
Abstract
Two cobalt(II) coordination polymers, [Co(L1)(tbi)(H2O)] n (1) and [Co(L2)(tbi)] n (2) (L1 = 1,4-bis(benzimidazole)butane, L2 = 1,4-bis(2-methylbenzimidazole)butane, H2tbi = 5-tert-butyl isophthalic acid) have been synthesized under hydrothermal conditions and characterized by physicochemical and spectroscopic methods as well as by single-crystal X-ray diffraction analysis. Both complexes exhibit similar 2D (4,4) layer structures, constructed from tbi2− and bis(benzimidazole)-based bridging ligands. The cobalt centers display different coordination environments, with an octahedral geometry in 1 and a distorted square-pyramidal configuration in 2. The thermal stabilities, fluorescence and catalytic properties of both complexes have been investigated.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The design and synthesis of metal–organic coordination polymers (MOCPs) have received much attention in recent decades due to their fascinating structures and potential applications in the fields of catalysis, luminescence, conductivity, magnetism and so on [1–5]. Although rapid progress in the construction of MOCPs has been made, it is still a challenge to control their final architectures and properties because the self-assembly of these compounds is influenced by many factors, such as organic ligands, pH, choice of metal, temperature, solvent systems [6–9]. In particular, the appropriate choice of organic ligands is crucial to obtain target MOCPs [10–12]. Therefore, significant interest has arisen into the structural tuning of MOCPs by the rational selection of bridging ligands. Organic aromatic carboxylates have been widely used as O-donor bridging ligands to construct novel metal–organic coordination polymers, owing to their versatile coordination modes and good stability. In particular, derivatives of 1,3-benzenedicarboxylic acid (isophthalic acid, H2ip), such as H2tbi (5-tert-butyl isophthalic acid), are of interest. The latter has a sterically hindered substituent, and the resulting subtle difference with H2tip may significantly influence the resulting MOCPs [13, 14]. Flexible bis(benzimidazole) derivatives have attracted much attention within crystal engineering and supramolecular chemistry by our and other groups, due to their good coordination ability and versatile conformations [15–20]. However, the incorporation of flexible bis(benzimidazole) ligands into Co(II)-5-tert-butylisophthalic MOCPs has been rarely reported.
Synthetic azo dyes such as Congo red and methyl orange have versatile applicability in the textile, pharmaceutical, food and cosmetics industries. Estimates indicate that approximately 1–15 % of synthetic textile dyes are lost in wastewater streams during manufacturing or processing operations [21]. Since most azo dyes are non-biodegradable, as well as toxic and/or carcinogenic, the effective removal of dyes from waste effluents is of significant environmental importance [22]. Fenton oxidation technologies utilize H2O2 which generates hydroxyl radicals that are very effective in degrading organic pollutants, but the uncatalyzed reaction rates are generally slow at ambient temperature, and activation of H2O2 is necessary to accelerate the process. MOCPs are potential candidates to catalyze the degradation of such organic pollutants in Fenton-like processes.
In this work, we have selected two related flexible bis(benzimidazole) ligands (L1 and L2 are bis(benzimidazole) ligands with aliphatic cores that only differ by a methyl group) and H2tbi (see Scheme 1) as co-ligand to construct two new Co(II) MOCPs, namely [Co(L1)(tbi)(H2O)] n (1) and [Co(L2)(tbi)] n (2) (L1 = 1,4-bis(benzimidazole)butane, L2 = 1,4-bis(2-methylbenzimidazole)butane, H2tbi = 5-tert-butyl isophthalic acid). In addition, the fluorescence properties and catalytic performances of both complexes for the degradation of Congo red dye have been investigated.

Two major reactions of this paper
Experimental
Materials and measurements
All reagents and solvents were obtained from commercial sources and used without further purification. L1 and L2 were prepared according to the literature procedure [23]. Elemental analyses were obtained on a Perkin-Elmer 240C elemental analyzer. FTIR spectra were recorded from KBr pellets in the range of 4,000–400 cm−1 on an Avatar 360 (Nicolet) spectrophotometer. Thermogravimetric analysis (TGA) was carried out on a NETZSCH TG 209 thermal analyzer from room temperature to 800 °C with a heating rate of 10 °C/min under N2 atmosphere. Luminescence spectra for the powdered solid samples were measured at room temperature on a Hitachi F-7000 fluorescence spectrophotometer. X-ray powder diffraction (XRPD) measurements were made on a Rigaku D/Max-2500PC X-ray diffractometer using Cu-Kα radiation (λ = 0.1,542 nm) and ω − 2θ scan mode at 293 K.
Synthesis of complex 1
A mixture of CoCl2·6H2O (0.1 mmol, 23.8 mg), L1 (0.1 mmol, 29.0 mg) and H2tbi (0.1 mmol, 22.2 mg) were dissolved in distilled water (10 mL). The mixture was sealed in a 25-mL Teflon-lined stainless steel vessel and heated at 140 °C for 3 days under autogenous pressure. After the mixture was cooled to room temperature at a rate of 5 °C/h, pink block-shaped crystals suitable for single-crystal X-ray diffraction were obtained with a yield of 35 % (based on CoCl2·6H2O). Calcd. for C30H32CoN4O5 (Fw = 587.54): C 61.3, H 5.5, N 9.5 %; found: C 61.0, H 5.1, N 9.7 %. FTIR (KBr pellet, cm−1): 3,327 vs, 2,945 m, 1,616 s, 1,548 m, 1,457 m, 1,372 s, 1,255 m, 915 w, 734 s, 659 w.
Synthesis of complex 2
The synthetic method for complex 2 was similar to that for 1, except that L2 (0.1 mmol, 31.8 mg) was used instead of L1. Purple block crystals of complex 2 suitable for single-crystal X-ray diffraction were obtained in 52 % yield (based on CoCl2·6H2O). Calcd. for C32H34CoN4O4 (Fw = 597.58): C 64.3, H 5.7, N 9.4 %; found: C 64.6, H 5.3, N 9.1 %. FTIR (KBr pellet, cm−1): 2,953 m, 1,627 s, 1,575 m, 1,474 w, 1,342 s, 1,020 w, 749 s, 625 w.
X-ray crystallography
Crystallographic data for complexes 1 and 2 were collected on a Bruker Smart 1000 CCD diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 293 K. Semi-empirical absorption corrections were applied using the SADABS program [24]. The structures were solved by direct methods and refined on F 2 by full-matrix least-squares using the SHELXTL program package [25]. All non-hydrogen atoms were refined anisotropically. Complex 2 showed disorder in C30, C31 and C32 of the 5-tert-butyl substituent groups; these atoms were refined with a split model with site occupation factor 0.56, SADI for restraining distances with related disordered atoms. The crystallographic data for complexes 1 and 2 are listed in Table 1, and selected lengths and angles are presented in Table 2, respectively.
Results and discussion
Synthesis and spectral characterization of the complexes
Polycarboxylate-bis(benzimidazole) systems provide an important and flourishing branch of mixed-ligand MOCPs. Acid and base ligands are ideal partners that can compensate charge balance as well as coordination deficiency. Such MOCPs are generally synthesized by using solvent to induce the self-assembly of a regular framework. Such reactions can be carried out by mixing a solution of the required metal ion with a solution of the ligands at room temperature, or under hydrothermal/solvothermal conditions. Hydrothermal reactions have recently been demonstrated to be a versatile technique for the construction of MOCPs, and a series of flexible bis(benzimidazole) complexes have been synthesized by this method [15–18]. In the present work, the two title complexes were synthesized by the reaction of cobalt chloride with 5-tert-butyl isophthalic acid and flexible bis(benzimidazole in a 1:1:1 molar ratio under hydrothermal conditions. Both complexes 1 and 2 were obtained independent of the reactant molar ratio and the hydrothermal reaction temperature, as confirmed by IR spectra and elemental analysis.
In the IR spectra of the complexes, the main features are assigned to the carboxylates, water molecules and N-containing ligands. A broad band centered at 3,327 cm−1 for complex 1 may provide some evidence for the participation of water molecules in strong hydrogen bonding. Characteristic bands at 1,548 cm−1 for 1, and 1,575 cm−1 for 2 are assigned to the ν C=N stretching of the benzimidazole ring. There are no strong absorption peaks around 1,700 cm−1 for –COOH, demonstrating that all carboxyl groups of the organic moieties are deprotonated in both complexes. Strong characteristic bands at 1,616 and 1,457 cm−1 for complex 1 and at 1,627 and 1,474 cm−1 for complex 2 are attributed to the asymmetric and symmetric vibrations of the carboxyl groups, respectively. The values of Δυ[υ as(COO)–υ s(COO)] are 149 and 153 cm−1, respectively, indicating bridging coordination of the carboxylate group to the metal centers [26]. The slightly different separations may suggest the coexistence of mixed binding modes of the carboxylate groups in the anionic tbi2− ligands, which is also in agreement with the single-crystal X-ray diffraction results.
Structural analysis of the complexes
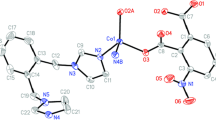
The structural analysis reveals that complex 1 crystallizes in the triclinic space group Pī. The asymmetric unit contains one Co(II) center, one L1 ligand, one anionic tbi2− ligand and one coordinated water ligand. As shown in Fig. 1a, the cobalt atom is six-coordinated by two N atoms from different L1 ligands, three O atoms from two tbi2− ligands and one water ligand to give an octahedral geometry. The best equatorial plane is formed by one chelating tbi2− carboxylate oxygen atom (O3 and O4), one bis-monodentate tbi2− carboxylate oxygen atom (O2A) (symmetry code: A = −1 + x, y, z) and the oxygen atom from the aqua ligand (O1 W). The apical positions are occupied by two nitrogen atoms (N1 and N3) from different L1 ligands. The Co–O bond distances vary in the range of 2.043(12)–2.236(13) Å, and the Co–N bond distances are 2.148(16) and 2.205(16) Å, respectively. The coordination angles range from 59.98(5) to 176.82(6)°, which are comparable to values observed for similar cobalt(II) complexes [19].

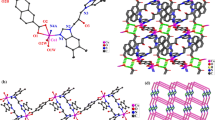
a Coordination environment of the Co atom in 1: Hydrogen atoms were omitted for clarity, and symmetry transformation was used to generating equivalent atoms: \( A = - 1+ x,y,z; \, B \, = 1+ x,y, \, - 1+ z \). b 2D (4,4) supramolecular structure in 1
In complex 1, the L1 ligand adopts trans-conformation and bridges adjacent Co(II) centers to assemble into infinite 1D [Co(L1)] n cation chains. The adjacent 1D chains are cross-connected by neighboring tbi2− ligands in monodentate and chelating coordination fashions to generate a 2D (4,4) grid structure, in which each 38-member parallelogram consists of four Co atoms at the corners connected by two L1 ligands and two tbi2− ligands, generating a [Co4(L1)2(tbi)2] unit for L1 and tbi2− bridging with the Co⋯Co distances of 13.988(9) and 10.156(9) Å, respectively (Fig. 1b). In addition, the O–H···O hydrogen bonds (O1 W–H1WA···O3B: 2.744(2) Å, 170°, symmetry codes: C = 1 − x, 2 − y, 2 − z) between the water ligands and carboxylate oxygen atoms and π–π stacking interactions between the imidazole rings from distinct L1 ligands (the center-to-center separations of 3.820 Å and slipping angles of β (γ) of 28.05°; Cg1: N3–C10–N4–C17–C11, symmetry codes: D = 1 − x, 2 − y, 1 − z) further enhance the stability of the final framework.
Complex 2 crystallizes in the orthorhombic space group Pbca and exhibits a 2D coordination network structure. The asymmetric unit of 2 consists of one Co(II) center, one L1 ligand and one tbi2− ligand. As shown in Fig. 2a, the Co atom is five-coordinated by three oxygen atoms (O1E, O2E, O4) (symmetry code: E = 0.5 − x, 0.5 + y, z) from different carboxylic groups of tbi2− ligands and two nitrogen atoms (N1, N3) from two distinct L2 ligands, furnishing a distorted square-pyramidal geometry, with a τ index of 0.42 [27]. The Co–O bond lengths vary from 1.955(14) to 1.968(15) Å, and the Co–N bond lengths are 2.027(17) and 2.046(17) Å, respectively. The bond angles at the Co center are in the range of 96.04(6)–121.16(7)°.

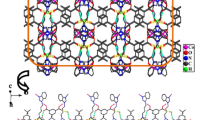
a Coordination environment of the Co atom in 2: Hydrogen atoms were omitted for clarity, and symmetry transformation was used to generating equivalent atoms: \( E = 0. 5- x, \, 0. 5+ y,z; \, F = - 0. 5+ x, \, 0. 5- y, - z \). b 1D zigzag chain connected by Co atoms and L2 ligands in 2. c 2D (4,4) supramolecular structure in 2. d The uninodal 4-connected 2D net with a point symbol of {44.62} topology network in 2
In complex 2, each L2 ligand adopts a twisted cis-conformation with a Ndonor⋯N–Csp3⋯Csp3 torsion angle of 163.62°, and the two benzimidazole arms bend in opposite directions to link two neighboring Co atoms, giving rise to a 1D [Co(L2)] n zigzag chain with a Co⋯Co distance of 9.185(6) Å (Fig. 2b). Furthermore, the carboxylic groups act in both monodentate and chelating coordination modes to connect two neighboring 1D zigzag chains, resulting in a 2D (4,4) network with a through-ligand Co–Co distance of 10.301(9) Å (Fig. 2c, d). The corrugated 2D network consists of one type of parallelograms, where the dimensions are 9.19 × 10.30 Å defined by Co⋯Co separations.
Similar to the present work, some related complexes, namely {[Co2(L3)2(tbi)2]·2H2O} n (3) [Co2(L4)(tbi)2(H2tbi)(bpp)(H2O)] n (4) and [Co(L5)(tbi)(H2O)] n (5) (L3 = 1,4-bis(imidazole)butane, L4 = 1,3-bis(4-pyridyl)propane, L5 = 4,4′-dipyridylamine) have been described previously [28–30]. In the present complexes 1 and 2, the tbi2− anions adopt the same coordination modes, involving one chelating and one bridging bidentate carboxylate groups. However, the two complexes show different coordination geometries (octahedral for 1, distorted square pyramidal for 2). The Co(II) centers are bridged by both tbi2− and flexible N-donor ligands to generate a 2D grid layer network for complex 1. When the H substituent is replaced by –CH3, as in complex 2, the result is a 2D corrugated layer structure. It is possible that the additional Me-group in 2 sterically prevents water from coordinating to the metal atom and the existence of hydrogen bonds or π–π stacking interactions in 1 may also have an effect on the final crystal structure.
In all of the complexes 1–5, the flexible N-containing ligands uniformly behave as bis-monodentate linkers to connect the Co centers, but exhibit different bending and rotating ability, which leads to different non-bonding Co⋯Co distances and distinct coordination geometries. As shown in Table 3, complex 1 features a 2D grid layer, while 2 displays a 2D corrugated layer, and 3, 4, 5 have a 2D helical layer, 2D bilayer, 2D twofold parallel layer, respectively. These results suggest that the different structures of 1–5 mainly stem from the different N-donor ligands used in these complexes.
Thermal behaviors and XRPD analysis
To characterize the present complexes more fully in terms of thermal stability, their thermal behaviors were examined by TGA (Fig. 3). For complex 1, two weight loss steps were observed. The first weight loss of 2.7 % can be attributed to the release of coordination water ligands in the temperature range of 112–139 °C (calcd. 3.1 %). A sharp weight loss is observed in the range 366–560 °C, corresponding to the decomposition of the organic ligands to leave a residue of CoO (calcd. 12.8 %; found. 13.7 %). In the range 420–590 °C, a weight loss of 88.3 % (calcd. 87.5 %) corresponds to decomposition of the organic components, leaving a CoO residue of 11.6 % (calcd. 12.5 %) for complex 2.

TG curves of compounds 1 and 2
To confirm the phase purity of both complexes, the XRPD patterns were obtained at room temperature and they are comparable to the corresponding simulated patterns calculated from the single-crystal X-ray diffraction data (Fig. 4).

a X-ray powder diffraction patterns of 1. b X-ray powder diffraction patterns of 2
Photoluminescence properties
The solid-state photoluminescence properties of complexes 1 and 2 and free ligands were investigated at room temperature. As depicted in Fig. 5, the free ligands L1 and L2 exhibit emission peaks at 399 and 375 nm under excitation at 335 nm, which can probably be assigned to the π → π* transitions [31]. Both complexes exhibit fluorescent emission under excitation at 250 nm with maxima at 356 nm for 1 and 361 nm for 2, respectively. For both complexes, a blue-shifted (43 nm for 1, 14 nm for 2) emission band is observed compared with the free L1 and L2 ligands. Therefore, the fluorescence behaviors of both complexes can be tentatively ascribed to ligand-to-metal charge transfer [32]. However, the emission bands of the carboxylate ligands originating from n − π* transitions are weak, and it is considered that the carboxylate ligands have no significant contribution to the fluorescence emission of complexes 1 and 2 in the presence of the N-donor ligand [33].

a Solid-state photoluminescence spectra of free L1 ligand and 1. b Solid-state photoluminescence spectra of free L2 ligand and 2
Catalytic properties
The catalytic degradation experiments were carried out according to the literature [34], and the results for decolorization of Congo red dye by using four different treatment processes are shown in Fig. 6. In the control experiment, when hydrogen peroxide was added to a solution of Congo red, there was no evident change for reaction times up to 130 min, showing that H2O2 alone was unable to decompose the azo dye effectively. In the presence of H2O2 and CoCl2, a degradation efficiency of 56 % was observed within 130 min. When complex 1 or 2 was introduced into the system, about 85 % or 72 % of the dye was decolored, respectively, after 130 min. A possible degradation mechanism can be suggested as follows [35];

Experimental results of the catalytic degradation of Congo red
On comparing to the degradation efficiencies of the four different treatment processes, it is obvious that free Co2+ ions have some catalytic effect, but with a lower efficiency than both complexes 1 and 2. The higher activity of complex 1 compared to complex 2 may arise from the presence of the water ligand in the former, which provides a labile substitution site for the production of hydroxyl radicals.
Conclusion
In summary, two new Co(II) metal–organic frameworks with tbi2− ligands and two flexible bis(benzimidazole) ligands have been synthesized and characterized. The subtle differences between complexes 1 and 2 in terms of structure and catalytic properties may result from the flexible N-bridging ligands bearing different substituent groups. Both complexes, especially 1, show promising catalytic activities in the decomposition of Congo red dye.
Supplementary materials
CCDC 993150 and 993151 contain the supplementary crystallographic data for the complexes 1 and 2. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Kreno LE, Leong K, Farha OK, Allendorf M, Van Duyne RP, Hupp JT (2012) Chem Rev 112:1105–1125
Masoomi MY, Morsali A (2012) Coord Chem Rev 256:2921–2943
Haldar R, Maji TK (2013) CrystEngComm 15:9276–9295
Du M, Li CP, Liu CS, Fang SM (2013) Coord Chem Rev 257:1282–1305
Xiao SL, Liu YG, Ma PJ, Cui GH (2013) Transit Met Chem 38:793–799
Liu GC, Huang JJ, Zhang JW, Wang XL, Lin HY (2013) Transit Met Chem 38:359–365
Zhang Y, Yang J, Yang Y, Guo J, Ma JF (2012) Cryst Growth Des 12:4060–4071
Kan WQ, Ma JF, Liu YY, Yang J (2012) CrystEngComm 14:2316–2326
Ming CL, Zhang H, Li GY, Cui GH (2014) Bull Korean Chem Soc 35:651–654
Piromchom J, Wannarit N, Boonmak J, Pakawatchai C, Youngme S (2014) Inorg Chem Commun 40:59–61
Liu YY, Jiang YY, Yang J, Ma JF (2011) CrystEngComm 13:6118–6129
Zhao Y, Zhai LL, Sui WY (2014) J Inorg Chem 30:90–105
Zhang SY, Zhang ZJ, Shi W, Zhao B, Cheng P (2010) Inorg Chim Acta 363:3784–3789
Qin JH, Chang XH, Ma LF, Wang LY (2014) Inorg Chim Acta 41:92–95
Qin L, Wang LN, Ma PJ, Cui GH (2014) J Mol Struct 1059:202–207
Geng JC, Liu LW, Xiao SL, Cui GH (2012) Transit Met Chem 38:143–148
Jiang H, Liu YY, Ma JF, Zhang WL, Yang J (2008) Polyhedron 27:2595–2602
Zhang JW, Gong CH, Hou LL, Tian AX, Wang XL (2013) J Solid State Chem 205:104–109
Wang XX, Liu YG, Li YH, Cui GH (2014) Transit Met Chem 39:461–467
Pochodylo AL, LaDuca RL (2011) Inorg Chem Comm 14:722–726
Zhu HY, Jiang R, Xiao L, Chang YH, Guan YJ, Li XD, Zeng GM (2009) J Hazard Mater 169:933–940
Mane VS (2013) Vijay Babu PV. J Taiwan Inst Chem Eng 44:81–88
Aakeroy CB, Desper J, Elisabeth E, Helfrich BA, Levin B, Urbina JF (2005) Z Kristallogr 220:325–332
Sheldrick GM (1996) SADABS. University of Göttingen, Germany
Sheldrick GM (2008) Acta Cryst A 64:112–122
Deacon GB, Phillips RJ (1980) Coord Chem Rev 33:227–250
Addison AW, Rao TN, Reedijk J, Rijn JV, Verschoor GC (1984) J Chem Soc Dalton Trans 7:1349–1356
Ma LF, Meng QL, Wang LY, Liang FP (2010) Inorg Chim Acta 363:4127–4133
Ma LF, Wang LY, Du M, Batten SR (2010) Inorg Chem 49:365–367
Lucas JS, Bell LD, Gandolfo CM, LaDuca RL (2011) Inorg Chim Acta 378:269–279
Niu D, Yang J, Guo J, Kan WQ, Song SY, Du P, Ma JF (2012) Cryst Growth Des 12:2397–2410
Ren H, Song TY, Xu JN, Jing SB, Yu Y, Zhang P, Zhang LR (2009) Cryst Growth Des 9:105–112
Li YW, Ma H, Chen YQ, He KH, Li ZX, Bu XH (2012) Cryst Growth Des 12:189–196
Geng JC, Qin L, He CH, Cui GH (2012) Transit Met Chem 37:579–585
Cui GH, He CH, Jiao CH, Geng JC, Blatov VA (2012) Cryst Eng Comm 14:4210–4216
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, X.X., Zhao, Y.N., Li, G.Y. et al. Self-assembly of two 2D cobalt(II) coordination polymers constructed from 5-tert-butyl isophthalic acid and flexible bis(benzimidazole)-based ligands. Transition Met Chem 39, 653–660 (2014). https://doi.org/10.1007/s11243-014-9846-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-014-9846-5