
Abstract
Poor adventitious root (AR) formation poses a major constraint to the propagation of tea cuttings. The present study identified two tea cultivars with different rooting behavior: Camellia sinensis cv. Taixuan 0310 (TX), with strong and rapid rooting; and C. sinensis cv. Qianmei 601 (QM), with poor and slow rooting. To elucidate the molecular events underlying AR formation in tea cuttings, transcriptomic analysis was performed during early root formation and root elongation. Differentially expressed genes (DEGs) between TX and QM were identified. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed that phenylpropanoid biosynthesis, plant hormone signal transduction, plant MAPK signaling and plant-pathogen interaction, all play important roles in AR information in C. sinensis. Among the DEGs, two GRETCHEN HAGEN3 (GH3) genes, CsGH3.2 and CsGH3.3, were isolated and characterized. Quantitative real-time PCR (qRT-PCR) and molecular analysis suggest that CsGH3.2 and CsGH3.3 may have specific functions in different organs. Moreover, overexpression of CsGH3.2 and CsGH3.3 inhibited root growth and development in transgenic rice plant. These results give new insights into the possible role of CsGH3.2 and CsGH3.3 in adventitious rooting, and also provide an interesting starting point to discover new genes involved in AR formation in tea cuttings.
Key message
Transcriptomic analysis revealed the molecular events underlying adventitious root formation in tea cuttings, and facilitated the discovery of genes involved in this process. CsGH3.2 and CsGH3.3 appear to function in adventitious rooting in tea cuttings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tea (Camellia sinensis L.) is a commercial cash crop used to produce one of the most popular and economically valuable beverages worldwide. It is primarily cultivated in China, India, and other tropical and subtropical regions with suitable climates (Xia et al. 2020a). Since tea offers enormous economic, social, health, and cultural benefits, research into tea cultivation has rapidly increased over the past few years, particularly in China. Moreover, the introduction of new tea cultivars with higher yields, reliable quality, improved aroma, and attractive appearance, such as purple tea(Wei et al. 2019) and albino tea(Wei et al. 2015), has led to the widespread commercialization of improved varieties. The propagation of new tea cultivars is crucial for continued development of the modern tea industry (Seth et al. 2021).
Tea plants are traditionally propagated using seeds or excised plant parts (cuttings) (Mondal et al. 2004). Seed-grown plants are highly variable and often unsuitable for mechanized harvesting and processing. Therefore, the most efficient and economical way to produce large numbers of homogeneous clonal tea plants are to through nodal cuttings (Lima et al. 2013). Most tea plantations developed over the past few decades are planted with improved clonal teas. However, the widespread adoption of these varieties are hampered by the lack of rapid propagation methods. Single-node semi-hardwood cuttings are usually used for the large-scale propagation of tea (Lima et al. 2013; Rout 2006; Wei et al. 2013). To survive independently of the mother plant, these cuttings must regenerate and develop new roots. These de novo roots, which form from non-root tissues spontaneously or in response to certain stimuli, are called adventitious roots (ARs). Tea cuttings are often lost due to slow or no rooting, which seriously restricts the development of tea tree plantations (Lima et al. 2013; Pacurar et al. 2014; Rout 2006; Wei et al. 2013). AR formation is thus essential for the successful propagation and long-term survival of tea plants.
De novo AR formation can be divided into four stages generally: parenchyma cell dedifferentiation, root initials formation, root primordium formation, and root elongation (Davies 2017; Davies et al. 2018). This complex process is influenced by internal and external factors including the tea variety, the use of easy- or hard-to-root tea genotypes, the nutritional status of stem cuttings, environmental conditions such as season-dependent rooting ability, and endogenous and exogenous phytohormones (Davies 2017; Wilson et al. 2018). Auxin is a central regulator of AR formation (Pacurar et al. 2014). In the absence of treatment with plant growth regulators such as the auxins indole-3-acetic acid (IAA) and indole-3-butyric acid (IBA), partial tea cuttings are not easily rooted. AR formation in cuttings involves dramatic changes in the transcription of auxin signaling cascade genes (Druege et al. 2016; Xiao and Zhang 2020). IBA-regulated genes might directly promote AR induction (Quan et al. 2017; Wei et al. 2014), although high auxin levels have an inhibitory effect during later stages of AR formation (Pacurar et al. 2014; Wei et al. 2013). Despite the mechanisms underlying this network during AR formation in cuttings are increasingly being revealed, the endogenous developmental signals that function during rooting in tea cuttings with different rooting abilities are still unclear.
Transcriptomic analysis has been widely employed to reveal functional differentially expressed genes (DEGs) involved in regulating root development (Ahkami et al. 2014; Li et al. 2018; Wei et al. 2014). In current study, transcriptomic analysis was performed during AR formation in tea cuttings, from root callus formation to root initiation to root elongation. More DEGs were observed during phase transitions than during the various growth stages. The differences in gene expression patterns were examined in primary calli and re-differentiated roots in two tea cultivars with different rooting abilities. GRETCHEN HAGEN3 (GH3) genes play crucial roles in root formation in rice and Arabidopsis; however, the functions of GH3 in tea plants are unclear. Among the DEGs, two putative GH3 genes CsGH3.2 and CsGH3.3 were characterized and investigated. The organ-specific expression analyses and protein localization assay suggest that CsGH3.2 and CsGH3.3 may have specific functions in different organs. Quantitative real-time PCR (qRT-PCR) analysis revealed that both of them were highly responsive to exogenously applied IBA, and CsGH3.2 and CsGH3.3 overexpression represses transgenic rice growth and development. Overall, the data show that CsGH3.2 and CsGH3.3 are functional GH3 family genes and may play crucial roles in root development of tea cuttings.
Materials and methods
Plant materials and growth conditions
Eight clonal tea cultivars of China from Guizhou Academy of Agricultural Sciences were involved in this study, including var. sinensis cv. Taixuan0310 (TX, GPD2019-520,010), var. sinensis cv. QianCha1 (QC1, GPD2019-520,007), var. sinensis cv. QiqnCha8 (QC8, GPD2019-520,007), var. sinensis cv. Qianmei 601 (QM601, GPD2020-330,024), var. sinensis cv. Qianmei 419(QM419, GSCT31), var. sinensis cv. Qianmei 809 (QM809, GSCT2002007), var. sinensis cv. zhongcha 108 (ZC108, GPD2021-330,016), and var. sinensis cv. Fudingdabaicha (FD, GSCT1). Freshly growing twigs of these tea cultivars were collected from nearly 10-year-old parental tea cultivars, which grown in the tea garden of Meitan in Guizhou, China (latitude: 27.78; longitude:107.48; altitude: 765 m). Tea cutting experiment was conducted three times, from October10 to May10 (2019–2021). The single-node semi-hardwood cuttings with full leaf were collected from the parental plants at the nearly same growth state, without treatment of any plant growth regulators, and then rooted in perlite and soil mixed medium at the propagated glasshouse in Guiyang, Guizhou, China (latitude: 26.34; longitude:106.69; altitude: 1055 m). The medium pH was around pH 5. Cuttings were grown at 20 ºC day/16 ºC night under a 16-h light/8-h dark photoperiod at 1000 lx, with 50–80% relative humidity. The rectangle containers for cuttings cultivation are about 50 cm long, 25 cm wide and 30 cm height. The planting management processes were kept the same for each cultivar during cultivation.
RNA preparation, cDNA library construction, and transcriptome sequencing
The new meristem (callus) around the basal parts of the cuttings stem (S0) were taken as sample S1, the new meristem with callus and root-like zone were taken as sample S2, the new roots about 0.5 cm with the residual callus/root zone were taken as sample S3. All the samples were taken after four months of cultivation on March20 (2020). Tissue segments were collected from 10 individual cuttings. The sample was frozen immediately in liquid nitrogen and stored at− 80 °C for subsequent analysis. Total RNAs were extracted using CTAB-PBIOZOL reagent and detected using NanoDrop and Agilent 2100 bioanalyzers. cDNA library and sequencing were generated on the BGIseq500 platform (BGI-Shenzhen, China) (Huang et al. 2017a).
Gene assembly, functional annotation, and DEG identification
The clean reads were obtained using SOAPnuke and trimmomatic to remove adaptor sequences, unknown reads (reads with unknown base ‘N’ more than 5%), and low-quality sequences. The clean reads were assembled into non-redundant transcripts using HISAT and Bowtie2 (Kim et al. 2015; Langmead and Salzberg 2012) and then were mapped to the reference genome sequence from tea plants (Xia et al. 2020a, 2019, 2020b) (TPIA, http://tpdb.shengxin.ren/). Next, RSEM was used to calculate the expression levels of genes and transcripts. DEGs were identify based on the DEseq2 method (Love et al. 2014), and FPKM (fragments per kilobase of exon model per million mapped fragments) was used as a value of normalized gene expression. The DEGs were identified as those FPKM values having log2 ratios greater than 1.00 or less than − 1.00 and a P-value < 0.01. According to the Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation results and the official classification, the differential genes were classified into biological pathways, and the phyper function in R software was used for enrichment analysis (Chen et al. 2020). False discovery rate correction was used to determine the P-value, and Q-value ≤ 0.01 was regarded as significant enrichment.
Plasmid construction and rice plant transformation
According to the predicted cDNA sequence from transcriptome data and the released genome data (Xia et al. 2019), the full-length open reading frames (ORFs) of CsGH3.2 and CsGH3.3 were amplified using the primers CsGH3.2-1F/CsGH3.2-1R and CsGH3.3-1F/CsGH3.3-1R from TX cultivars. The amplification product was cloned into pMD19-T vector (Takara), and three constructs were verified by sequencing. To construct overexpression vectors, the cDNA fragments of CsGH3.2 and CsGH3.3 were amplified and inserted into the pU1301 vector (Bio-Transduction) under the control of the ubiquitin-1 promoter. To generate the CsGH3.2 and CsGH3.3 green fluorescent protein (GFP) vectors, fragments without the termination codons were amplified and inserted into the SmaI and XbaI sites of pSuper1300-GFP vector. All these constructs were introduced into Agrobacterium tumefaciens strain EHA105. The strain carrying the overexpression vector was used to transform rice cultivar Zhonghua 11 by A. tumefaciens–mediated transformation method (Puhan et al. 2012). A. tumefaciens carrying p35S::CsGH3.2-GFP, p35S::CsGH3.3-GFP, and p35S::GFP plasmids was transiently infiltrated into N. benthamiana leaves (Huang et al. 2009).
Quantitative reverse transcription PCR (qRT-PCR)
Total RNA was isolated from tea or rice plants using an RNA extraction kit (Takara, Fruit-mate™, Japan) according to the Plant RNA Protocol. Reverse transcription was conducted using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) with 4 µg RNA as the template. qRT-PCR was performed using a Bio-Rad Real-Time PCR System and the SYBR® Green PCR Master Mix Kit (Applied Biosystems) as described previously (Huang et al. 2017b). Three reactions were repeated independently using GAPDH (for tea plants) or UBI5 (for rice plants) as an internal control; all the gene-specific primers are available in Table S4.
Microscopy observation
Root tips of ZH11 and CsGH3.3 transgenic plants were fixed in 75% FAA solution for more than 24 h. The treated samples were dehydrated through a graded ethanol series, embedded in paraffin (Sigma, Germany), sliced into sections, stained with annona red and fast green double dye (service-bio) (Zhong et al. 2018). Paraffin sections were examined and photographed using a CaseViewer digital microscopy application (3DHISTECH).
Results
Isolation of two tea cultivars with different rooting abilities
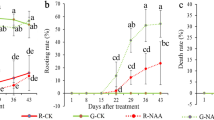
To investigate the molecular mechanisms underlying AR formation during the propagation of tea cuttings, The rooting capacity of eight clonal tea cultivars from Guizhou province were examined. Detailed information about the samples and rooting data are listed in Table S1. The AR morphology of nearly 1-year-old plants clonally propagated from cuttings of tea cultivars C. sinensis cv. QianCha1 (QC1), C. sinensis cv. QiqnCha8 (QC8), C. sinensis cv. Qianmei 601 (QM), and C. sinensis cv. Taixuan 0310 (TX) is shown in Fig. 1A, B. The biomass of TX roots was significantly higher than that of the other cultivars (Table S1). Notably, the fresh and dry weights of ARs in QM plants were only ~ 62% and 64% those of TX plants (Fig. 1C, D), respectively. Therefore, TX and QM601 were selected as candidate cultivars to study the AR initiation process. To describe the initiation of rooting more clearly, the rooting process was divided into four stages (S0–S3) as follows: S0, an original cutting prior to rooting (Fig. 2A); S1, root callus formation stage (Fig. 2B); S2, juvenile root initiation stage (Fig. 2C); and S3, large root formation stage (Fig. 2D). The rooting status of nodal cuttings at 1 month, 2 months, and 4 months after rooting were observed. Based on statistical analysis of the data, root initiation and root formation occurred significantly later in QM601 than in TX (Table S2 and Fig. 2E).

Comparison of the rooting capacities of two clonal tea cultivars from Guizhou province, China. A, B AR morphology of 1-year-old plants clonally propagated from cuttings of four tea cultivars. Bar = 10 cm. C, D The fresh and dry weights, respectively, of ARs in QM601 and TX plants. Data are means ± SD (n = 20). *Significant difference compared to TX plants (P < 0.05, Student’s t-test). Bar = 10 cm

The four stages of AR development. A S0, an initial cutting prior to rooting. B S1, root callus formation stage. C S2, juvenile root initiation stage. D S3, large root formation stage. E AR phenotypes of 4-month-old plants clonally propagated from cuttings of TX (left) and QM (right) plants grown in soil. Bar = 1 cm
Transcriptome analysis of TX and QM601 roots tissue at stages S1–S3
To reveal the molecular mechanism regulating the AR initiation process, transcriptome analysis of TX and QM601 new meristem (callus/root zone) at stages S1–S3 was performed. The corresponding cDNA libraries were subjected to sequencing on the BGIseq500 platform. After filtering, 42 million clean reads were obtained: The proportion of effective reads was ~ 95.98–96.87%. The Q20 and Q30 values (percentage of sequences with sequencing error rates < 1% and 0.1%, respectively) were ~ 96.54 and 91.32%, respectively (Table 1). De novo assembly of these clean reads yielded 47,676 unigenes, among which 36,150, 37,276, 36,790, 36,288 37,288, and 37,468 genes were expressed in TX-S1, TX-S2, TX-S3, QM601-S1, QM601-S2, and QM601-S3, respectively (Table 1), providing sufficient information for investigating root development.
Transcriptome analysis of clean reads from samples containing two young leaves and a bud (the tissue collected from each shoot of a tea plant during commercial harvesting), followed by de novo assembly, yielded ~ 51,143 unigenes. While 5693 genes were expressed only in buds/leaves, 2226 genes were expressed only in ARs (Fig. 3A), indicating that the number and expression patterns of genes in leaves vs. root tissues are different. Moreover, Pearson correlation analysis of transcriptome data from all samples versus replicates of the same tissue revealed that there was little variation among experimental replicates but significant differences among samples from different stages of root development (Fig. 3B). Therefore, the transcriptome data were reliable and could be used for further analysis.

Global identification of differentially expressed genes. A Venn diagram showing the number of DEGs between samples containing two leaves and a bud (left) and ARs (right). B Pearson correlation coefficients of transcriptome data from all samples. C Venn diagram showing the number of unique and overlapping DEGs in each comparison of the three stages of AR development in TX cuttings. D KEGG pathway enrichment analysis of the 2907 DEGs commonly expressed in TX-S1 vs. TX-S2 and TX-S1 vs. TX-S3
Global identification of differentially expressed genes
To explore DEGs involved in AR formation in tea cuttings, the current study analyzed the expression of unigenes in each library via FPKM. The results of statistical analysis of DEGs from nine comparisons (TX-S1 vs. TX-S2, TX-S1 vs. TX-S3, TX-S2 vs. TX-S3, QM-S1 vs. QM-S2, QM-S1 vs. QM-S3, QM-S2 vs. QM-S3, TX-S1 vs. QM-S1, TX-S2 vs. QM-S2, and TX-S3 vs. QM-S3) are shown in Fig. S1. The present study focused on specific DEGs from different comparison groups. The DEGs from TX-S1 vs. TX-S2, TX-S1 vs. TX-S3, and TX-S2 vs. TX-S3 exhibited similar KEGG enrichment patterns to the DEGs from QM-S1 vs. QM-S2, QM-S1 vs. QM-S3, and QM-S2 vs. QM-S3, respectively, suggesting that similar mechanisms are employed to regulate root initiation in different cultivars. There were more DEGs in S1 vs. S2 and S1 vs. S3 than in S2 vs. S3, as shown by the overlap in the Venn diagram (Fig. 3C), demonstrating the importance of analyzing DEGs during root callus formation. Numerous DEGs were detected during stage S1 of root development, highlighting the complex regulatory network that functions during this stage.
The current study then performed a detailed comparison of the DEGs between TX-S1 vs. TX-S2 and TX-S1 vs. TX-S3 were then performed. There were 2,907 DEGs common to both comparisons (Fig. 3C), which we analyzed using the KEGG database; the enriched pathways, including the top 4, are shown in Fig. 3D. They are phenylpropanoid biosynthesis (Ko00940, 109 unigenes, P-value = 3.48e−7), plant hormone signal transduction (Ko04075, 112 unigenes, P-value = 2.20e−5), plant MAPK signaling pathway (Ko04016, 115 unigenes, P-value = 7.69e−5) and plant-pathogen interaction pathway (Ko04626, 159 unigenes, P-value = 5e−3). A comparison between QM-S1 vs. QM-S2 and QM-S1 vs. QM-S3 yielded similar results. Moreover, a comparison of TX-S1 vs. QM-S1, TX-S2 vs. QM-S2, and TX-S3 vs. QM-S3 found that the respective DEGs were also mainly enriched in these four pathways. This global analysis provided an overall view of the DEGs and suggest that the differential expression of genes in these four pathways may be the key factor in the different capacities for AR formation in various tea cultivars.
DEGs mapped to hormone signal transduction, plant MAPK signaling, phenylpropanoid biosynthesis and plant–pathogen interaction pathways
The expression levels of DEGs involved in the plant hormone signal transduction pathway varied between the two tea cultivars (TX and QM601; Fig. S2-S3). DEGs involved in signal transduction pathways of various hormones showed great changes in expression at different stages of AR development, confirming their importance. For example, in the TX-S1 vs. TX-S3 comparison, 40 auxin genes were significantly differentially expressed. Most of these genes encode proteins involved in the transcriptional regulation of auxin responses, including indole-3-acetic acid-amido synthetases (GH3s), flavin monooxygenase (YUCCA)-like enzymes, auxin response factors (ARFs), and small auxin-up RNAs (SAURs) (Fig. S2). Notably, many CsGH3 and YUCCA family genes exhibited similar trends in expression from stages S1 to S3 (Table S3). Such as, the transcripts of CsGH3.3, CsGH3.4, CsYUCCA2 and CsYUCCA8 were highly expressed at stages S2 and S3 but were expressed at much lower levels at stage S1 (Fig. 4). These results suggest that many genes related to auxin biosynthesis were expressed at the initial stage of AR development and were upregulated during the formation of AR system architecture. However, CsGH3.2 exhibited the opposite trend: It was highly expressed at stage S1 but was expressed at much lower levels at stages S2 and S3 (Fig. 4). This result suggests that different mechanisms may simultaneously regulate the expression of specific CsGH3 genes involved in auxin storage or degradation during AR formation.

Differential expression of partial genes was analyzed based on the transcriptome data. Statistical analysis was performed by the least significant difference (LSD) test (P < 0.05); means do not differ if they are labeled with the same letter
Interesting, many genes that function in the ethylene and BR pathways, such as the ethylene receptor gene ETR1 (CSS0000918), BR receptor kinase gene BRI1 (CSS0010595), BRASSINOSTEROID RESISTANT1 (BZR1; CSS0034485), and BRASSINOSTEROID-6-OXIDASE1 (BR6OX1; CSS0004050), were expressed at higher levels in TX-S1 compared to TX-S2 and TX-S3 (Fig. 4). However, the expression levels of these key regulatory genes did not appear to fluctuate greatly during different stages of development in QM tea. In summary, the expression levels of multiple hormone signal transduction pathway-related genes exhibit dynamic changes during AR initiation and formation to satisfy the requirements for different proteins during AR development.
The DEGs during different stages of AR development, such as TX-S1 vs. TX-S3 and QM-S1 vs. QM-S3, were also significantly enriched in MAPK signaling, phenylpropanoid biosynthesis and plant–pathogen interaction pathways. Further analysis revealed that many genes, such as MITOGEN-ACTIVATED PROTEIN KINASE KINASE4 (MKK4; CSS0009636), MITOGEN-ACTIVATED PROTEIN KINASE4 (MPK4; CSS0013507), and the transcription factor genes WRKY22 (CSS0021156), which function downstream of MAPKs, were significantly upregulated at S1 but were expressed at low levels at S3. In addition, several peroxidase genes involved in phenylpropanoid metabolism, including CsPerox4 (CSS0008526), CsPerox16 (CSS0035213), and CsPerox27 (CSS0032700), were all expressed at low levels at stage S1 but at high levels at stage S3, perhaps reflecting the different demands for these phenylpropanoids during specific developmental stages. Moreover, many genes related to plant-pathogen interactions, such as the pathogenesis-related transcriptional activator gene PTI6 (CSS0000882), the disease resistance gene RPS2 (CSS0011038), and the LRR receptor-like serine/threonine-protein kinase gene FLS2 (CSS0014658), were significantly upregulated at S1 and downregulated at S3 (Fig. 4).
The above results suggest that plant hormone signal transduction, phenylpropanoid biosynthesis, the plant MAPK signaling pathway, and plant-pathogen interactions all play critical roles in AR formation in C. sinensis.
Characterization of the DEGs CsGH3.2 and CsGH3.3
The transcriptomic analysis described above identified numerous functional DEGs. To verify the reliability of the data and to investigate the physiological roles of candidate genes, the current study identified and characterized the coding regions of CsGH3.2 and CsGH3.3 from TX plants. The open reading frame of CsGH3.2 contains 1833 nucleotides encoding a protein of 611 amino acids. The open reading frame of CsGH3.3 contains 1794 nucleotides encoding a protein of 598 amino acids (Fig. 5A). Motif analysis and BLAST searches revealed that both CsGH3.2 and CsGH3.3 contain the conserved domain PF03321, an auxin-responsive domain found in GH3 gene promoters (Fig. 5B). The sequences of this domain in these genes contain several SNPs compared to the reference genome (http://tpia.teaplant.org/). However, the identified SNPs in these coding regions do not alter the amino acid sequences of the encoded proteins, pointing to the functional conservation of CsGH3.2 and CsGH3.3 in tea plants. Phylogenetic analysis showed that CsGH3.2 belongs to Group III of the GH3 family, while CsGH3.3 belongs to Group II. These genes share high sequence similarity with homologs from other species (Fig. 5C). CsGH3.2 shares 60.80% identity with AtGH3.9 (AT2G47750) from Arabidopsis and 53.29% similarity with OsGH3.11 (LOC_Os07g47490) from rice (Oryza sativa). CsGH3.3 shares 79.00% similarity with AtGH3.1(AT2G14960) from Arabidopsis and 53.29% similarity with OsGH3.2 (LOC_Os01g55940) from rice.

Gene structures, protein sequence analysis, and molecular characterization of CsGH3.2 and CsGH3.3. A Exon–intron structures of CsGH3.2 and CsGH3.3; exons are indicated by black boxes, and introns are indicated by black lines. B Auxin-responsive GH3 promoter domains were detected in the N-terminal regions of CsGH3.2 and CsGH3.3. C Phylogenetic analysis of GH3 protein in Camellia sinensis, Arabidopsis thaliana, and rice (Oryza sativa). CsGH3.2 and CsGH3.3 are highlighted in red. The phylogenetic tree was constructed using the neighbor-joining method and a bootstrap test with 1,000 iterations, performed in MEGA7. D Relative expression levels of CsGH3.2 and CsGH3.3 in AR stages S1–S3. **Significant difference compared to TX plants (P < 0.01, Student’s t-test). E Relative expression levels of CsGH3.2 and CsGH3.3 in different tissues and organs. R represents root, S stems, YL young leaves, OL old leaves, B buds, and F flowers
The current study performed qRT-PCR to examine the expression patterns of CsGH3.2 and CsGH3.3 in ARs at stages S1–S3. The qRT-PCR results were consistent with those of transcriptome analysis (Fig. 5D). The expression patterns of CsGH3.2 and CsGH3.3 in different tissues and organs were then examined. The expression level of CsGH3.2 was highest in buds, while the expression level of CsGH3.3 was highest in roots (Fig. 5E). The different expression patterns of CsGH3.2 and CsGH3.3 in tea plants suggest that they have specific functions in different organs. To examine the subcellular localizations of CsGH3.2 and CsGH3.3, the green fluorescent protein gene (GFP) was fused to CsGH3.2 and CsGH3.3, transiently transformed Nicotiana benthamiana leaves with the 35S::CsGH3s::GFP constructs, and examined the subcellular localization of the fusion proteins by confocal microscopy. The control protein (35S::GFP) was observed in the nucleus and throughout the cytoplasm, whereas CsGH3.2::GFP was detected throughout the cytoplasm and CsGH3.3::GFP was detected in chloroplasts (Fig. 6).

Subcellular localization of CsGH3.2 and CsGH3.3 in N. benthamiana leaves. p35S::CsGH3.2/CsGH3.3-GFP and p35S::GFP were transformed into N. benthamiana by A. tumefaciens–mediated infiltration. Signals from intact leaf mesophyll cells were detected by confocal laser scanning microscopy
Heterologously expressing CsGH3.2 and CsGH3.3 in rice represses root and shoot growth and development
Since most GH3 family members are upregulated by auxin (Staswick et al. 2005), the current study analyzed CsGH3.2 and CsGH3.3 expression in tea plants following exogenous IBA treatment. Both CsGH3.2 and CsGH3.3 were rapidly upregulated in roots after treatment with IBA (Fig. 7A, B), indicating that CsGH3.2 and CsGH3.3 are responsive to auxin and may be involved in auxin-dependent modulation of root architecture. To investigate their roles in plants in more detail, a gain-of-function approach by exogenously expressing these genes in rice was used. Of the 32 and 36 independent transgenic lines identified for CsGH3.2 and CsGH3.3, respectively, ~ 60 and 70% exhibited a morphology that differed from the control plants (empty vector).

Phenotypic characterization of transgenic CsGH3.2/CsGH3.3-overexpressing and control plants. A, B CsGH3.2 and CsGH3.3 expression in tea plants treated with 15 µM IBA for the indicated periods. h, hour. Expression levels were measured using qRT-PCR. C, D qRT-PCR analysis of the relative expression levels of CsGH3.2 and CsGH3.3 in CsGH3.2/CsGH3.3-overexpressing rice plants (OE1-OE10). Data are means ± SD (n = 4). **Significant difference compared to TX plants (P < 0.01, Student’s t-test). E Morphological phenotypes of CsGH3.2/CsGH3.3-overexpressing and control rice plants grown in soil for 2.5 months. F Morphological phenotypes of CsGH3.2/CsGH3.3-overexpressing and control rice plants grown in tissue culture medium for 1 month. G Longitudinal sections of root tips of control and CsGH3.3-overexpressing rice plants. Bar = 25 µm
The current study analyzed CsGH3.2 and CsGH3.3 expression in 10 independent transgenic rice lines via qRT-PCR (Fig. 7C, D). Both the aboveground and belowground parts of transgenic plants with high levels of CsGH3.2 and CsGH3.3 expression were smaller than the controls, with dwarf phenotypes and tufted shapes (Fig. 7E). Overall, the abnormal phenotypes were more severe in CsGH3.3- vs. CsGH3.2-expressing plants. It was difficult to induce callus formation from the CsGH3.3-expressing plants during tissue culture, and these plants showed poor survival on rooting culture medium. Moreover, CsGH3.3-expressing plants had significantly shorter and fewer roots than control and CsGH3.2-expressing plants (Fig. 7F). Cytological observation revealed significant decreases in cell size and cell number in the roots of CsGH3.3-expressing plants vs. the control (Fig. 7G). These results suggest that maintaining proper CsGH3.2 and CsGH3.3 expression levels is crucial for normal plant growth and development.
Discussion
AR formation is an organogenic and complex biological process, involves sophisticated regulatory cascade (Betti et al. 2019; Diaz-Sala 2020; Lee et al. 2019). At present, so as to reveal how stem cells switch to root meristem cells, combining of transgenic plant with fluorescent reporters and single-cell mRNA-sequencing profiling can be adopted in tomato or rice(Omary et al. 2020; Zhang et al. 2021), but not tea plants. Therefore, transcriptome and proteome analyses were generally used to uncover the mechanisms underlying AR formation in some tree species (Li et al. 2018; Wang et al. 2019; Wei et al. 2014). However, despite increasing knowledge of important players involved in AR development, the molecular mechanisms behind the successive, irreversible steps of the rooting process of tea cuttings are still unclear. The current study focused on the dynamic changes in gene expression during different stages of AR formation. Comparative transcriptome analysis was performed during callus formation, root initiation, and root elongation in two tea cultivars with different rooting abilities; more genes were detected during each stage of AR development vs. root elongation. These findings may provide a more comprehensive view of AR formation in tea cuttings than previously reported.
KEGG enrichment analysis primarily mapped the DEGs to four pathways: phenylpropanoid biosynthesis, plant hormone signal transduction, plant MAPK signaling, and plant-pathogen interaction (Fig. 3). It is not surprising that the first two pathways were enriched among the DEGs, since many studies have already revealed their important roles in root development (Druege et al. 2016; Wang et al. 2016; Wei et al. 2014). However, the plant MAPK signaling pathway and plant-pathogen interaction pathway were rarely reported to involve in AR formation of tea plants. These results give new insights into the molecular regulatory mechanisms underlying AR formation in tea plants. Notably, DEGs from TX-S1 vs. QM-S1 were enriched in these four pathways and were the same as DEGs identified in the other comparisons. These findings emphasize the important roles of these four KEGG pathways. In addition, they suggest that the differential expression of these genes might be the key factor affecting the rooting ability of the two tea cultivars.
The successive stages of AR development are governed by complex interactions among many phytohormones, together with other internal and external core factors (Druege et al. 2016; Xiao and Zhang 2020). Auxin is likely to be the “master regulator” in almost all these interactions (Druege et al. 2016; Pacurar et al. 2014; Xiao and Zhang 2020). Endogenous auxin content is tightly controlled by internal developmental signals. GH3 family genes function in auxin homoeostasis and are key regulators of root development (Fu et al. 2011; Mellor et al. 2016; Zhao et al. 2020). In present study, the expression of five GH3 genes dynamically changed during AR formation in tea (Table S3). Two of these genes, CsGH3.2 and CsGH3.3, were isolated and characterized. Notably, both of them were expressed at significantly lower levels in the hard-to-root cultivar QM compared to the easy-to-root cultivar TX (Fig. 5D). This result indicated that the transcript levels of GH3 genes may be related to rooting ability. Further analysis of their organ-specific expression (Fig. 5) and protein localizations (Fig. 6) suggested that CsGH3.2 and CsGH3.3 have distinct and specific functions at the organ and subcellular levels. In addition, qRT-PCR analysis revealed that both CsGH3.2 and CsGH3.3 are highly responsive to auxin (Fig. 7A, B) and may be involved in modulating root development. Consistent with this conclusion, transgenic rice plants expressing CsGH3.2 and CsGH3.3 showed auxin-deficient developmental defects to different degrees (Fig. 7). These results indicate that both CsGH3.2 and CsGH3.3 are functional and that endogenous auxin content may tightly regulate by specific GH3 proteins during the successive steps of the rooting process.
Although expressing CsGH3 genes inhibited root development, this study could not conclude that they are negative regulators of AR formation based on this finding. There are three ways to induce the accumulation of CsGH3 transcripts in plants: transgenic expression (Fig. 7D), exogenous IBA treatment (Fig. 7B), and regulation by internal developmental signals (Fig. 5D). The first type of treatment involves constitutive induction, supporting the notion that functional CsGH3 regulates plant development by affecting endogenous auxin content. However, the two other methods could be used to uncover the roles of CsGH3 expression as a developmental signal. The current study hypothesize that auxin over-accumulation induces endogenous CsGH3 (e.g. GH3.3) expression during AR formation. To some extent, the expression level of CsGH3.3 serves as an indicator of the requirement for endogenous active auxin in tea cuttings. The current study proposes that in TX, ARs are well developed at stage S3, at which point endogenous auxin levels are excessive and must be limited; therefore, CsGH3.3 is expressed at high levels in this cultivar. By contrast, in QM, AR development still needs to be promoted at this stage, and endogenous auxin might still need to be produced to satisfy this developmental requirement.
Based on the transcriptome data, the changes in the expression of hormone signal transduction pathway genes during AR development are generally the same in these two cultivars, but the changes are delayed or exhibit less fluctuation in QM vs. TX. These observations suggest that differences in the levels of important hormonal regulators during successive developmental stages might underly the differences in rooting ability of different tea varieties. Combined, these results underscore the important roles of hormone signaling in AR formation in tea plants, which helps build a comprehensive view of AR formation in tea cuttings.
Conclusion
Overall, our findings provide a comprehensive view of genes that potentially participate in internal developmental signal-induced AR formation in tea cuttings, and broaden our understanding of the rooting phase transition rather than certain stages of AR formation. The functional verification of CsGH3.2 and CsGH3.3 in transgenic rice help to validate the effectiveness of transcriptomic analysis and give new insights into the essential role of them in adventitious rooting. Further analysis of DEGs from other comparison groups may provide additional information related to AR formation. The present study would provide an interesting starting point to discover new genes involved in AR formation in tea cuttings.
Data availability
Supplementary material related to this article can be found in the online version.
Abbreviations
- AR:
-
Adventitious root formation
- DEGs:
-
Differentially expressed genes
- GFP:
-
Green fluorescent protein
- GH3:
-
Gretchen hangen3
- IAA:
-
Indole-3-acetic acid
- IBA:
-
Indole-3-butyric acid
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Ahkami A, Scholz U, Steuernagel B, Strickert M, Haensch KT, Druege U, Reinhardt D, Nouri E, von Wiren N, Franken P, Hajirezaei MR (2014) Comprehensive transcriptome analysis unravels the existence of crucial genes regulating primary metabolism during adventitious root formation in Petunia hybrida. PLoS One 9:e100997
Betti C, Della Rovere F, Ronzan M, Fattorini L (2019) EIN2 and COI1 control the antagonism between ethylene and jasmonate in adventitious rooting of Arabidopsis thaliana thin cell layers. Plant Cell Tiss Org 138:41–51
Chen M, Niu L, Zhao M, Xu C, Pan B, Fu Q, Tao Y, He H, Hou C, Xu Z (2020) De novo genome assembly and Hi-C analysis reveal an association between chromatin architecture alterations and sex differentiation in the woody plant Jatropha curcas. Giga Sci 9(2):giaa009
Davies F, Geneve R, Wilson S (2018) Hartmann and Kester’s plant propagation-principles and practices, 9th edn. Pearson
Davies F, 2017. What's new in the biology of cutting propagation, Proceedings of the 2017 annual meeting of the international plant propagators' society, pp. 321–330
Diaz-Sala C (2020) A perspective on adventitious root formation in tree species. Plants 9:1789
Druege U, Franken P, Hajirezaei MR (2016) Plant hormone homeostasis, signaling, and function during adventitious root formation in cuttings. Front Plant Sci 7:381
Fu J, Yu H, Li X, Xiao J, Wang S (2011) Rice GH3 gene family: regulators of growth and development. Plant Signal Behav 6:570–574
Huang XZ, Zhang XY, Yang SH (2009) A novel chloroplast-localized protein EMB1303 is required for chloroplast development in Arabidopsis. Cell Res 19:1225–1225
Huang J, Liang X, Xuan Y, Geng C, Li Y, Lu H, Qu S, Mei X, Chen H, Yu T, Sun N, Rao J, Wang J, Zhang W, Chen Y, Liao S, Jiang H, Liu X, Yang Z, Mu F, Gao S (2017a) A reference human genome dataset of the BGISEQ-500 sequencer. Giga Sci 6:1–9
Huang XZ, Zhang Y, Zhang XY, Shi YT (2017b) Long-chain base kinase1 affects freezing tolerance in Arabidopsis thaliana. Plant Sci 259:94–103
Kim D, Landmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357-U121
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357-U354
Lee HW, Cho C, Pandey SK, Park Y, Kim MJ, Kim J (2019) LBD16 and LBD18 acting downstream of ARF7 and ARF19 are involved in adventitious root formation in Arabidopsis. BMC Plant Biol 19(1):1–11
Li K, Liang Y, Xing L, Mao J, Liu Z, Dong F, Meng Y, Han M, Zhao C, Bao L, Zhang D (2018) Transcriptome analysis reveals multiple hormones, wounding and sugar signaling pathways mediate adventitious root formation in apple rootstock. Int J Mol Sci 19(8):2201
Lima JD, Bolfarini ACB, da Silva SHMG, Moraes WD (2013) Propagation of Camellia sinensis: effect of genotype, cuttings, substrate, recipient and indolebutyric acid. Hortic Bras 31:74–79
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):1–21
Mellor N, Band LR, Pencik A, Novak O, Rashed A, Holman T, Wilson MH, Voss U, Bishopp A, King JR, Ljung K, Bennett MJ, Owen MR (2016) Dynamic regulation of auxin oxidase and conjugating enzymes AtDAO1 and GH3 modulates auxin homeostasis. P Natl Acad Sci USA 113:11022–11027
Mondal TK, Bhattacharya A, Laxmikumaran M, Ahuja PS (2004) Recent advances of tea (Camellia sinensis) biotechnology. Plant Cell Tiss Org 76:195–254
Omary M, Gil-Yarom N, Yahav C, Steiner E, Hendelman A, Efroni I (2020) A conserved superlocus regulates above- and belowground root initiation. bioRxiv. https://doi.org/10.1101/2020.11.11.377937
Pacurar DI, Perrone I, Bellini C (2014) Auxin is a central player in the hormone cross-talks that control adventitious rooting. Physiol Plant 151:83–96
Puhan P, Vipparla A, Vemireddy LR, Anuradha G, Siddiq EA (2012) An efficient and universal Agrobacterium-mediated transformation protocol in rice. J Plant Biochem Biotechnol 21:252–260
Quan JE, Meng S, Guo EH, Zhang S, Zhao Z, Yang XT (2017) De novo sequencing and comparative transcriptome analysis of adventitious root development induced by exogenous indole-3-butyric acid in cuttings of tetraploid black locust. BMC Genomics 18(1):1–14
Rout GR (2006) Effect of auxins on adventitious root development from single node cuttings of Camellia sinensis (L.) Kuntze and associated biochemical changes. Plant Growth Regul 48:111–117
Seth R, Maritim TK, Parmar R, Sharma RK (2021) Underpinning the molecular programming attributing heat stress associated thermotolerance in tea (Camellia sinensis (L.) O. Kuntze). Hortic Res 8:99
Staswick PE, Serban B, Rowe M, Tiryaki I, Maldonado MT, Maldonado MC, Suza W (2005) Characterization of an Arabidopsis enzyme family that conjugates amino acids to indole-3-acetic acid. Plant Cell 17:616–627
Wang P, Ma L, Li Y, Sa W, Li L, Yang R, Ma Y, Wang Q (2016) Transcriptome profiling of indole-3-butyric acid-induced adventitious root formation in softwood cuttings of the Catalpa bungei variety ‘YU-1’at different developmental stages. Genes Genom 38:145–162
Wang Z, Hua J, Yin Y, Gu C, Yu C, Shi Q, Guo J, Xuan L, Yu F (2019) An integrated transcriptome and proteome analysis reveals putative regulators of adventitious root formation in Taxodium “Zhongshanshan.” Int J Mol Sci 20(5): 1225
Wei K, Wang LY, Cheng H, Zhang CC, Ma CL, Zhang LQ, Gong WY, Wu LY (2013) Identification of genes involved in indole-3-butyric acid-induced adventitious root formation in nodal cuttings of Camellia sinensis (L.) by suppression subtractive hybridization. Gene 514:91–98
Wei K, Wang LY, Wu LY, Zhang CC, Li HL, Tan LQ, Cao HL, Cheng H (2014) Transcriptome analysis of indole-3-butyric acid-induced adventitious root formation in nodal cuttings of Camellia sinensis (L.). PLoS One 9:e107201
Wei K, Wang L, Zhang C, Wu L, Li H, Zhang F, Cheng H (2015) Transcriptome Analysis Reveals Key Flavonoid 3’-Hydroxylase and Flavonoid 3’,5’-Hydroxylase Genes in Affecting the Ratio of Dihydroxylated to Trihydroxylated Catechins in Camellia sinensis. PLoS One 10:e0137925
Wei K, Wang L, Zhang Y, Ruan L, Li H, Wu L, Xu L, Zhang C, Zhou X, Cheng H, Edwards R (2019) A coupled role for CsMYB75 and CsGSTF1 in anthocyanin hyperaccumulation in purple tea. Plant J 97:825–840
Wilson S, Geneve R, Davies F, Sotala A (2018) New online supplemental resources for Hartmann and Kester’s plant propagation: principles and practices, 9th edn. Hortscience, pp S85–S85
Xia EH, Li FD, Tong W, Li PH, Wu Q, Zhao HJ, Ge RH, Li RP, Li YY, Zhang ZZ, Wei CL, Wan XC (2019) Tea Plant Information Archive: a comprehensive genomics and bioinformatics platform for tea plant. Plant Biotechnol J 17:1938–1953
Xia E, Tong W, Hou Y, An Y, Chen L, Wu Q, Liu Y, Yu J, Li F, Li R, Li P, Zhao H, Ge R, Huang J, Mallano AI, Zhang Y, Liu S, Deng W, Song C, Zhang Z, Zhao J, Wei S, Zhang Z, Xia T, Wei C, Wan X (2020a) The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mol Plant 13:1013–1026
Xia EH, Tong W, Wu Q, Wei S, Zhao J, Zhang ZZ, Wei CL, Wan XC (2020b) Tea plant genomics: achievements, challenges and perspectives. Hortic Res 7(7). https://doi.org/10.1038/s41438-019-0225-4
Xiao G, Zhang Y (2020) Adaptive growth: shaping auxin-mediated root system architecture. Trends Plant Sci 25:121–123
Zhang TQ, Chen Y, Liu Y, Lin WH, Wang JW (2021) Single-cell transcriptome atlas and chromatin accessibility landscape reveal differentiation trajectories in the rice root. Nat Commun 12:2053
Zhao D, Wang YT, Feng C, Wei Y, Peng X, Guo X, Guo XW, Zhai ZF, Li J, Shen XS, Li TH (2020) Overexpression of MsGH3.5 inhibits shoot and root development through the auxin and cytokinin pathways in apple plants. Plant J 103:166–183
Zhong J, Peng Z, Peng Q, Cai Q, Peng W, Chen M, Yao J (2018) Regulation of plant height in rice by the Polycomb group genes OsEMF2b, OsFIE2 and OsCLF. Plant Sci 267:157–167
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 31960615).
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
XH and RC: designed the study; XH and RC: conducted the research; XH: analyzed the data; XH, RC and DZ: wrote and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Communicated by Heidi Halbwirth.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, R., Zhao, D. & Huang, X. Transcriptome analysis of easy- and hard-to-root tea plants uncovers roles for CsGH3.2 and CsGH3.3 in adventitious root formation. Plant Cell Tiss Organ Cult 150, 385–398 (2022). https://doi.org/10.1007/s11240-022-02291-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-022-02291-8