1640 (s), 1610 (s), 1577 (s), 1536 (m), 1455 (s), 1414 (s), 1376 (s), 1335 (s), 1312 (s), 1294 (m), 1280 (s), 1269 (s), 1234 (s), 1129 (m), 1079 (m), 1053 (m), 1006 (m), 939 (m), 910 (m), 885 (m), 825 (m), 800 (m), 765 (s), 747 (s), 701 (m), 669 (m), 645 (m), 589 (m), 499 (m), 475 (m), 447 (m), 405 (m).
Synthesis of complex 2
A mixture of CoCl2·6H2O (0.240 g, 1 mmol), bbop (0.278 g, 1 mmol), H3L (0.226 g, 1 mmol), NaOH (3 mL 1 mol L−1) and 10 mL of H2O was placed in a 20-mL Parr Teflon-lined stainless steel vessel. The vessel was sealed and heated to 180 °C for 72 h. Then, the reactant mixture was cooled to room temperature, leading to the formation of blue crystals 2. Yield 56 % (based on CoCl2·6H2O). Anal. Calcd for C26H20N4O8Co (%): C, 54.27; H, 3.50; N, 9.74. Found: C, 54.35; H, 3.44; N, 9.66. IR (KBr, cm−1): 3431 (s), 3075 (s), 2922 (s), 2796 (m), 1741 (m), 1692 (s), 1610 (s), 1580 (s), 1537 (s), 1490 (m), 1455 (s), 1416 (s), 1386 (s), 1331 (s), 1315 (s), 1274 (s), 1239 (s), 1143 (s), 1131 (s), 1082 (s), 1043 (m), 997 (m), 942 (m), 911 (m), 879 (m), 843 (m), 762 (s), 752 (m), 703 (s), 632 (m), 590 (m), 489 (m), 427 (m).
X-ray crystallography
Single-crystal X-ray diffraction data of the complexes 1 and 2 were collected on a Bruker SMART 1000 CCD diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å) using ω scan mode at room temperature. Data reduction and cell refinement were performed using SAINT [26]. Intensity data were corrected for Lp factors and empirical absorption. The structures of 1 and 2 were solved by direct methods and expanded by using Fourier differential techniques with SHELXTL [27]. All non-hydrogen atoms were located with successive difference Fourier syntheses. The structure was refined by full-matrix least-squares methods on F 2 with anisotropic thermal parameters for all non-hydrogen atoms. The hydrogen atoms of free COOH 1 and 2 were located from the E-maps. The other hydrogen atoms were geometrically fixed and allowed to ride on the parent atoms to which they are attached. The main crystallographic data and structural refinements for complexes 1 and 2 are presented in Table 1. The selected bond distances and angles, and the hydrogen bond data for complexes 1 and 2 are listed in Tables 2 and 3, respectively.
Electrochemical studies
The electrochemical experiments were performed on a CHI 832B electrochemical analyzer (Shanghai CHI Instrument Company, China) using a three-electrode system composed of a carbon paste electrode (CPE) as a working electrode, SCE/(saturated) KCl as a reference electrode, and Pt wire as an auxiliary electrode. A 0.2-mol L−1 Britton–Robinson buffer solution (B-R buffer) was used as buffer solution. The pH value was measured with a PHS-3D pH meter (Shanghai LeiCi Device Works, Shanghai, China) with a combined glass-calomel electrode. The cyclic voltammetry measurements were taken on a CPE in B-R buffer solution (pH 4.5) using 0.1 mol L−1 KCl as supporting electrode at 25 °C with a scan rate of 2–200 mV s−1. The solutions of complex were prepared by first dissolving the complex in the appropriate amount of DMSO and then diluting with B-R buffer.
Results and discussion
Syntheses
The hydrothermal method is an effective synthetic route to prepare polymeric solids with better crystallinity. In this work, two new coordination polymers were prepared by changing the molar ratio of starting materials to solvent. Compound 1 was obtained from the hydrothermal reaction of CoCl2, bbe, H3L, NaOH and H2O in a molar ratio of 1:1:1:3:2 at 160 °C. Compound 2 was generated from the hydrothermal reaction of CoCl2, bbop, H3L, NaOH and H2O in the molar ratio of 1:1:1:3:1 at 180 °C. The results show that this system is solvent and temperature dependent.
IR spectral studies
In the IR spectra of complexes 1 and 2, the characteristic bands at 1610 and 1376 cm−1 for 1, and 1610 and 1386 cm−1 for 2 are, respectively, attributed to the asymmetric vibration and the symmetric vibration of carboxylate groups of HL2− ligand. The separations between ν as(COO−) and ν s(COO−) are 234 and 224 cm−1 for 1 and 2, respectively, indicating that the carboxylate groups adopt unidentate coordination modes. The broad bands at 3424 cm−1 for 1 and 3431 cm−1 for 2 are assigned to the O–H stretching vibrations of COOH groups. The presence of strong peaks at 1695 cm−1 for 1 and 1692 cm−1 for 2 suggests the H3L ligand adopts the partly deprotonated HL2− form. The Co–O and Co–N vibrations are also observed in the far infrared regions of IR spectra of 1 and 2 [28]. These are consistent with the X-ray crystal structural analysis.
Crystal structure of [Co(HL)(bbe)] n (1)
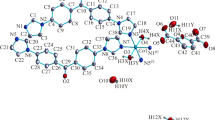
Single-crystal X-ray diffraction analysis reveals that complex 1 crystallizes in the triclinic crystal system of Pī space group in which the asymmetric unit contains one Co(II) ion, one HL2− anion and one bbe ligand. The central ion Co(II) is four-coordinated and exhibits a tetrahedral geometry defined by one rigid carboxylate oxygen atom from one HL2− ligand and one flexible carboxylate oxygen atom from another HL2− ligand, and two nitrogen atoms from one bbe ligand (Fig. 1). All coordinate bond lengths (Co–O 1.946(2) and 1.963(2) Å, Co–N 2.004(2) and 2.027(2) Å) fall in the normal range found in the other related complexes [11–17]. The angles around atom Co1 fall in the normal range of 100.21(8)–121.37(8)°. There exist three carboxylate groups in H3L molecule, but only two are deprotonated. Each HL2− anion as a μ 2-bridge connects two metal ions to form a 1D infinite chain in the b direction with the Co–Co distances in the chain 11.354(2) Å. These 1D chains are linked with each other via intermolecular O4–H4B···O3 [O4···O3 2.627(3) Å, O4–H4B–O3 176°] hydrogen bonds to form a 1D ladder structure (Fig. 2). The 1D ladders are further connected by N2–H2A···O1 (N2···O1 2.748(3) Å, N2–H2A–O1 150°) and N4–H4A···O6 (N4···O6 2.677(3) Å, N4–H4A–O6 160°) hydrogen bonds to form the 2D network architecture (Fig. 3). The related hydrogen bond data are shown in Table 3. Moreover, there exist three modes of π–π stacking interactions in complex 1 (Fig. 4). The centroid–centroid distances between N1-containing imidazole rings, N3-containing imidazole rings and benzene rings in HL2− ligands are 3.536(2), 3.706(2) and 3.702(1) Å, respectively. These strong π–π stacking interactions further extend the 2D layers into a 3D supramolecular structure.

Molecular structure and coordination environment of 1. The hydrogen atoms are omitted for clarity. Symmetry codes: #1 = x, y + 1, z

1D ladder structure of 1 formed by hydrogen bonds (dotted lines)

2D structure of 1 formed by hydrogen bonds (dotted lines). Benzene rings in bbe molecules are omitted for clarity

π–π stacking interactions in complex 1
Crystal structure of [Co(HL)(bbop)] n (2)
Compared with bbe, the bbop molecule adds an oxygen atom in the alkyl chain. When using bbop as an auxiliary ligand, complex 2 was obtained. Complex 2 crystallizes in the triclinic Pī space group, and the asymmetric unit contains one Co(II) ion, one HL2− anion and one bbop ligand (Fig. 5). The central ion Co(II) is five-coordinated by two nitrogen atoms (N1, N3) and three oxygen atoms [O8, O2, O7#1 (symmetry code #1 = x, y, z + 1)]. N1, N3 and O8 are from one bbop ligand, O2 from rigid carboxylate of one HL2− ligand and O7#1 from flexible carboxylate of another HL2− ligand. The Co1–O8 has a longer bond distance (2.588 Å), while N1–Co1–O8 (69.55°) and N3–Co1–O8 (69.04°) show smaller bond angles. This is due to chelation of bbop to the metal. The other coordination bond lengths [Co–O 1.979(2) and 2.011(2) Å, Co–N 2.043(2) and 2.049(2) Å] and angles [90.17(8) to 167.73(8)°] are in the normal ranges and comparable to those in the structure-related complexes [11–17]. The coordination geometry of the metal atom in 2 can be described as a distorted tetragonal pyramid.

Molecular structure and tetragonal pyramidal coordination environment of 2. The hydrogen atoms are omitted for clarity. Symmetry codes: #1 = x, y, z + 1
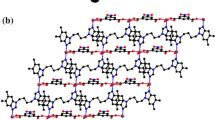
In complex 2, bbop as a tridentate ligand coordinates with the metal to form two stable five-membered rings. Two benzimidazoles are coplanar with a dihedral angles of 19.58(11)°. Two deprotonated carboxylate groups bridge two metal ions to form a 1D chain. The 1D chains are linked into a 1D ladder via O–H···O hydrogen bonds. The 1D ladders are further connected with the 2D architecture by N–H···O hydrogen bonds. The 2D structures are extended into a 3D supramolecular structure by the π–π stacking interactions. These structural characteristics are similar to those in complex 1. The related hydrogen bond data are shown in Table 3. The π–π stacking data are 4.053(2), 4.025(1) and 3.419(2) Å, respectively. The 2D network of 2 is shown in Fig. 6.

2D network of 2 formed by hydrogen bonds
Thermal analysis
Thermogravimetric and differential thermogravimetric analyses were carried out for compounds 1 and 2 in order to characterize the compounds more fully in terms of thermal stability (Figs. 7). For compound 1, no weight loss is observed from room temperature to 340 °C, and after this temperature, the compound decomposes. The weight loss corresponding to the release of two organic ligands (two endothermic peaks at 411 and 480 °C, respectively) is observed from 340 to 500 °C (obsd 83.78 %, calcd 85.17 %). The remaining residue corresponding to the formation of Co2O3 is 16.23 % (calcd 14.83 %). Compound 2 does not show any weight loss from room temperature to 260 °C and shows the 84.71 % weight loss from 260 to 400 °C, corresponding to the decomposition of two organic ligands (calcd 85.58 %, two endothermic peaks at 304.2 and 325.6 °C, respectively). The remaining residue is 15.29 %, corresponding to the formation of Co2O3 (calcd 14.41 %).

a TG and DTG curves of complex 1. b TG and DTG curves of complex 2
DNA binding studies
Electronic absorption spectra of complexes 1 and 2 were recorded in the range of 200–600 nm. In the absorption spectra, the broad absorption band at 240 nm for 1 and 239 nm for 2 could be assigned to intraligand π → π* transitions of the benzene ring in HL2−. The two sharp absorption bands at 271 and 277 nm for 1, and 270 and 276 nm for 2 could be assigned to intraligand π → π* transitions of benzimidazole in bbe or bbop, respectively. It can be seen from Figs. 8 and 9 that the absorption spectra of both 1 and 2 show obvious decrease after adding dsDNA to the complex solution, but these absorptions had no redshift for both 1 and 2. This suggests that the interaction between complexes and DNA might be electrostatic [29].

a UV–Vis absorption spectra of complex 1 (7.475 × 10−5 mol L−1) and 1 + DNA (2.213 × 10−5 mol L−1). b Plot of [DNA]/(ε a − ε f) versus [DNA] for absorption titration of DNA with complex 1

a UV–Vis absorption spectra of complex 2 (7.475 × 10−5 mol L−1) and 2 + DNA (2.213 × 10−5 mol L−1). b Plot of [DNA]/(ε a − ε f) versus [DNA] for absorption titration of DNA with complex 2
DNA binding properties of complexes 1 and 2 were studied by UV–Vis titration. For the UV–Vis titration experiment, 20 μL solutions of DNA (2.213 × 10−5 mol L−1 in water) were added to a 3.0-mL solution of complexes (7.475 × 10−5 mol L−1 in water) by a micropipette. Because of the limited solubility of complexes in water, the titration experiments were carried out in the presence of a small amount of DMSO. The intrinsic binding constant K b was determined from the collected absorbance data of the titration experiments at the specified wavelength and the following equation [30]:
where [DNA] is the concentration of DNA, ε f, ε a and ε b refer to the extinction coefficients for the free complex, for each addition of DNA to the complex and for the complex in the fully bound form, respectively. When [DNA]/(ε a − ε f) is plotted against [DNA], a straight line is obtained. The binding constant K b obtained by the ratio of slope to intercept is 4.03 × 104 L mol−1 for 1 (Fig. 8) and 1.48 × 104 L mol−1 for 2 (Fig. 9).
In order to clarify what species interact with the DNA, compound 2 was measured by high-resolution mass spectra (HRMS) using a Bruker Maxis UHR-TOF with Ion Source APCI system (Fig. 10). The signals of m/z > M+, e.g., 621.31 and 1194.82, in mass spectra show that compounds are polymers in the solution used for the DNA binding studies. This shows further that the complexes interact with DNA in the form of polymers.

Mass spectra of 2 in the DMSO–water solution
Electrochemical studies
Cyclic voltammograms of complex 1 and 2 in 0.04 mol L−1 B-R buffer solution at pH 4.5 are shown in Figs. 11 and 12. The complexes show similar electrochemical behaviors. Complex 1 showed an oxidation peak at 0.217 V with a peak current 3.614 × 10−7 A, and complex 2 showed an oxidation peak at 0.246 V with a peak current 5.873 × 10−7 A. This indicates that the electrochemical behaviors of both complexes in electrode involve an irreversible process [31]. For this process, the electron transfer number n can be estimated according to the equation \(\left| {{E_{{\rm{pa}}}} - {E_{{\rm{pa}}}}/2} \right| = 47.7/n\alpha\) [32]. When α is assumed to be 0.5, n = 1.36 for 1 and 1.18 for 2 are obtained. Thus, about one electron is involved in the redox of both complexes 1 and 2, corresponding to the process CoII↔CoIII.

a Cyclic voltammogram of complex 1 (7.475 × 10−5 mol L−1 complex in 0.04 mol L−1 B-R buffer solution at pH 4.5). b Plot of peak current (I pa) against square root of scan rate (v 1/2)

a Cyclic voltammogram of complex 2 (7.475 × 10−5 mol L−1 complex in 0.04 mol L−1 B-R buffer solution at pH 4.5). b Plot of peak current (I pa) against square root of scan rate (v 1/2)
The effect of varying the scan rate (0.04–0.16 V s−1) on the oxidation peak current of complexes 1 and 2 was also studied. The results showed that the oxidation peak currents of both complexes increased linearly with the square root of scan rate (see Fig. 11 for 1 and Fig. 12 for 2), suggesting that oxidation of the complexes at the CPE is diffusion controlled [33]. This observation is different from {[Co(L)(mip)(H2O)]H2O} n and [Co(L)(oba)] n , [L = N,N′-bis(pyridine-3-yl)pyridine-3,5-dicarboxamide, H2mip = 5-methylisophthalic acid, H2oba = 4,4′-oxybis(benzoic acid)], where the surface-controlled process was reported [34]. The difference might be attributed to the different structures of the complexes.
Conclusion
Two new cobalt(II) coordination polymers were synthesized by the hydrothermal reactions of two structurally similar bis(benzimidazole) ligands with 5-hydroxyisophthalic acid and cobalt dichloride, and their structures were characterized by elemental analyses, IR spectra, thermogravimetric analyses, electronic absorption spectra, cyclic voltammetry and single-crystal X-ray diffraction. In both hydrogen-bonded 3D supramolecular architectures, the different coordination modes of HL2− anion and benzimidazole ligands lead to diverse coordination geometries of the central ions, and these slight structural differences further lead to the difference in the DNA binding and electrochemical properties of these complexes.
Supplementary material
CCDC 1049400 and 1049401 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Li JR, Kuppler RJ, Zhou HC (2009) Chem Soc Rev 38:1477–1504
Farha OK, Malliakas CD, Kanatzidis MG, Hupp JT (2009) J Am Chem Soc 132:950–952
Zhu SL, Ou S, Zhao M, Shen H, Wu CD (2015) Dalton Trans 44:2038–2041
Ahmad M, Sharma MK, Das R, Poddar P, Bharadwaj PK (2012) Cryst Growth Des 12:1571–1578
Allendorf MD, Bauer CA, Bhaktaa RK, Houka RJT (2009) Chem Soc Rev 38:1330–1352
Gong Y, Zhang MM, Hua W, Sun JL, Shi HF, Jiang PG, Liao FH, Lin JH (2014) Dalton Trans 43:145–151
Hu JS, Shang YJ, Yao XQ, Qin L, Li YZ, Guo ZJ, Zheng HG, Xue ZL (2010) Cryst Growth Des 6:2676–2684
Kan WQ, Liu YY, Yang J, Liu YY, Ma JF (2011) CrystEngComm 13:4256–4269
Chen J, Feng YL, Jiang ZG, Cheng JW (2011) CrystEngComm 13:6071–6076
Wang GY, Yang LL, Li Y, Song H, Ruan WJ, Chang Z, Bu XH (2013) Dalton Trans 42:12865–12868
Wen YH, Dou RT, Yao K, Xu GF (2015) J Coord Chem 68:38–54
Li HH, Ma YJ, Zhao YQ, Cui GH (2015) Transit Met Chem 40:21–29
Wen YH, Xu GF, Yao K, Dou RT, Guo JX (2014) Z Anorg Allg Chem 640:2091–2096
Karmakar A, Titi HM, Goldberg I (2011) Cryst Growth Des 11:2621–2636
Liu JQ, Wang YY, Huang YS (2011) CrystEngComm 13:3733–3740
Wang XX, Ma YJ, Li HH, Cui GH (2015) Transit Met Chem 40:99–108
Wang GH, Lei YQ, Wang N, He RL, Jia HQ, Hu NH, Xu JW (2010) Cryst Growth Des 10:534–540
Hao JM, Zhao YN, Yu BY, Hecke KV, Cui GH (2014) Transit Met Chem 39:741–753
Wang XX, Zhao YN, Li GY, Cui GH (2014) Transit Met Chem 39:653–660
Wang XX, Zhao YN, Li GY, Cui GH (2014) Transit Met Chem 39:875–882
Cao XY, Zhang J, Cheng JK, Kang Y, Yao YG (2004) CrystEngComm 6:315–317
Liu HY, Wu H, Yang J, Liu YY, Liu B, Liu YY, Ma JF (2011) Cryst Growth Des 11:2920–2927
Marmur J (1961) J Mol Biol 3:208–218
Reichmann ME, Rice SA, Thomas CA, Doty P (1954) J Am Chem Soc 76:3047–3053
Pang DW, Abruna HD (1998) Anal Chem 70:3162–3169
Bruker AXS (2006) APEX2, SAINT. Bruker AXS Inc., Madison
Sheldrick GM (2008) Acta Crystallogr A 64:112–122
Nakamoto K (1986) Infrared and Raman spectra of inorganic and coordination compounds, 4th edn. Wiley-Interscience, New York
Long EC, Barton JK (1990) Acc Chem Res 23:271–273
Baguley BC, Bret ML (1984) Biochemistry 23:937–943
Li W, Li CH, Yang YQ, Chen ZM, Wang Y (2008) Chin J Inorg Chem 24:1360–1364
Galus Z (1976) Fundamentals of electrochemical analysis. Ellis Horwood, Chichester
Bard AJ, Faulkner LR (1980) Electrochemical methods fundamentals and applications. Wiley, New York
Wang XL, Chen NL, Liu GC, Luan J, Qu Y (2014) Transit Met Chem 39:681–690
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 20971076).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wen, HL., Wang, DM., Mu, XW. et al. Syntheses, crystal structures, DNA binding and electrochemical properties of two mixed-ligand cobalt(II) coordination polymers constructed from 5-(carboxymethoxy)isophthalic acid and bis-benzimidazole ligands. Transition Met Chem 40, 509–517 (2015). https://doi.org/10.1007/s11243-015-9943-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-015-9943-0