
Abstract
Three new Co(II) coordination polymers, [Co(L1)(bpdc)] n (1), [Co(L2)(ndc)(H2O)·2H2O] n (2) and [Co(L3)(ndc)(H2O)·H2O] n (3) (L1 = 1,2-bis(5,6-dimethylbenzimidazole)ethane, L2 = 1,3-bis(5,6-dimethylbenzimidazole)propane, L3 = 1,4-bis(5,6-dimethylbenzimidazole)butane, H2bpdc = 4,4′-biphenyldicarboxylic acid, H2ndc = 2,6-naphthalenedicarboxylic acid) have been synthesized under hydrothermal conditions and structurally characterized by X-ray crystallography. All three complexes feature (4,4) networks that extend into 3D supramolecular frameworks via hydrogen bonding interactions. The luminescence properties and catalytic activities of these complexes with respect to the degradation of methyl orange in a Fenton-like process have been investigated.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The rational design and construction of coordination polymers (CPs) continue to attract much interest, due to the endless possibilities and nearly inexhaustible synthesis options for tailoring their structures and properties [1–4]. Much of the effort toward this goal was concentrated on crystal engineering, and there has been an increasing trend toward the synthesis of coordination polymers containing benzimidazole ligands [5–7]. In particular, the flexible bis(5,6-dimethylbenzimidazole) ligands with two N-donor sites have freely rotating methylene groups, so they can adopt various conformations to coordinate to different metal centers, which is beneficial for the construction of extended architectures [8–11]. A number of CPs with interesting structures and properties derived from these ligands have been prepared and characterized. However, the systematic synthesis of such compounds still presents a significant long-term challenge [12–15].
In general, it has been recognized that intermolecular forces such as hydrogen bonding interactions can be used as structure directing tools in the formation of coordination polymers of higher dimensionality [16–18]. The π–π stacking interaction is also an important and powerful non-covalent intermolecular interaction for the direction of supramolecular architectures [19–21]. A successful strategy for the construction of such coordination polymers is to employ appropriate functional ligands having strong coordination ability as well as hydrogen bond acceptors/donors and π-conjugated systems for extension of their networks. Multifunctional aromatic dicarboxylic acids with various coordinating modes and strong coordination capability are usually chosen as co-ligands with N-heterocyclic neutral ligands; the corresponding carboxylate conjugate bases are excellent hydrogen bond acceptors [22–25].
In this report, we introduce bis(5,6-dimethylbenzimidazole)-based ligands with different –(CH2) n – (n = 2, 3, 4) spacers, with a view to an exploration of the influence of spacer length on the structures of their ternary mixed-ligand coordination polymers. Three new cobalt(II) coordination polymers have been hydrothermally synthesized and characterized, namely [Co(L1)(bpdc)] n (1), [Co(L2)(ndc)(H2O)·2H2O] n (2) and [Co(L3)(ndc)(H2O)·H2O] n (3) (L1 = 1,2-bis(5,6-dimethylbenzimidazole)ethane, L2 = 1,3-bis(5,6-dimethylbenzimidazole)propane, L3 = 1,4-bis(5,6-dimethylbenzimidazole)butane, H2bpdc = 4,4′-biphenyldicarboxylic acid, H2ndc = 2,6-naphthalenedicarboxylic acid). The structures of these ligands are shown in Scheme 1. Considerations relevant to their stepwise assembly and structural correlations are discussed. The luminescence properties and catalytic activities of the complexes are also presented.

The structural formulae of L1, L2, L3, H2bpdc, H2ndc
Experimental
Materials and methods
All the chemicals and reagents employed were purchased from Sinopharm Chemical Reagent Co., Ltd. and used as received without further purification. L1, L2 and L3 were synthesized according to the literature procedure [26]. Elemental analyses (C, H and N) were performed on a Perkin-Elmer 240C Elemental Analyzer. IR spectra (KBr pellets) were recorded on an Avatar 360 (Nicolet) spectrophotometer in the range of 4000–400 cm−1. X-ray powder diffraction (XRPD) investigations were carried out on a Rigaku D/Max-2500PC X-ray diffractometer at 40 kV, 40 mA with Cu-Kα radiation (λ = 1.542 Å). Thermogravimetric analyses (TGA) were carried out on a NETZSCH TG 209 thermal analyzer in flowing N2 with a heating rate of 10 °C/min. A Hitachi F-7000 fluorescence spectrophotometer was used for powdered solid samples at room temperature to obtain luminescence spectra. The absorptivity value of methyl orange was recorded with a Shanghai Jingke 722 N spectrophotometer at the maximum wavelength of 506 nm.
Synthesis of [Co(L1)(bpdc)] n (1)
A mixture of Co(OAc)2·4H2O (49.2 mg, 0.2 mmol), L1 (32.3 mg, 0.1 mmol), H2bpdc (48.4 mg, 0.2 mmol), NaOH (8.0 mg, 0.2 mmol), ethanol (4 mL) and water (10 mL) was heated at 140 °C for 3 days in a 25 mL Teflon-lined vessel container. The mixture was then cooled to room temperature at a rate of 5 °C/h. Purple crystals suitable for single-crystal X-ray diffraction were obtained by filtration and washed with distilled water in 58 % yield (based on Co(OAc)2·4H2O). Calcd. for C34H30CoN4O4 (Fw = 617.55): C 66.1, H 4.9, N 9.1 %. Found: C 65.9, H 5.1, N 9.3 %. IR (KBr, cm−1): 1605 s, 1560 m, 1510 m, 1430 m, 1300 m, 1178 w, 847 w, 758 m.
Synthesis of [Co(L2)(ndc)(H2O)·2H2O] n (2)
A mixture of CoSO4·7H2O (56.7 mg, 0.2 mmol), L2 (33.8 mg, 0.1 mmol), 2,6-H2ndc (43.7 mg, 0.2 mmol), NaOH (8.0 mg, 0.2 mmol) and water (10 mL) was heated at 140 °C for 3 days in a 25 mL Teflon-lined vessel container. The mixture was then cooled to room temperature at a rate of 5 °C/h. Purple block crystals of 2 were separated in 46 % yield based on CoSO4·7H2O. Calcd. for C33H36CoN4O7 (Fw = 659.59): C 60.1, H 5.5, N 8.5 %. Found: C 59.9, H 5.2, N 8.7 %. IR (KBr, cm−1): 3437 s, 1605 m, 1570 s, 1510 m, 1472 m, 1400 s, 1220 m, 1060 w, 1053 m, 997 w, 795 s, 621 w, 480 w.
Synthesis of [Co(L3)(ndc)(H2O)·H2O] n (3)
The synthesis of 3 was similar to that of 2, but using L3 (0.1 mmol, 35.7 mg) in place of L2. Purple block crystals of 3 suitable for single-crystal X-ray diffraction were isolated in 54 % yield based on CoSO4·7H2O. Calcd. for C34H36CoN4O6 (Fw = 655.60): C 62.3, H 5.5, N 8.6 %. Found: C 62.0, H 5.1, N 8.9 %. IR (KBr, cm−1): 3389 s, 3110 w, 1605 m, 1560 s, 1520 m, 1470 m, 1390 s, 1353 s, 1273 m, 1229 m, 933 w, 791 s, 459 w.
X-ray crystallography
Crystallographic data for the complexes were collected on an Agilent SuperNova Dual diffractometer using mirror-monochromated Cu-Kα radiation (λ = 1.54178 Å) at room temperature with an Atlas detector in ω scan mode. All structures were solved by direct methods and refined on F 2 by full-matrix least-squares procedures with the SHELXTL program package [27]. Atoms were located from iterative examination of difference F-maps following least-squares refinements of the earlier models. Anisotropic parameters were used for non-hydrogen atoms, hydrogen atoms of water were located on a difference Fourier map, while other H atoms were included in calculated positions and refined with fixed thermal parameters, each riding on a corresponding parent atom. The two lattice water molecules in complex 2 and one water molecule in complex 3 are disordered; these structures were refined by the SQUEEZE routine of PLATON [28]. The crystallographic data are listed in Table 1, and selected bond lengths and angles are presented in Table 2.
Catalysis experiments
The catalytic reactions were performed through a typical process as follows: 60 mg powder of the required complex and 2 mL of 30 % (w/w) H2O2 were mixed together with 200 mL of an aqueous solution of methyl orange (20 mg/L). The solution was kept continuously stirring with a magnetic stirrer, the temperature was constant at 40 °C and the pH was adjusted to 6.0. At given time intervals, 2 mL aliquots were removed and centrifuged to remove the residual catalyst, then measured for absorbance at 506 nm using a Shanghai Jingke 722 N visible spectrophotometer. A control experiment was also carried out under the standard conditions without any catalyst. The degradation efficiency of methyl orange was evaluated based on the following formula:
where C 0 (mg/L) is the initial concentration of methyl orange and C t (mg/L) is the concentration of methyl orange at the reaction time, t (min).
Results and discussion
Synthesis of the polymers
The hydrothermal method has been extensively explored as a powerful tool in the self-assembly of CPs. By varying the molar ratio/composition of the initial reactants, reaction time and/or temperature, single crystals of complexes 1–3 suitable for single-crystal X-ray diffraction analysis were successfully obtained. It is noteworthy that NaOH solution was used in the preparation of these complexes, in order to deprotonate the carboxylic group, while the ethanol solvent in complex 1 may help to control the crystal shape. Without it, only unformed powders were obtained under hydrothermal conditions. These compounds are stable in air and insoluble in water and general organic solvent, such as benzene, alcohol, dichloromethane and acetonitrile.
Crystal structure of complex 1
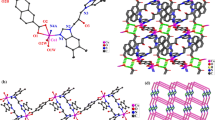
X-ray single-crystal structure analysis reveals that complex 1 crystallizes in the orthorhombic Pcca space group and its asymmetric unit contains half a [Co(L1)(bpdc)] molecule. As shown in Fig. 1a, the Co(II) center is six-coordinated by two nitrogen atoms (N1, N(1)A) from two separate L1 ligands and four oxygen atoms (O1, O2, O1A, O2A) from two anionic bpdc2− ligands to form a {CoN2O4} trigonal prismatic geometry [29] (symmetry code: A: −x + 3/2, −y + 1, z). The Co–N bond length is 2.091(2) Å, and the Co–O bond lengths range from 2.1535(2) to 2.2177(2) Å. The bond angles around the Co1 center are well matched to those observed in similar complexes [30]. The ligand L1 employs trans-conformation such that the mean dihedral angle between the two benzimidazole rings is 59.06(5)°; by acting as a μ 2-bridging ligand, L1 bridges adjacent Co(II) centers to form a 1D infinite linear [Co(L1)] n chain. The benzimidazole rings of L1 are slightly twisted over each other by a torsion angle of 43.11(3)°. The bpdc2− ligands also connect neighboring Co(II) atoms to shape a 1D linear [Co(bpdc)] n chain, in which the bpdc2− ligands adopt the bis-chelating coordination mode. The two different types of 1D chain cross each other, resulting in a 2D grid structure. To better understand the structure of complex 1, topological analysis was used [31]. All Co(II) centers can be simplified to 4-connected nodes and the shortest circuit is a four-membered ring, such that the 2D layer can be simplified to a 44-sql net with a mesh size of 7.8836 × 14.9962 Å (Fig. 1b). Weak intermolecular C–H···O hydrogen bonding interactions between the imidazole groups and carboxylate oxygen atoms of the bpdc2− ligands generate the overall 3D supramolecular framework of complex 1 [H8–O1B = 2.44 Å, C8–H8···O1B = 123°, symmetry code: B: −x + 2/3, y, z − 1/2].

a Coordination environment around the Co(II) centers in complex 1. Hydrogen atoms are omitted for clarity (symmetry code: A: −x + 3/2, −y + 1, z; B: −x + 1, y, −z + 1/2; C: −x + 3/2, −y, z). b The 2D (4,4) layer structure of 1. c View of the 3D supramolecular framework formed through C–H···O interactions. The dashed lines indicate C–H···O interactions
Crystal structure of complex 2
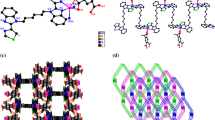
Complex 2 crystallizes in the triclinic Pī space group, and the asymmetric unit is composed of a cobalt atom, one neutral L2 ligand, one ndc2− ligand, one coordinated water ligand and two lattice water molecules. As depicted in Fig. 2a, the Co(II) center is six-coordinated by three O atoms (O2A, O3, O4) from two anionic ndc2− ligands, one O1W from a water ligand and two N atoms (N1, N3B) from two different L2 ligands (symmetry codes: A: x, y + 1, z − 1; B: x, y + 1, z). A distorted {CoO4N2} octahedral coordination environment is observed at the cobalt atom, with two nitrogen atoms in the axial positions and four oxygen atoms in the equatorial plane. The Co–N bond lengths are 2.108(5) and 2.127(5) Å, and the Co–O distances fall in the range of 2.031(4)–2.300(4) Å. For the ndc2− ligand, the coordination modes of the two carboxyl groups are different: one coordinates to a Co(II) center in a monodentate mode, while the other coordinates to a different Co(II) center in a chelating fashion. The ndc2− ligands bridge neighboring Co(II) atoms to give a [Co(ndc2−)] n straight chain in which the Co···Co distance is 13.2210 Å. In addition, L2 acts as a bidentate µ 2-N,N′ bridging ligand to link the adjacent two Co(II) centers into a [Co(L2)] n zigzag chain, and the dihedral angle between two benzimidazole rings is 82.71(9)°. The contiguous [Co(L2)] n chains are bridged by ndc2− ligands forming a (4,4) grid structure (Fig. 2b). These neighboring layers are extended into a 3D supramolecular network (Fig. 2c) through hydrogen bonds between the carboxyl oxygen atoms (O1, O3) from ndc2− ligands and the O1W from the water ligand [O1W–H1WA···O1C = 2.636(7) Å, O1W–H1WB···O3D = 2.636(7) Å, symmetry codes: C: x − 1, y + 1, z − 1; D: x − 1, y, z].

a Coordination environment around the Co(II) centers in complex 2. Hydrogen atoms are omitted for clarity (symmetry code: A: x, y + 1, z − 1; B: x, y + 1, z). b A schematic drawing of the 2D layer-like structure in 2. c The 3D supramolecular network of 2, constructed by C–H···O hydrogen bonding interactions. The dashed lines indicate C–H···O bonds
Crystal structure of complex 3
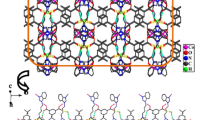
Single-crystal X-ray diffraction analysis shows that complex 3 crystallizes in the monoclinic P 21 space group. The asymmetric unit contains one Co(II) atom, a complete L3 ligand, one ndc2− ligand, one water ligand and one lattice water. Similar to 2, complex 3 possesses a distorted octahedral {CoO4N2} coordination geometry (Fig. 3a), in which each Co(II) atom is coordinated by four oxygen atoms from two different ndc2− ligands in the equatorial plane [Co(1)–O(4)A = 2.069(4) Å, Co(1)–O(1W) = 2.093(4) Å, Co(1)–O(1) = 2.137(3) Å, Co(1)–O(2) = 2.268(3) Å] and two nitrogen atoms from L3 ligands in the axial position [Co(1)–N(4)B = 2.104(6) Å, Co(1)–N(1) = 2.121(6) Å] (symmetry codes: A: x, y, z + 1; B: −x, y − 1/2, −z + 2).

a Coordination environment around the Co(II) centers in complex 3. Hydrogen atoms are omitted for clarity (symmetry code: A: x, y, z + 1; B: −x, y − 1/2, −z + 2). b View of the 2D layer-like structure in 3. c The 3D supramolecular network of 3, constructed by C–H···O hydrogen bonding interactions. The dashed lines indicate C–H···O bonds
The N-donor ligands connect cobalt(II) atoms in a bis(monodentate) binding mode to form a 1D Ω-like chain along the b-axis such that the dihedral angle between the benzimidazole rings of the L3 ligand is 7.06(2)°. Each ndc2− moiety serves as a chelating-monodentate ligand to join two Co(II) atoms, such that the ndc2− ligands span a Co···Co internuclear distance of 13.1638 Å and form a neutral [Co(ndc2−)] n coordination polymer chain arranged along the c direction. These two chains propagate into a 2D 44 grid-like layer, arranged in an offset way with the grid occupied by groups from the adjacent chains (Fig. 3b). Complex 3 displays a uninodal 2D layer with a (4,4) topology, and the rectangular window of the 2D motif has dimensions of ca. 11.7235 × 13.1638 Å. Aggregation between layers is promoted by classical hydrogen bonding interactions provided by the oxygen atom of the lattice water molecule, which serves as a hydrogen bonding donor to the oxygen atom from ndc2− [O1W–H1WA···O3C = 2.672(6) Å, O1W–H1WB···O2D = 2.771(5) Å, symmetry codes: C: x − 1, y, z + 1; D: x − 1, y, z]. In this manner, the 2D network is further extended into a 3D supramolecular architecture.
To summarize, three flexible bis(benzimidazole) ligands (L1, L2, and L3) with different –(CH2) n – spacers (n = 2, 3, 4) gave three cobalt(II) complexes. All three complexes have 2D layered structures and exhibit typical 44 sql topology. In complex 1, the bpdc2− ligand adopts the bis-chelating coordination mode, while the ndc2− ligand employs the same chelating-monodentate coordination mode for complexes 2 and 3. For all three complexes, all the bis(benzimidazole) ligands adopt the bidentate bridging mode, connecting the Co(II) centers with Co···Co distances of 7.8836(2) Å for 1, 11.4289(15) Å for 2, 11.7235(16) Å for 3 (Table 3). All the three complexes are self-assembled into 3D networks through C–H···O (for 1) and O–H···O (for 2 and 3) hydrogen bonding interactions.
IR spectra and X-ray powder diffraction studies
The IR spectra exhibit the main characteristic bands of water molecules, the carboxylate ligands and nitrogen-containing organic ligands expected for the title complexes. The bands at 3437 and 3389 cm−1 for complexes 2 and 3, respectively, are attributed to the lattice water. There is no absorption band around 1700 cm−1 for any of these complexes, indicating that all carboxyl groups are completely deprotonated [32]. Strong peaks at 1605 and 1560 cm−1 for 1, 1605, 1570, 1474 and 1400 cm−1 for 2, 1605, 1560, 1470 and 1390 cm−1 for 3, may be attributed to the asymmetric and symmetric vibrations of carboxylato groups. The separations (∆ν[ν as(COO)–ν s(COO)]) between these bands indicate the presence of chelating (45 cm−1 for 1) and chelate/monodentate (205 and 96 cm−1 for 2; 215 and 90 cm−1 for 3) carboxyl groups. The bands at ca. 1500 cm−1 for all three complexes are assigned to the ν (C=N) stretches of the benzimidazole rings.
To determine whether the crystal structures are truly representative of the bulk materials used for property studies, X-ray powder diffraction (XRPD) experiments were carried out for the complexes. The XRPD experimental and as-simulated patterns are shown in Fig. 4. The crystalline samples of complexes 1–3 gave a positive match between the experimental and simulated XRPD patterns, indicating that the bulk synthesized material and the crystals are homogeneous.

The powder X-ray diffraction patterns calculated from the single-crystal data and that obtained from the experiments for polymer 1-3, respectively
Thermal and luminescence properties
To characterize the stabilities of complexes 1–3, their thermal behaviors were investigated under nitrogen atmosphere by TGA. As depicted in Fig. 5, complex 1 remains stable up to 405 °C, finally decomposing to CoO as a residue (obsd 12.3 %, calcd 12.1 %). The TG curve for complex 2 shows a gradual weight loss between 95 and 160 °C, which can be ascribed to the loss of three water molecules (obsd 8.3 %, calcd 8.2 %). The loss of organic ligands occurs within the range of ca. 336–604 °C. The remaining weight corresponds to the formation of CoO (obsd 11.2 %, calcd 11.4 %). For complex 3, the loss of two water molecules is observed within the range of 80–170 °C (obsd 5.6 %; calcd 5.5 %) and the decomposition of the compound occurs at 365 °C to give CoO residue (obsd 11.4 %; calcd 11.3 %).

The TGA curves of polymers 1–3
The solid-state luminescence properties of both the complexes and free ligands have been investigated at room temperature. As shown in Fig. 6, free L1, L2, and L3 display luminescence with emission maxima at 389, 369, and 365 nm, respectively (λ ex = 315 nm for L2 and L3, λ ex = 230 nm for L1), attributed to π* → π transitions [33, 34]. Recent studies have shown that solid H2bpdc exhibits weak emission at 404 nm (λ ex = 335 nm) [35]. Comparing with L1, the blue-shift of ca. 48 nm for complex 1 may be assigned to metal-to-ligand charge-transfer (MLCT). Red-shifts of ca. 33 nm for complex 2 and 35 nm for 3 are observed compared with L2 and L3, respectively, which can probably be assigned to intra-ligand (π* → n) luminescence emissions [36].

Emission spectra of complexes 1–3 and the free ligands
Catalytic degradation properties
In recent years, much work has been carried out on advanced oxidation processes (AOPs) for the treatment of azo dye wastewaters. The Fenton-like reaction appears to be the most promising such AOP in terms of both cost-effectiveness and ease of operation. It involves the production of strongly oxidizing agents, mainly ·OH radicals, which react rapidly and non-selectively with most inorganic and organic compounds. The ·OH radical is sufficiently active as to destroy organic compounds into CO2, H2O and other products [37, 38]. In this work, we have chosen methyl orange (MO), as a model dye contaminant, in order to evaluate their catalytic effectiveness for AOP. The degradation efficiencies of MO against reaction time (t) for the title complexes are shown in Fig. 7. In the absence of any catalyst, there is no noticeable change in the absorbance of dye, and the self-degradation efficiency of control experiments in the presence of H2O2 only reaches 18.3 % after 120 min. When each of the complexes is added, a reaction takes place leading to loss of the dye color. Thus, approximately 81.3 % of MO has decomposed after 120 min for complex 3 as catalyst, 72.8 % for complex 1, and 64.9 % for complex 2. The catalytic activity of complex 3 is thus higher than complexes 1, 2 and many other CPs that have been investigated [39, 40]. The differences in catalytic activities may be attributed to the different dicarboxylic acids and the various spacer lengths in the N-containing ligands, as well as the overall structures of these polymeric cobalt(II) complexes.

Experimental results of the catalytic degradation of methyl red dye
Conclusions
In conclusion, by introducing H2bpdc and H2ndc as mixed ligands, three Co(II) 2D coordination polymers based on flexible bis(5,6-dimethylbenzimidazole) ligands have been successfully synthesized. All three complexes present similar 2D grid structures, which are extended into 3D supramolecular frameworks through hydrogen bonding interactions. Complex 3 shows higher catalytic activity than 1, 2 and many other CPs in the Fenton-like process for decomposition of dye [41].
Supplementary materials
CCDC 1427142-1427144 contain the supplementary crystallographic data for the complexes 1-3. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Zhu QL, Xu Q (2014) Chem Soc Rev 43:5468–5512
Zhang T, Lin W (2014) Chem Soc Rev 43:5982–5993
Li M, Li D, O’Keeffe M, Yaghi OM (2014) Chem Rev 114:1343–1370
Lin HY, Luan J, Wang XL, Zhang JW, Liu GC, Tian AX (2014) RSC Adv 4:62430–62445
Yang FF, Yu XY, Luo YH, Wang XF, Sun DH, Zhang H (2015) Polyhedron 85:337–346
Zhang JW, Gong CH, Hou LL, Tian AX, Wang XL (2013) J Solid State Chem 205:104–109
Yang YQ, Yang J, Kan WQ, Yang Y, Guo J, Ma JF (2013) Eur J Inorg Chem 2013:280–292
Li M, Zhao S, Peng YF, Li BL, Li HY (2013) Dalton Trans 42:9771–9776
Li M, Ling Q, Yang Z, Li BL, Li HY (2013) CrystEngComm 15:3630–3639
Nobakht V, Beheshti A, Proserpio DM, Carlucci L, Abrahams CT (2014) Inorg Chim Acta 414:217–225
Meng W, Xu Z, Ding J, Wu D, Han X, Hou H, Fan Y (2014) Cryst Growth Des 14:730–738
Wang XX, Yu BY, Hecke KV, Cui GH (2014) RSC Adv 4:61281–61289
Hao JM, Yu BY, Hecke KV, Cui GH (2015) CrystEngComm 17:2279–2293
Qin L, Liu LW, Du X, Cui GH (2012) Transit Met Chem 38:85–91
Xiao SL, Li YH, Ma PJ, Cui GH (2013) Inorg Chem Commun 37:54–58
Wang XX, Liu YG, Ge M, Cui GH (2015) Chin J Inorg Chem 31:2065–2072
Atwood JL, Barbour LJ, Jerga A (2004) Angew Chem Int Ed 116:3008–3010
Sherrington DC, Taskinen KA (2001) Chem Soc Rev 30:83–93
Yang J, Ma JF, Liu YY, Ma JC, Batten SR (2007) Inorg Chem 46:6542–6555
Liu LL, Huang JJ, Wang XL, Liu GC, Yang S, Lin HY (2013) Inorg Chim Acta 394:715–722
Shi WJ, Hou L, Li D, Yin YG (2007) Inorg Chim Acta 360:588–598
Han J, Yau CW, Chan CW, Mak TCW (2012) Cryst Growth Des 12:4457–4465
Okuno T, Sakoda Y, Kinuta T, Sato T, Tokutome H, Tajima N, Nakano Y, Fujiki M, Kuroda R, Imai Y (2012) CrystEngComm 14:4819–4825
Wang XX, Yu YM, Huan DH, Hecke KV, Cui GH (2015) Inorg Chem Commun 61:24–26
Shiota N, Okuno T, Kinuta T, Sato T, Kuroda R, Matsubara Y, Imai Y (2011) CrystEngComm 13:1683–1686
Aakeroy CB, Desper J, Elisabeth E, Helfrich BA, Levin B, Urbina JF (2005) Z Kristallogr 220:325–374
Sheldrick GM (2008) Acta Cryst A 64:112–122
Spek AL (2000) PLATON. Utrecht University, Utrecht
Banerjee S, Ghosh A, Wu B, Lassahn PG, Janiak C (2005) Polyhedron 24:593–599
Wang XL, Sui FF, Lin HY, Zhang JW, Liu GC (2014) Cryst Growth Des 14:3438–3452
Blatov VA (2012) Struct Chem 23(4):955–963
Liu GC, Huang JJ, Zhang JW, Wang XL, Lin HY (2013) Transit Met Chem 38:359–365
Liu HY, Wu H, Ma JF, Liu YY, Liu B, Yang J (2010) Cryst Growth Des 10:4795–4805
Allendorf MD, Bauer CA, Bhakta RK, Houk RJT (2009) Chem Soc Rev 38:1330–1352
Dai JC, Wu XT, Cui CP, Hu SM, Du WX, Wu LM, Zhang HH, Sun RQ (2002) Inorg Chem 41:1391–1396
Ugale B, Singh D, Nagaraja CM (2015) J Solid State Chem 226:273–278
Wu XY, Qi HX, Ning JJ, Wang JF, Ren ZG, Lang JP (2015) Appl Catal B 168:98–104
Qi HX, Wang JF, Ren ZG, Ning JJ, Lang JP (2015) Dalton Trans 44:5662–5671
Zhang X, Meng XL, Huang CM, Cui GH (2015) J Mol Struct 1100:94–99
Hao JM, Li YH, Li HH, Cui GH (2013) Transit Metal Chem 39:1–8
Wang CC, Li JR, ZhangYQ LuXL, Guo GS (2014) Energy Environ Sci 7:2831
Acknowledgments
The project was supported by the National Natural Science Foundation of China (51474086), Natural Science Foundation-Steel and Iron Foundation of Hebei Province (B2015209299), the Graduate Student Innovation Fund of North China University of Science and Technology (2015S13) and the Hercules Foundation (project AUGE/11/029 “3D-SPACE: 3D Structural Platform Aiming for Chemical Excellence”) and the Research Fund-Flanders (FWO) for funding.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, X., Liu, Y.G., Yu, B. et al. Three 2D mixed-ligand Co(II) coordination polymers containing flexible bis(benzimidazole) ligands with different spacers. Transition Met Chem 41, 213–223 (2016). https://doi.org/10.1007/s11243-015-0013-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-015-0013-4