
Abstract
In vitro regeneration of Viola stagnina Kit., endangered in most part of its European distribution range, was successfully obtained based on the newly developed protocol. Adventitious shoots via direct and indirect organogenesis were induced on leaf blade and petiole fragments on solidified Murashige and Skoog (MS) medium supplemented with 0.5 or 1 mg l−1 thidiazuron, respectively. Shoots were rooted on half-strength MS medium with 2% sucrose and 0.5 mg l−1 indole-3-acetic acid (IAA), and plantlets were successfully acclimatized. Sixty-five of the regenerated plants (72% of isolated shoots cultured on rooting medium) survived at the experimental plot conditions. Among recovered via organogenesis plants, individuals ‘true-to-type’ derived from initial plant G1 and also plants genetically distant from initial plants were detected by ISSR markers. In all groups of clones genetic indices (number of genotypes, polymorphic markers, gene diversity, total gene diversity, mean gene diversity) were lower than in natural populations. Regenerated plantlets had the same genome size estimated by flow cytometry as initial material and plants from natural populations. They developed chasmogamous flowers with highly viable pollen grains (over 90%), cleistogamous flowers, and set seeds from both flower types in the first and second seasons cultivated at experimental plots. This is the first report of a successfully developed micropropagation protocol of V. stagnina, and the first detailed genetic analysis of recovered plants with the use of ISSR markers and genome size measurements allowing to discuss the advantageous role of somaclonal variation in ex situ plant conservation with the use of in vitro micropropagation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Somaclonal variation (alteration of nuclear and organelle genomes) is an usual phenomenon accompanying in vitro micropropagation. It leads to the formation of individuals genetically differing from the initial plant (Bayliss 1973; Larkin and Scowcroft 1981; D’Amato and Bayliss 1985; Lee and Phillips 1988; Rodriguez-Enriquez et al. 2011; Neelakandan and Wang 2012; Shan et al. 2012; Wang and Wang 2012). The genetic diversity of regenerated plants versus the initial material should be evaluated with the use of molecular markers along with their ploidy and generative reproduction (pollen viability, seed set) before reintroduction (Karp 1994; Phillips et al. 1996; Bairu et al. 2011; Haque and Ghosh 2013; Nybom et al. 2014; Slazak et al. 2015a; Sebastiani and Ficcadenti 2016; Avila-Treviño et al. 2017; Bhattacharyya et al. 2017).
Punctual mutations in DNA are easily detectable by molecular markers but represent only one type of genetic changes accompanying in vitro propagation; changes of whole genome number (polyploidization) can also occur. The ploidy of regenerated plants strongly depends on explants’ polysomaty, which is an effect of endoreduplication, a process of DNA replication that is not followed by mitosis. Endoreduplication intensity in an individual plant varies among different tissue types (Kolano et al. 2009; Maluszynska et al. 2012). It usually coincides with an increasing of the size of some cells, which, in turn, correlates with a given tissue function (Barow 2006). A very useful method to estimate ploidy/endopolyploidy of plant material cultured in vitro is flow cytometry (FCM) (Thiem and Sliwinska 2003; Bairu et al. 2011; Ochatt et al. 2011; Viehmannova et al. 2014; Slazak et al. 2015a). In comparison with classical cytological methods like chromosome counting, FCM is faster, more accurate, and allow the establishment of nuclear DNA content in larger number of cells (Dhooghe et al. 2011). FCM analysis is especially important when polysomatic species are micropropagated. Polysomatic plant tissues are composed of a heterogeneous population of cells with different ploidy and morphogenetic potential, which could provide great instability of in vitro propagated plants (Pontaroli and Camadro 2005; Slazak et al. 2015a). It has been reported that the more 2C cells (C = DNA content of a holoploid genome with chromosome number n, Greilhuber et al. 2005) in the explant, the higher the frequency of 2C regenerated plants (Gilissen et al. 1994; Kubaláková et al. 1996; Doležel 1997). However, regenerated individuals with multiplied genomes (polyploids) can also occur. They cannot be used for reintroduction in ex situ conservation programs because they could violate genetic integrity of preserved species (Mallon et al. 2010).
Viola stagnina Kit. (fen violet) belongs to sect. Viola of subsect. Rostratae Küpffer and is considered to be a paleotetraploid (2n = 4x = 20, Kuta 1976; Marcussen and Nordal 1998); genome size 2C = 1.31 pg (Krasne near Rzeszów, S Poland. Kuta unpublished data). It is distributed in Central and Eastern Europe. V. stagnina is declining throughout its whole range due to the loss of its typical base-rich fen peat resulting from a lowering of water table in meadows, mainly by drainage but also by competition from taller-growing vegetation, and changes in agricultural practices (Danihelka et al. 2009). The species is endangered in many countries (Curtis and McGough 1988; Korneck et al. 1996; Niklfeld and Schratt-Ehrendorfer 1999; Wiggington 1999; Feráková et al. 2001; Zarzycki and Szeląg 2006; Grulich 2012; Kaźmierczakowa et al. 2014) and requires active conservation management for its preservation in the whole European distribution range.
Viola stagnina can spread by adventitious root buds or from the persistent seed bank after major, locally restricted disturbance events (Eckstein et al. 2006a). The seeds are formed from either cleistogamous (CL), obligatory self-pollinated flowers or from chasmogamous (CH) self- and cross-pollinated flowers. CL seeds have poorer germination percentage than CH ones. The share of CH seeds is dependent on the year and can be as low as 1% and as high as 61% but actually the production of CL seeds may be underestimated in this species due to the very long period of CL flowers forming (Eckstein et al. 2006a). The clonal propagation and autogamy of self-pollinated CL flowers are dominant reproduction modes in this species (Eckstein et al. 2006b). The seeds of V. stagnina germinate poorly in outdoor conditions, only 18% of seeds germinated. According to information from seed bank of the Botanical Garden—Centre for Biological Diversity Conservation, Polish Academy of Sciences, Warsaw its germination can even not reach 1%. Fresh seeds of V. stagnina in laboratory conditions show almost no germination, reducing the chance for easy plant multiplication. There is a need to develop ex situ preservation of this species using in vitro plant multiplication. Taking into account that natural populations are genetically differentiated, which results with the type of propagation, it would be unfavorable to introduce into the studied populations genetically uniform individuals (clones) obtained as a result of in vitro multiplication. Genetic diversity of micropropagated plants caused by somaclonal variation can be considered a beneficial process if introduced diversity overlaps genetic diversity of the species natural populations.
Only a few publications address in vitro culture of Violaceae. Most of those studies focus on micropropagation or callus induction of several rare and medicinal plants of the genera Hybanthus Jacq. and Viola L. (Prakash et al. 1999; Chalageri and Babu 2012; Naeem et al. 2013; Vishwakarma et al. 2013; Soni and Kaur 2014; Mokhtari et al. 2015; Slazak et al. 2015a, b) or micropropagation of heavy metal hyperaccumulators (Bidwell et al. 2001; Li et al. 2010). In vitro culture has also proved useful for propagation of ornamental pansy (Wang and Bao 2007) and in experimental embryology (Wijowska et al. 1999).
The goal of the study was to develop the efficient in vitro micropropagation protocol for the endangered V. stagnina and to evaluate the genetic diversity of regenerants in order to ex situ protection of this species.
We assumed the potential prevalence of somaclonal variability generated by in vitro culture conditions in the conservation practice. Introduction to the nature of regenerated plants covering the genetic variation of V. stagnina has an advantage over genetically uniform clones which might reduce genetic differentiation of the species.
Materials and methods
Plants from natural sites
Plants of V. stagnina were collected in May 2011 in North-eastern Poland under a permit from the Regional Directorate for Environmental Protection and originated from two localities: Piaśnickie Łąki (PL) nature reserve near Władysławowo (coll.&leg. Drs. Ryszard Marecki and Tomasz S. Olszewski; N54°49′26.95″, E18°03′35.23″; specimens are deposited in the Herbarium of Gdańsk University—UGDA, Gdańsk, Poland) and Garbas (G) near Suwałki (coll.&leg. dr. Artur Pliszko; N54°08′52.77″, E22°36′30.12″; specimens are deposited in the Herbarium of the Jagiellonian University—KRA, Cracow, Poland). From each population 4 or 5 plants were harvested and cultivated in pots under natural light at room temperature (22–25 °C) in the Department of Plant Cytology and Embryology (Jagiellonian University, Cracow) and used for studies.
Explants for in vitro culture
Young leaves with petioles were collected from plants and washed in water with addition of commercial detergent (Ludwik, Grupa INCO S.A., Warsaw, Poland) to remove any dust or soil from the surface. The sterilization procedure included immersion in 70% ethanol for 1.5 min. and 15% commercial bleach (Ace; Procter & Gamble DS Polska, Warsaw, Poland) with sodium hypochlorite (5% w/v) for 13–16 min. subsequently. The sterilizing solution was removed by washing material thrice with autoclaved distilled water under a laminar flow hood. Fragments of petiole (~ 10 mm long) and leaf blades (squares and rectangles ~ 8–10 mm) were used as explants. After excision with a sterilized surgical blade in sterile conditions explants were placed on medium-filled Petri dishes, diameter 90 mm (five explants per dish).
Culture media and conditions
Media were based on MS (Murashige and Skoog 1962) medium containing 3% sucrose (w/v), solidified with 0.8% agar (w/v), supplemented with plant growth regulators (PGRs) in different combinations and concentrations to select most effective for induction of organogenesis. 310 explants (220 leaf blade fragments and 90 petiole) were used in this test experiment (Table S1). One PGR, thidiazuron (TDZ) at two concentrations (0.5 mg l−1, 1 mg l−1) was selected as giving the best efficiency of organogenesis and further 712 explants (284 petioles and 428 leaf blade fragments) were cultured. The pH of media was adjusted at 5.7–5.8.
Cultures were maintained in a growth chamber at 25 ± 3 °C under a 16 h photoperiod (cool-white fluorescent lamps, 60–90 µmol m−2 s−1).
Rooting of shoots was achieved on ½ MS + 0.5 mg l−1 indole-3-acetic acid (IAA) + 2% (w/v) sucrose; plantlets were transplanted to plastic pots with autoclaved commercial organic garden soil and kept in a plastic mini-greenhouse for 2 weeks to harden. The hardened plants were cultivated at an experimental plot in Modlnica near Cracow and monitored for two seasons. The whole experiment was repeated five times in years 2011–2014.
Flow cytometry for estimation explants endopolyploidy and plants genome size
Samples of initial explants, leaf blades, and petioles were prepared as previously described (Thiem and Sliwinska 2003), using two-step procedure (Otto 1990). 4′,6-Diamidino-2-phenylindole (DAPI; 2 µg/ml) was used for DNA staining. Analyses were performed on five biological replicates by a Partec CCA (Partec GmbH, Münster, Germany) flow cytometer, equipped with a HBO mercury arc lamp, using a logarithmic amplification. For each sample, DAPI fluorescence of 3000–7000 nuclei was analyzed. Histograms were evaluated using the DPAC v. 2.2 program (Partec GmbH, Münster, Germany). The proportion of nuclei with different DNA contents, the (4C + 8C)/2C ratio, and the mean C-value (mean ploidy; Lemontey et al. 2000) were established. Nuclei having at least 8C DNA content were considered to be endopolyploid, since it is not possible to distinguish by FCM the 4C nuclei in cells that have just entered endoreduplication (i.e. being in the G2 phase of the first endocycle) from those within cells in the G2 phase of the mitotic cycle.
For genome size estimation, leaf samples of initial plants from two natural locations and of selected regenerated in vitro plantlets were prepared as described above; propidium iodide (PI; 50 µg/ml) instead of DAPI was used for DNA staining. Solanum lycopersicum cv. Stupicke (2C = 1.96 pg; Doležel et al. 1992) was used as internal standard. For each sample, PI fluorescence was measured in at least 5000 nuclei, using a CyFlow SL Green (Partec GmbH, Münster, Germany) flow cytometer, equipped with a high-grade solid-state laser with green light emission at 532 nm, as well as with SSC and FSC scatters. Analyses were performed using linear amplification. Histograms were evaluated using FloMax software (Partec GmbH, Münster, Germany). CV of the G0/G1 peak of V. stagnina ranged between 3.69 and 8.87%. Nuclear DNA content was calculated using the linear relationship between the ratio of the 2C peak positions of Viola/internal standard on a histogram of fluorescence intensities. Totally, for genome size estimations, 12 plants from natural habitats (PL, G) and 31 plants regenerated via direct and indirect organogenesis were analyzed.
FCM results were statistically analyzed using a one-way ANOVA (preceded by test of homogeneity of variance) and a Tukey HSD tests post hoc (P = 0.05) (Statistica 10 software, StatSoft, Cracow, Poland). The percentage data were subjected to analysis of variance after angular transformation, but actual percentages are presented in the Table 1.
Histological examination of organogenesis
Petiole and leaf blade fragments after 4 weeks of culture on MS + 1 mg l−1 or 0.5 mg l−1 TDZ were fixed in the mixture of glutaraldehyde and paraformaldehyde overnight, washed four times in 0.1 M phosphate buffer (pH 7.2) and then dehydrated in an ethanol series (10%, 30%, 50%, 70%, 96%; 15 min each) and kept overnight in absolute ethanol. Fixed tissue samples were embedded in Technovit 7100, sectioned 5 µm thick, stained with toluidine blue, mounted in Entellan (Merck, Darmstadt, Germany) as described in Slazak et al. (2015a). LM sections were photographed with a Nikon DS-Fi2 digital camera on Nikon Eclipse E400 light microscope using NIS-Elements D software (Nikon, Tokyo, Japan).
Adventitious shoot initials and callus in scanning electron microscopy
The explants (petiole and leaf blade fragments) were prefixed in 5% buffered glutaraldehyde (0.1 M phosphate buffer, pH 7.2) for 2 h at room temperature. After dehydration in a graded ethanol series, samples were CO2 critical-point dried (EMITECH K850 system), sputter-coated with gold (SPI SUPPLIES ion sputtering system) and observed with a HITACHI S-4700 scanning electron microscope.
Examination of flower elements and pollen grains viability (stainability) and size by histochemical test
Chasmogamous flowers of plants from natural population (G) and regenerants were fixed in a mixture of ethanol and acetic acid (3:1 v/v) and flower elements were observed with stereo microscope.
Dry pollen of herbarium specimens of plants from natural population, and fixed of regenerants was used for analysis. Viability test was performed with Alexander’s dye. In this staining viable pollen grains stain purple, non-viable are green (Singh 2003). A total of 1036 pollen grains from 10 flowers of five herbarium specimens (KRA 0421202, 0421203, 0421207, 0421208, 0421209) originating from G population and 3825 pollen grains of 19 regenerated in vitro plants were analyzed under light microscope. The diameter of 50 pollen grains from each plant from natural population (G) and regenerated in vitro plantlets was measured using NIS-Elements D program (Nikon). Statistical analysis of pollen viability and size data was performed in Statistica 10 software using Kruskal–Wallis ANOVA with multiple comparisons.
Evaluation of genetic diversity of regenerants versus initial and plants from natural sites
Twenty-five individuals originating from two localities in northern Poland (G and PL) were used to estimate intra- and inter-populational genetic variation. 55 regenerants obtained via organogenesis were subjected to ISSR analysis.
For isolation of DNA, fully developed leaves were used, showing no damage symptoms caused by insects or fungi. Leaves were freshly collected and placed in silica gel (Sigma-Aldrich, Saint Louis, MO, USA) to dry. DNA was extracted with DNeasy Plant Kit (Qiagen, Hilden, Germany) according to instructions provided by manufacturer of isolation kit. The quality of genomic DNA was checked using electrophoresis on 1% agarose gel and concentration of DNA was measured by NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). PCR reactions of ISSR markers used six primers (Table S2) (Stepansky et al. 1999). 25 µl of reaction mixture used for amplification comprised: a 2.5 µl tenfold concentrated reaction buffer supplied by the DNA polymerase manufacturer (Thermo Scientific, Waltham, MA, USA), 1.4 units of Taq polymerase (recombinant), 1.5 mM MgCl2, 0.19 mM of each dNTPs (Thermo Scientific), 27 pmol of a primer and 100 ng of a template DNA. 2720 thermal cycler (Applied Biosystems, Waltham, MA, USA) was used to conduct reactions. The annealing temperature for primers ISSR1, ISSR3, ISSR5 was 47 °C, and for ISSR2, ISSR4, ISSR6 was 44 °C. For optimal reaction conditions, thermal cycler was programmed as follows: initial denaturation: 94 °C—5 min, 42 amplification cycles: denaturation 94 °C—59 s, annealing 47 °C (44 °C)—59 s, elongation 72 °C—59 s, final elongation 72 °C—7 min. Reaction without a template DNA was included in each amplification as a negative control. PCR products were separated during electrophoresis in 1.5% agarose gel in 1× TBE buffer, stained with ethidium bromide (500 µl l−1) for about 1.5 h at 100 V. Band patterns were observed and archived with an Imagemaster VDS with Liscap Capture 1.0 software (Amersham Biosciences, Uppsala, Sweden).
Analysis of obtained band patterns involved GelScan 1.45 software (Kucharczyk TE, Warsaw, Poland). Using calibration curve based on the band pattern of markers length (GeneRuler 100 bp, Thermo Scientific) the molecular weights of the amplification products were determined. ISSR reproducibility tests (Bonin et al. 2004) were conducted for the DNA extracts.
POPGENE 1.31 (Yeh et al. 1999) and FAMD 1.25 (Schlüter and Harris 2006) software were used to calculate genetic parameters of populations and analyzed groups of individuals according to Nei (1973). Calculations of genetic distance (FST) and its statistical significance were performed with 1000 permutations in ARLEQUIN v.3.5 (Excoffier et al. 2005). Neighbour Net (NN) was computed with SplitsTree v.4.6 (Huson and Bryant 2006) using a matrix of Nei–Li coefficients (Nei and Li 1979); bootstrap was performed with 1000 replicates. Principal coordinate analysis (PCoA) was performed in FAMD 1.25 and based on Nei–Li genetic distance matrix. To measure the degree of somaclonal variation, frequency of maternal plant genotype and number of private markers not present in ancestral natural population were calculated for each group of regenerated plants. Calculations were done in Arlequin v. 3.5.2 (Excoffier et al. 2005) and FAMD v. 1.31 (Schlüter and Harris 2006). In order to show relationships between regenerated and genotypes, as well as among genotypes from natural populations, a minimum spanning network (MSN) was constructed (Bandelt et al. 1999) with forcibly imposed ancestry between maternal and regenerated genotypes. MSN was calculated and visualized in PopART v. 1.7 (Leigh and Bryant 2015).
Results
Endopolyploidy of explants
In the leaf and petiole of V. stagnina nuclei with 2C, 4C, and 8C were detected, indicating their polysomaty (Table 1). Only in leaves originated from the PL population endopolyploid nuclei (8C) were not present, although relatively high proportion of 4C nuclei (22%), similar to that found in the remaining plant material, was detected. The leaves of plants from both populations (PL, G) possessed higher frequency of 2C nuclei (75–78%) than petioles (65–67%). Moreover, in leaves none or a few (from G; 1.86%) endopolyploid (8C) nuclei occurred, whereas in petioles 7.01–9.71% of 8C nuclei were detected. Consequently, the mean C-value in the petiole was higher than in the leaf, which was especially evident in the material from PL. The (4C + 8C)/2C ratio did not differ significantly, regardless of the organ and population location (Table 1).
Shoots multiplication
The highest frequency (70.5–84.9%) of shoot development was on MS + TDZ (0.5 mg l−1 or 1.0 mg l−1), both on petiole and leaf fragments. Combinations of auxins (NAA, 2,4-D) and cytokinins (BAP, KIN) in different sets and concentrations resulted in low percentage (0–45%) of explants producing shoots (Table S1). For further investigation exclusively media supplemented with TDZ were used.
Shoots were formed directly on petiole and leaf fragments on both TDZ concentrations and also indirectly via callus on 1 mg l−1 TDZ (Table 2; Fig. 1a). The efficiency of organogenesis ranged from 13 to 75.9% depending on the explant, concentration of TDZ and type of organogenesis (direct, indirect). The percentage of responding explants on higher (1 mg l−1) TDZ concentration was lower when analyzing separately the data of both types of organogenesis and explants (Table 2).

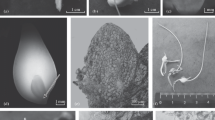
Micropropagation, rooting, acclimatization of V. stagnina (a–g), chasmogamous flower and pollen of plants from natural population (h–j) and of regenerant (k–m). Indirect shoot formation on petiole (a) after 8 weeks of culture on MS medium with 0.5 and 1 mg l−1 TDZ, longitudinal section of adventitious shoot forming directly from leaf blade after four weeks of culture (b), young shoots (asterisks) developing on MS + 0.5 mg l−1 TDZ directly from the leaf explant after 7 weeks of culture (c), callus (arrowheads) proliferating on MS + 1 mg l−1 TDZ covered by fibrous (arrows) ECM (d), regenerated shoot rooted on ½ MS + 0.5 mg l−1 IAA + 2% sucrose (e), regenerated plants grown at experimental plot in Modlnica developing chasmogamous flowers (f) and capsules (arrows in g), chasmogamous flower of plant from natural population (h), pistil (i) and five stamens, two with anther nectaries (asterisks), visible anther appendages (arrow) (j), chasmogamous flower (k), pistil (l), anthers (m) of regenerated plant, Alexander stained pollen grains from plant taken from natural population (n) and from regenerant (o). Bar 1 cm (a), 100 µm (b, d), 300 µm (c), 1 mm (i, j, m), 500 µm (l), 25 µm (n, o)
Shoots (small shoot buds) started to develop after 3-weeks-culture, on cut edges of petiole and leaf fragments (Fig. 1a, b). In subsequent days of culture adventitious shoots covered almost whole surface of the explants and their real numbers were uncountable (Fig. 1a). Surface of adventitious shoots was smooth, with regular epidermis cells (Fig. 1c). Induction of organogenesis had been continued till 10th week of culture even after excision of mature shoots from the explants.
Remarkable callus proliferation was noticed on medium with 1 mg l−1 TDZ on the cut edges of explants but not on 0.5 mg l−1 TDZ (Table 2). Callus was compact, with large cells and partly covered with fibrous extra cellular matrix (ECM) (Fig. 1d).
Rooting and acclimatization of regenerants
Ninety shoots induced on petiole and leaf fragments on MS media supplemented with TDZ (0.5 mg l−1, 1 mg l−1) were removed from the explants and transferred to rooting medium. Seventy-seven (86%) developed roots (35 from petiole and 42 from leaf blade) on 1/2 MS + 0.5 mg l−1 IAA + 2% sucrose (Fig. 1e). Seventy-two (94% of rooted plants) were acclimatized indoor in pots with organic soils. Finally, sixty-five (90% of rooted plants) survived for two vegetative seasons at the experimental plots.
Regenerants versus plants from natural sites
Flower and pollen characteristic, reproduction
Regenerated plants developed CH (Fig. 1f) and CL flowers, fruits (capsules) (Fig. 1g), and set seeds from both flower types, in the first and second season after the introduction to the experimental plots in the same seasonal cycle as plants from natural populations (CH flowers in May, CL June–September/October). There was no difference in morphology of CH flowers and flower organs (pistil, anthers, anther nectariferous appendages on two out of five stamens), pollen size and viability between plants from natural populations (Fig. 1h–j, n) and regenerated in vitro (Fig. 1k–m, o). The frequency of viable pollen grains estimated by Alexander test of plants from natural population (G) was very high (over 96%) and their mean diameter − 26.44 µm (Table 3). Pollen viability of 16 out of 19 regenerated plants was also very high (mean for both populations 93.18%) with no significant differences between pollen viability of plants from natural locality (Table 3). Pollen of three regenerated plants (15%) showed reduced viability (below 80%) and these result were excluded from statistical analysis. The mean diameters of pollen grains of 16 regenerants was almost equal being 28.37 µm and 28.71 µm, on 1 mg l−1 and 0.5 mg l−1, respectively and they did not differ from pollen diameter of plants from natural site (Table 3).
Genetic diversity of regenerants versus initials and plants from natural populations
Genetic variation in natural populations of V. stagnina was considered as a reference for somaclonal variation assessment in recovered in vitro plants. It yielded a total of 157 clearly resolved bands. The number of polymorphic bands within natural populations of V. stagnina was high and ranged from 50 (31.85%) in PL to 94 (59.87%) in G. The average number of polymorphic bands within regenerated plants was much lower and ranged from 0 in regenerants of G1 and PL3 initial plants to 56 (35.67%) in regenerants of PL2 maternal plant. Total number of polymorphic bands in natural populations was slightly higher (103; 65.61%) than among all regenerated plants (96; 61.15%). The highest number of private markers was in G population (23) and there was a lack of private markers in PL and regenerants of G1 and PL3 maternal plants. Discriminating markers were absent in all analyzed groups. The highest number (19) of genotypes in groups were noted in G natural population. The number of genotypes in each population was almost as high (19) as the number of analyzed individuals (20) in G or equal (5/5) in PL. The number of genotypes in groups of regenerated plants was evidently lower. Regenerants originated from initial plants G1 and PL3 represented low genetic variability with only one genotype. Eleven genotypes in a group of regenerants of initial plant PL2 represented 50% of total number of analyzed individuals and was the highest within the whole regenerants (Table 4).
Nei’s gene diversity (Hj) in all plants from PL and G grouped jointly was higher (0.20) than in group of all regenerants regardless of the initial plants (0.18). Hj values in groups of regenerants (0–0.10) were lower than values recorded for V. stagnina plants in natural populations (0.12–0.17). In natural populations total gene diversity (HT) and mean gene diversity within populations (HS) were higher than in regenerants (0.20 vs. 0.16 and 0.15 vs. 0.04, respectively), while Nei’s gene diversity between natural populations (GST) and the variance among populations (FST) was lower than between plants recovered in vitro (0.28 vs. 0.77 and 0.34 vs. 0.74, respectively). Values of FST were highly statistically significant (P < 0.001) (Table 4).
PCoA (Fig. S1) showed three distinct groups which did not correspond to division on two natural populations and had the highest variance percentage (53.07%) along the first component. The first group comprises only G population individuals, the second group some individuals of G population along with regenerants (RPL2) of initial plant PL2, the third group, strongly diversified, contains initial plants (G1, PL1, PL2, PL3), some regenerants (RPL1, RPL3, RG1) and several plants from natural populations (PL4, PL5 and G2–G20) not used for in vitro culture. Regenerants of PL1, PL3 and G1 grouped together with their initial plants, whereas all regenerants of PL2 jumped out of their maternal plant (Fig. S1).
The NN analysis (Fig. 2) including initial plants and regenerants showed two clearly diverged groups with strongly supported split (bootstrap: 95.7): one represented by all regenerants of PL2 initial plant and the second by initial plants PL1, PL3 and G1 and their regenerants.

NeighborNet of the ISSR analysis based on matrix of Nei–Li genetic distances performed on V. stagnina plants from natural populations and in vitro propagated clones. Bootstrap was performed on 1000 replicates
Of the four regenerated plant groups visible in MSN (Fig. 3), only RG1 had the frequency of maternal genotype at 1, and an absence of private bands, while the rest of the groups exhibited some degree of somaclonal variation. The lowest variation was shown by RPL1 (freq = 0.61; Nprt = 8), while RPL2 plants exhibited highest somaclonal variation, as no maternal genotype was present in any of the regenerated plants and the number of private markers was also the highest (Nprt = 33). Two regenerants derived from PL3 had identical genotypes, however they differed from PL3 genotype. The overall distance between regenerant genotypes of RPL2 and maternal plant was also the highest, with the greatest number of mutations (50) between PL2 and R33PL2, which is much higher than maximal number of changes between wild genotypes (32). In RPL1 the distance was considerably smaller, ranging from 1 to 15 mutations, while more than 60% of regenerants had genotypes identical with PL1 (Fig. 3). In total, 22 (40%) regenerants were genetically uniform with their initial plants and 33 (60%) regenerants were distant from initial plants.

MSN of 21 wild V. stagnina genotypes (black circles) along with four maternal plant and 22 regenerated genotypes (grey circles). Size of circles represents genotype frequency. Number of mutations between genotypes is given in the brackets
DNA content/ploidy of regenerants versus plants from natural populations
Thirty-one selected regenerated plants, regardless of culture media composition and origin of initial plants, possessed genome size 2C = 1.30 pg. There were no significant differences between 2C DNA values of regenerated plantlets and plants from natural sites. All regenerated plantlets represented the same ploidy level as plants from two (G, PL) natural populations (Table 5).
Discussion
Efficient and reproducible protocol for V. stagnina micropropagation
Sixty-five regenerants were obtained from petiole and leaf blade fragments of initial plants via direct and indirect organogenesis, confirmed by SEM and histological observations, on MS medium supplemented with TDZ, and successfully acclimatized at the experimental plot. The morphogenic nature of the callus was confirmed by the presence of extracellular matrix (ECM) covering the surface of the tissue similarly as in endosperm-derived callus (Popielarska-Konieczna et al. 2008, 2013) and Arabidopsis thaliana callus (Żabicki et al. 2013).
In the majority of papers on Violaceae micropropagation, indirect organogenesis was achieved using more complex medium supplemented with different PGRs (TDZ, 2,4-D, BAP, NAA, IBA, GA3) in various concentrations and combinations (Bidwell et al. 2001; Wang and Bao 2007; Li et al. 2010; Chalageri and Babu 2012; Naeem et al. 2013; Soni and Kaur 2014; Mokhtari et al. 2015). Similarly, in all previous studies on Violaceae the endoploidy of the explants cells’ was not estimated by FCM, with the exception of V. uliginosa (Slazak et al. 2015a). The knowledge about the frequency of 2C cells in the initial explants permits the ploidy of regenerated plants to be predicted, especially if plants that are obtained via direct organogenesis. It has been reported previously that 2C cells express higher totipotency than those with higher DNA content (Gilissen et al. 1994; Kubaláková et al. 1996). Nevertheless, polysomatic explants used for in vitro culture can also yield regenerants of increased ploidy. In the present study, despite that polysomatic petioles and polysomatic leaf blades being taken as explants none polyploid plantlets were obtained, confirming the higher totipotency of the 2C cells.
In vitro culture conditions generate somaclonal variation in V. stagnina regenerants?
There is a risk of genetic integrity loss due to stressful in vitro conditions inducing genetic and epigenetic alterations in regenerants. For the last several years, molecular markers were extensively used to verify the genetic fidelity of micropropagated plants and to identify genotypes resulting from somaclonal variation. In available for Violaceae micropropagation protocols, the genetic diversity of regenerated plants was not estimated by molecular markers (Sato et al. 1995; Prakash et al. 1999; Bidwell et al. 2001; Wang and Bao 2007; Li et al. 2010; Naeem et al. 2013; Mokhtari et al. 2015). In some papers the results of ISSR and RAPD was limited to simple comparison of band patterns (Chalageri and Babu 2012; Soni and Kaur 2014) or to a few basic statistical analyses (Slazak et al. 2015a).
Our present comprehensive analysis revealed genetic variations among regenerated plantlets. Genetic fidelity of regenerants with maternal plants was dependent on initial plant origin and was the lowest in the case of PL2 plant. Distribution of samples representing in vitro recovered plants in PCoA and NeigbourNet showed some groups of regenerants situated very close to their initial plants and some groups distant from them. These results correspond with low numbers of genotypes, private markers and genetic parameters (Hj and HT) for closely related somaclones and higher values for distant derivates. Hj and HT values in all micropropagated plants were lower than in natural populations. Results, especially MSN clearly revealed that somaclonal variation is present at some degree in nearly all of the regenerated plant groups but with relatively small number of punctual mutations in most cases. Regenerants derived from initial plant G1 maintained complete genetic stability and uniformity and showed no evidence of somaclonal variation at all.
Genome multiplication leading to polyploid (autopolyploid) plants recovered in tissue cultures is one of somaclonal alteration (Leva et al. 2012). All regenerated plantlets of V. stagnina possessed the same genome size (ploidy) as initial plants and plants from natural populations (G, PL). Lack of polyploid regenerants obtained via indirect shoot formation on MS + 1 mg l−1 TDZ stays in contrast to results on Viola uliginosa micropropagation under the same in vitro conditions and media composition; among regenerated plantlets, 35.7% represented 4× (Slazak et al. 2015a).
The range of genetic diversity of V. stagnina natural populations resulted from reproduction system
Genetic diversity of V. stagnina from two populations (G and PL) revealed genetic variation similar to Central European populations of this species estimated by AFLP method (Eckstein et al. 2006b) and other species belonging to the same subsection Rostratae of section Viola (V. riviniana and V. reichenbachiana) (Kuta et al. 2014; Migdałek 2015). Nei’s (1973) gene diversity between populations and the variance among populations computed in this study were similar to V. stagnina from German and Czech peripheral populations (Eckstein et al. 2006b). The genetic diversity of Polish natural populations of V. stagnina is characteristic for a mixed breeding system, formation of out-crossing CH and obligatory self-pollinated CL flowers. High numbers of genotypes in each population (G and PL), high numbers of private markers (23) in G population and also the presence of individuals from both populations in two strongly diverged groups in the NeigbourNet suggest high genetic diversity of V. stagnina. This might be explained by paleotetraploid origin of V. stagnina (Marcussen and Nordal 1998). Also pollination experiments indicate that there may be outbreeding depression among distant populations of V. stagnina resulting from low gene flow and high genetic differentiation among populations (L. Eckstein et al. unpublished data based on) Eckstein and Otte 2005.
Regenerants represent the same reproductive system as plants from natural populations
Flowering period of CH and CL flowers covers the cycle of V. stagnina from natural populations. The first flowers in a season are CH flowers (May) followed by CL long-flowering flowers (end of May–September/October). Floral morphology and generative organ structure of regenerated plants did not differ across plants from natural sites. The success of generative reproduction depends, among other things, on pollen viability and seed set. Low pollen viability and pollen grains differing in size (very small and giant) indicate disturbed microsporogenesis, typical in interspecific hybrids (Kuta 1990), aneuploids, autopolyploids, and also impact of environmental pollution (Dafni and Firmage 2000; Słomka et al. 2010, 2012; Migdałek et al. 2014). The pollen of 16 regenerated V. stagnina plants was highly viable and uniform in size, comparable to the pollen of plants from a natural site. Regenerated plants set seeds both from CH and CL flowers at experimental plot conditions.
Advantages of somaclonal variation excluding genome multiplication in conservation practice
The question arises: in conservation practice does the ploidy and genetic identity of micropropagated plants need to match those of the initial material?
Producing genetically identical somaclones (true-to-type plant material) by in vitro micropropagation, an ex situ conservation technique, might have a negative impact on population genetic structure. Uniform genotypes are sensitive to pathogens attacks and reduce the genetic diversity of the population which could be enhanced by inbreeding by self-compatible species or developing CL obligatory self-pollinated flowers as many violets. Introduction to the natural habitats of diverse genetic micropropagated plantlets of V. stagnina, covering genetic diversity and morphological variability of plants from natural sites, will maintain the genetic variation of this species populations. Sexually reproducing regenerating plants that produce seeds both from CH and CL flowers and have the same mean genome size/ploidy as plants from nature (4x/1.3 pg vs. 4x/1.3 pg) are a good source of material for re-introduction.
Somaclonal variation is an alternative tool to new technologies, such as genetic manipulation, to obtain relatively rapidly genetic variability and was successfully used in crops, medicinal and ornamental plants improvement (Abdellatif et al. 2012; Slazak et al. 2015a, b; Krishna et al. 2016).
Conclusions
Somaclonal variation influencing genetic diversity of regenerated plants without genome multiplication plays advantageous role in conservation practice of endangered species.
-
1.
Highly effective and reproducible protocol developed for V. stagnina micropropagation enables the production of regenerants to be introduced into natural sites as an ex situ conservation technique of endangered V. stagnina.
-
2.
Somaclonal variation although presented at some degree in nearly all of the regenerated plant groups introduced genetic differentiation which covered genetic variability of V. stagnina natural populations but did not influenced genome multiplication. All regenerants possessed the same genome size as initial plants and those originated from natural populations.
-
3.
Floral morphology and the reproductive system of regenerated in vitro plantlets are true-to-type plant material.
-
4.
In conservational practice, before reintroduction of any plants originated from in vitro cultures, genetic variation of propagated in vitro plants along with their genome size/ploidy evaluation, and reproductive system assessment (vs. plants originated from natural populations) should be performed routinely.
References
Abdellatif KF, Hegazy AE, Aboshama HM, Emara HA, El-Shahed AA (2012) Morphological and molecular characterization of somaclonal variation in tissue culture-derived banana plants. J Genet Eng Biotechnol 10:47–53
Avila-Treviño JA, Muñoz-Alemán JM, Pérez-Molphe-Balch E, Rodríguez-Sahagún A, Morales-Domínguez JF (2017) In vitro propagation from bud and apex explants of Moringa oleifera and evaluation of the genetic stability with RAMP marker. S Afr J Bot 108:149–156
Bairu MW, Aremu AO, van Staden J (2011) Somaclonal variation in plants: causes and detection methods. Plant Growth Regul 63:147–173
Bandelt H, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16(1):37–48
Barow M (2006) Endopolyploidy in seed plants. BioEssays 28:271–281
Bayliss MW (1973) Origin of chromosome number variation in cultured plant cells. Nature 246:529–530
Bhattacharyya P, Kumar V, van Staden J (2017) Assessment of genetic stability amongst micropropagated Ansellia africana, a vulnerable medicinal orchid species of Africa using SCoT markers. S Afr J Bot 108:294–302
Bidwell SD, Pederick JW, Sommer-Knudsen J, Woodrow IE (2001) Micropropagation of the nickel hyperaccumulator, Hybanthus floribundus (Family Violaceae). Plant Cell Tissue Organ 67:89–92
Bonin A, Bellemain E, Bronken Eidesen P, Pompanon F, Brochmann C, Taberlet P (2004) How to track and assess genotyping errors in population genetics studies. Mol Ecol 13:3261–3273
Chalageri G, Babu UV (2012) In vitro plant regeneration via petiole callus of Viola patrinii and genetic fidelity assessment using RAPD markers. Turk J Bot 36:358–368
Curtis TGF, McGough HN (1988) The Irish Red Data Book 1. Vascular plants. Wildlife Service Ireland, Stationary Office, Dublin
D’Amato FD, Bayliss MW (1985) Cytogenetics of plant cell and tissue cultures and their regenerates. Critical Rev Plant Sci 3:73–112
Dafni A, Firmage D (2000) Pollen viability and longevity: practical, ecological and evolutionary implications. In: Dafni A, Hesse M, Pacini E (eds) Pollen and pollination. Springer, Vienna, pp 113–132
Danihelka J, Niklfeld H, Šípošová H (2009) Viola elatior, V. pumila and V. stagnina in Austria, Czechia and Slovakia: a story of decline. Preslia 81:151–171
Dhooghe E, van Laere K, Eeckhaut T, Leus L, van Huylenbroeck J (2011) Mitotic chromosome doubling of plant tissues in vitro. Plant Cell Tissue Organ Cult 104:359–373
Doležel J (1997) Application of flow cytometry for the study of plant genomes. J Appl Genet 38:285–302
Doležel J, Sgorbati S, Lucretti S (1992) Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiol Plant 85:625–631
Eckstein RL, Otte A (2005) Effects of cleistogamy and pollen source on seed production and offspring performance in three endangered violets. Basic Appl Ecol 6(4):339–350
Eckstein RL, Hölzel N, Danihelka J (2006a) Biological flora of Central Europe: Viola elatior, V. pumila and V. stagnina. Perspect Plant Ecol 8:45–66
Eckstein RL, O’Neill RA, Danihelka J, Otte A, Köhler W (2006b) Genetic structure among and within peripheral and central populations of three endangered floodplain violets. Mol Ecol 15:2367–2379
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Feráková V, Maglocký Š, Marhold K (2001) Red list of ferns and flowering plants of Slovakia. In: Baláž D, Marhold K, Urban P (eds) Červený zoznam rastlín a živočíchov Slovenska. Red list of plants and animals of Slovakia. Ochr Prír 20(suppl 160):44–77 (In Slovak)
Gilissen LJW, van Staveren MJ, Hakkert JC, Smulders MJM, Verhoeven HA, Creemers-Molenaar J (1994) The competence of cells for cell division and regeneration in tobacco explants depends on cellular location, cell cycle phase and ploidy level. Plant Sci 103:81–91
Greilhuber J, Doležel J, Lysák MA, Bennett MD (2005) The origin, evolution and proposed stabilization of the terms ‘genome size’ and ‘C-value’ to describe nuclear DNA contents. Ann Bot 95(1):255–260
Grulich V (2012) Red List of vascular plants of the Czech Republic. Preslia 84(3):631–645
Haque SM, Ghosh B (2013) Micropropagation, in vitro flowering and cytological studies of Bacopa chamaedryoides, an ethno-medicinal plant. Environ Exp Bot 11:59–68
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23(2):254–267
Karp A (1994) Origins, causes and uses of variation in plant tissue cultures. In: Vasil IK, Thorpe TA (eds) Plant cell and tissue culture. Kluwer Academic Publishers, Dordrecht, pp 139–152
Kaźmierczakowa R, Zarzycki K, Mirek Z (2014) Polish Red Data book of plants. Pteridophytes and flowering plants. Instytut Ochrony Przyrody. Polska Akademia Nauk, Cracow
Kolano B, Siwinska D, Maluszynska J (2009) Endopolyploidy patterns during development of Chenopodium quinoa. Acta Biol Crac Ser Bot 51(2):85–92
Korneck D, Schnittler M, Vollmer I (1996) Red List of vascular plants (Pteridophyta et Spermatophyta) Germany. Schr Reihe Veg Kd 28:21–187 (In German)
Krishna H, Alizadeh M, Singh D, Singh U, Chauhan N, Eftekhari M, Sadh RK (2016) Somaclonal variations and their applications in horticultural crops improvement. 3 Biotech 6:54. https://doi.org/10.1007/s13205-016-0389-7
Kubaláková M, Doležel J, Lebeda A (1996) Ploidy instability of embryogenic cucumber (Cucumis sativus L.) callus culture. Biol Plant 38:475–480
Kuta E (1990) Biosystematic studies on the genus Viola L.; section Plagiostigma Godr. II. Embryological analysis of V. epipsila Ledeb. V. palustris L. and their hybrids from Poland. Acta Biol Crac Ser Bot 31:46–62
Kuta E (1976) Viola L. In: Skalińska M, Jankun A, Wcisło H et al (eds) Further studies in chromosome numbers of polish angiosperms. Eleventh contribution. Acta Biol Crac Ser Bot 19:107–148
Kuta E, Jedrzejczyk-Korycińsk M, Cieślak E, Rostański A, Szczepaniak M, Migdałek G, Wąsowicz P, Suda J, Combik M, Słomka A (2014) Morphological versus genetic diversity of Viola reichenbachiana and V. riviniana (sect. Viola, Violaceae) from soils differing in heavy metal content. Plant Biol 16(5):924–934
Larkin PJ, Scowcroft WR (1981) Somaclonal variation—a novel source of variability from cell cultures for plant improvement. Theor Appl Genet 60:197–214
Lee M, Phillips RL (1988) The chromosomal basis of somaclonal variation. Ann Rev Plant Physiol Plant Mol Biol 39:413–437
Leigh JW, Bryant D (2015) PopART: full-feature software for haplotype network construction. Methods Ecol Evol 6(9):1110–1116
Lemontey C, Mousset-Déclas C, Munier-Jolain N, Boutin JP (2000) Maternal genotype influences pea seed size by controlling both mitotic activity during early embryogenesis and final endoreduplication level/cotyledon cell size in mature seed. J Exp Bot 51:167–175
Leva AR, Petruccelli R, Rinaldi LMR (2012) Somaclonal variation in tissue culture: a case study with olive. In: Leva A (ed) Recent advances in plant in vitro culture. InTech, Rijeka
Li JT, Deng DM, Peng GT, Deng JC, Zhang J, Liao B (2010) Successful micropropagation of the cadmium hyperaccumulator Viola baoshanensis (Violaceae). Int J Phytoremediat 12(8):761–771
Mallon R, Rodriguez-Oubina J, Gonzalez ML (2010) In vitro propagation of the endangered plant Centaurea ultreiae: assessment of genetic stability by cytological studies, flow cytometry and RAPD analysis. Plant Cell Tissue Organ Cult 101:31–39
Maluszynska J, Kolano B, Sas-Nowosielska H (2012) Endopolyploidy in plants. In: Leitch IJ, Greilhuber J, Doležel J, Wendel J (eds) Plant genome diversity, vol 2, Springer, Vienna, pp 99–119
Marcussen T, Nordal I (1998) Viola suavis, a new species in the Nordic flora, with analyses of the relation to other species in the subsection Viola (Violaceae). Nord J Bot 18:221–237
Migdałek G (2015) Population genetic diversity and relationships between two closely related forest violets V. reichenbachiana Jordan ex Bor. and V. riviniana Rchb. (Violaceae) based on nuclear, plastid and AFLP markers. Dissertation, Jagiellonian University
Migdałek G, Kolczyk J, Pliszko A, Kościńska-Pająk M, Słomka A (2014) Reduced pollen viability and achene development in Solidago × niederederi Khek from Poland. Acta Soc Bot Pol 83(3):251–255
Mokhtari A, Otroshy M, Barekat T (2015) Plant regeneration through callus induction on medicinal herb Viola odorata—role of plant growth regulator and explants. Agric For 61(3):161–170
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Naeem M, Naveed I, Naqvi SMS, Mahmood T (2013) Standardization of tissue culture conditions and estimation of free scavenging activity in Viola odorata L. Pak J Bot 45:197–202
Neelakandan AK, Wang K (2012) Recent progress in the understanding of tissue culture-induced genome level changes in plants and potential applications. Plant Cell Rep 31:597–620
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci 70(12):3321–3323
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci 76(10):5269–5273
Niklfeld H, Schratt-Ehrendorfer L (1999) Red List of endangered ferns and flowering plants (Pteridophyta and Spermatophyta) In: Niklfeld H (ed) Rote Listen gefährdeter Pflanzen Österreichs. Red Lists of threatened plants in Austria, 2nd edn. Bundesministerium für Umwelt, Jugend und Familie, Wien, pp 33–130
Nybom H, Weising K, Rotter B (2014) DNA fingerprinting in botany: past, present, future. Investig Genet 5:1
Ochatt SJ, Patat-Ochatt EM, Moessner A (2011) Ploidy level determination within the context of in vitro breeding. Plant Cell Tissue Organ Cult 104:329–341
Otto F (1990) DAPI staining of fixed cells for high-resolution flow cytometry of nuclear DNA. Methods Cell Biol 33:105–110
Phillips RL, Kaeppler SM, Olhoft P (1996) Genetic instability of plant tissue cultures: breakdown of normal controls. Proc Natl Acad Sci USA 91:5222–5226
Pontaroli AC, Camadro EL (2005) Somaclonal variation in Asparagus officinalis plants regenerated by organogenesis from long-term callus cultures. Genet Mol Biol 28:423–430
Popielarska-Konieczna M, Kozieradzka-Kiszkurno M, Świerczyńska J, Góralski G, Ślesak H, Bohdanowicz J (2008) Ultrastructure and histochemical analysis of extracellular matrix surface network in kiwifruit endosperm-derived callus culture. Plant Cell Rep 27(7):1137–1145
Popielarska-Konieczna M, Kozieradzka-Kiszkurno M, Tuleja M, Ślesak H, Kapusta P, Marcińska I, Bohdanowicz J (2013) Genotype-dependent efficiency of endosperm development in culture of selected cereals: histological and ultrastructural studies. Protoplasma 250:361–369. https://doi.org/10.1007/s00709-012-0419-1
Prakash E, Sha Valli Khan PS, Sairam Reddy P, Rao KR (1999) Regeneration of plants from seed-derived callus of Hybanthus enneaspermus L. Muell., a rare ethnobotanical herb. Plant Cell Rep 18:873–878
Rodriguez-Enriquez J, Dickinson HG, Grant-Downton RT (2011) MicroRNA misregulation: an overlooked factor generating somaclonal variation? Trends Plant Sci 16:242–248
Sato T, Kwon OC, Miyake H, Taniguchi T, Maeda E (1995) Regeneration of plantlets from petiole callus of wild viola (Viola patrinii DC.). Plant Cell Rep 14:768–772
Schlüter PM, Harris SA (2006) Analysis of multilocus fingerprinting data sets containing missing data. Mol Ecol Notes 6(2):569–572
Sebastiani MS, Ficcadenti N (2016) In vitro plant regeneration from cotyledonary explants of Cucumis melo L. var. cantalupensis and genetic stability evaluation using RAPD analysis. Plant Cell Tissue Organ 124:69–79
Shan X, Li Y, Tan M, Zhao Q (2012) Tissue culture-induced alteration in cytosine methylation in new rice recombinant inbred lines. Afr J Biotechnol 11:4338–4344
Singh RJ (2003) Plant Cytogenetics. 2nd ed. CRC Press, Boca Raton
Slazak B, Sliwinska E, Saluga M, Ronikier M, Bujak J, Słomka A, Göransson U, Kuta E (2015a) Micropropagation of Viola uliginosa (Violaceae) for endangered species conservation and for somaclonal variation-enhanced cyclotide biosynthesis. Plant Cell Tissue Organ 120:179–190
Slazak B, Jacobsson E, Göransson U, Kuta E (2015b) Exogenous plant hormones and cyclotide expression in Viola uliginosa (Violaceae). Phytochemistry 117:527–536
Słomka A, Kawalec P, Kellner K, Jędrzejczyk-Korycińska M, Rostański A, Kuta E (2010) Was reduced pollen viability in Viola tricolor L. the result of heavy metal pollution or rather the tests applied? Acta Biol Crac Ser Bot 52(1):123–127
Słomka A, Jedrzejczyk-Korycinska M, Rostanski A, Karcz J, Kawalec P, Kuta E (2012) Heavy metals in soil affect reproductive processes more than morphological characters in Viola tricolor. Environ Exp Bot 75:204–211
Soni M, Kaur R (2014) Rapid in vitro propagation, conservation and analysis of genetic stability of Viola pilosa. Physiol Mol Biol Plants 20(1):95–101
Stepansky A, Kovalski I, Perl-Treves R (1999) Intraspecific classification of melons (Cucumis melo L.) in view of their phenotypic and molecular variation. Plant Syst Evol 271:313–332
Thiem B, Sliwinska E (2003) Flow cytometric analysis of nuclear DNA content in cloudberry (Rubus chamaemorus L.) in vitro cultures. Plant Sci 164:129–134
Viehmannova I, Bortlova Z, Vitamvas J, Hlasna Cepkova P, Eliasova K, Svobodova E, Travnickova M (2014) Assessment of somaclonal variation in somatic embryo-derived plants of yacon Smallanthus sonchifolius (Poepp. and Endl.) H. Robinson using inter simple sequence repeat analysis and flow cytometry. Electron J Biotechnol 17(2):102–106
Vishwakarma U, Gurav A, Sharma P (2013) Regeneration of multiple shoots from petiole callus of Viola serpens Wall. Pharmacogn Res 5(2):86–92
Wang J, Bao MZ (2007) Plant regeneration of pansy (Viola wittrockiana) ‘Caidie’ via petiole-derived callus. Sci Hortic 111:266–270
Wang QM, Wang L (2012) An evolutionary view of plant tissue culture: somaclonal variation and selection. Plant Cell Rep 31(9):1535–1547
Wiggington MJ (1999) British Red Data books 1. Vascular plants. Joint Nature Conservation Committee, Peterborough
Wijowska M, Kuta E, Przywara L (1999) Autonomous endosperm induction by in vitro culture of unfertilized ovules of Viola odorata L. Sex Plant Reprod 12:164–170
Yeh FC, Yang RC, Boyle T (1999) PopGene version 1.31: Microsoft Window-based freeware for population genetic analysis. Centre for International Forestry Research, University of Alberta, Edmonton, pp 11–23
Żabicki P, Kuta E, Tuleja M, Rataj K, Malec P (2013) Arabidopsis cyclin-dependent kinase gene Cdkg;2 is involved in organogenic responses induced in vitro. Acta Biol Crac Ser Bot 55(1):37–48
Zarzycki K, Szeląg Z (2006) Red list of vascular plants in Poland. In: Mirek Z, Zarzycki K, Wojewoda W, Szeląg Z (eds) Red list of plants and fungi in Poland. Szafer Institute of Botany, Polish Academy of Sciences, Cracow, pp 11–20
Acknowledgements
Authors would like to thank dr. Artur Pliszko from Institute of Botany, Jagiellonian University and Drs. Ryszard Marecki and Tomasz S. Olszewski from Gdansk University for their help in collecting of plant material. This work was funded in part by the Dean of the Faculty of Biology and Earth Sciences of Jagiellonian University (Project DS/MND/WBiNoZ/IB/25/2012 for P. Żabicki).
Author information
Authors and Affiliations
Contributions
All Authors made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data, participated in drafting the article or revising it critically. PŻ study conception and design, acquisition of data, analysis and interpretation of data, drafting of manuscript; EŚ performed and evaluated flow cytometric analyses and participated in writing the manuscript; JM conducted calculations of genetic indices; AS acquisition, analysis and interpretation of data; MT acquisition of data, participated in drafting the article; GM analysis and interpretation of data, participated in drafting the manuscript; JŻ analysis and interpretation of data, participated in revising the manuscript; AS acquisition of data, participated in revising the manuscript; MK participated in revising the manuscript and formatting it according to instructions for authors; EK contribution to interpretation of data, supervising experiments, critical revision of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Sergio J. Ochatt.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Żabicki, P., Sliwinska, E., Mitka, J. et al. Does somaclonal variation play advantageous role in conservation practice of endangered species?: comprehensive genetic studies of in vitro propagated plantlets of Viola stagnina Kit. (Violaceae). Plant Cell Tiss Organ Cult 136, 339–352 (2019). https://doi.org/10.1007/s11240-018-1519-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-018-1519-1