
Abstract
The insecticidal crystal protein derived from gram positive soil bacterium Bacillus thuringiensis plays an important role in controlling lepidopteran infestation. The present study seeks to protect chickpea plants from Helicoverpa armigera infestation by over expressing cry1Ac. Homologous Ubiquitin and RuBisCO small subunit (rbcS) promoters were used to transcribe cry1Ac in transgenic chickpea both constitutively and in a tissue specific manner through Agrobacterium mediated transformation of chickpea var. ICCV89314. Expressed Cry1Ac was specifically targeted to the chloroplast rich tissues using transit peptide sequence. After monitoring transgene integration by Southern hybridization, transgenic chickpea lines were further analyzed by western blot, ELISA and insect bioassay. Expression of cry1Ac in chickpea under the control of above two promoters conferred a high level of protection against pod borer infestation, where chloroplast targeting system was found to be more efficient in controlling this particular devastating lepidopteran pest.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chickpea is the third most important grain legume grown worldwide as a major source of dietary protein. It enhances soil fertility through nodular nitrogen fixation (Abu-Salem and Abou 2011; Maiti 2001). Chickpea yield is greatly affected due to several biotic constraints. A wide range of fungal pathogens, as well as insect pests affect chickpea productivity severely. More specifically, the polyphagous lepidopteran insect Helicoverpa Armigera, commonly known as pod borer is considered to be the most devastating one. In India, it accounts for almost 20-30 % annual yield loss (Gaur et al. 2010). The common measures used to combat this pest have relied mainly on chemical pesticides (Jouanin et al. 1998; Rondon et al. 2007). This might offer a convenient resource for plant protection but the indiscriminate applications of these insecticides enable insects to become resistant to such compounds. Additionally, high expense and bio-safety issues raised social concern regarding its long term utility (Meiyalaghan et al. 2004; Alvarez et al. 2005). Alternatively, development of resistant genotypes through conventional breeding in chickpea remained unsuccessful due to its narrow genetic base, sexual incompatibility and high degree of autogamy (Van Rheenen et al. 1993; Somers et al. 2003; Atif et al. 2013). Recent progress in draft genome sequences has allowed to identify candidate genes for crop improvements (Jain et al. 2013; Varshney et al. 2013).
For the last few decades, genetic engineering with Bacillus thuringiensis endotoxin (Bt) coding gene remained the most lucrative option for the researchers to effectively control the infestation of several lepidopteran and coleopteran insects (Koziel et al. 1993; Wunn et al. 1996; Nayak et al. 1997; Datta et al. 1998; Kota et al. 1999; Maqbool et al. 2001). Such high specificity of Bt toxins to these particular groups of insects had made it relatively insensitive for other non target organisms. This facilitated its use as a biologically safe and reliable molecular device for genetic improvements of various crops like rice, cotton, maize, brinjal, sugarcane, soybean, potato etc. (Ye et al. 2001; Kumar and Kumar 2004; Dutton et al. 2005; Arvinth et al. 2010; Khan et al. 2013).
The unique and selective mode of action of Bt toxin depends on its interaction with an array of specific receptors of the insect midgut. In last couple of years, several models have been proposed regarding its complex mechanism of action (Jurat-Fuentes and Adang 2006; Sarkar et al. 2009). According to the pore formation model α-loop of domain II of toxin binds to larval midgut membrane associated receptors, including cadherin, aminopeptidase N and alkaline phosphatase type receptors (Perera et al. 2009; Tabashnik et al. 2011; Sengupta et al. 2013), this results in opening of cation selective channels and subsequently causes larval death due to osmotic lysis of the midgut epithelium cells.
Transgenic plants offer many unique beneficial opportunities but they have encountered certain challenges; such as evolution of resistance among target insect population. To generate more sustainable and improved transgenic lines, several strategies, based on manipulation of Bt gene, have been employed by previous workers (Gatehouse 2008; Acharjee et al 2010). Different initiatives, including the introduction of different toxins in different varieties, expressing a mixture of toxins, and gene pyramiding approaches were taken by different scientific domains. Making use of chimeric Bt gene comprising different elements of other cry genes e.g. cry1Aa, cry1Ab, cry1Ac, cry2Aa etc was thought to become more fruitful for delaying the process of resistance development among insects than using a single toxin gene (Asharani et al. 2011; Mehrotra et al. 2011). Moreover, the level of toxin expression is likely to have a direct co-relation with the occurrence of resistance towards target insects. Bt crops producing a lower dose of Cry toxin might have induced resistance amongst target insects rapidly. Toxin expression greater than 0.02 % of total soluble protein (TSP) is required to kill insects that are heterozygous for resistant alleles (Bates et al. 2005). But one of the major concerns in engineering plant with cry genes is to regulate the expression level in such a manner that there remains a substantial balance in the ecosystem.
In such a scenario a rational approach involving the use of a suitable promoter element is taken to regulate the gene expression. Promoter plays the most crucial role in regulation of transcriptional control and gene expression in a spatio temporal manner. CaMV35S promoter is by far the most extensively studied promoter used in plant transformation (Canedo et al. 1999; Chakrabarti et al. 2000; Davidson et al. 2002, 2004; Douches et al. 2002; Meiyalaghan et al. 2004). However, its efficacy in gene expression has been reported to vary in different parts and species of plants (Narváez-Vásquez et al. 1992). In addition, there is a risk of multiple transformations with different gene cassettes using a single promoter which may lead to silencing of the genes in successive generations. In recent years, rapid progress has been made for development and evaluation of different plant promoters in genetic transformation studies.
The present study depicts an optimized and effective expression of cry1Ac both constitutively and tissue specifically, along with their stable inheritance in successive progenies by using homologous Ubiquitin promoter and green tissue specific rbcS promoter along with its N terminal transit peptide to target Cry1Ac efficiently into the chloroplast. Chloroplast targeting system not only restrains toxin expression in green tissues only, but it also reduces energy expenditure throughout the plant body. The quantitative estimation of Cry1Ac was done using indirect ELISA. Insect feeding bioassay of T2 transgenic plants expressing Cry1Ac exhibited an effective protection measure against H. armigera compared to wild type plants.
Materials and methods
Plant materials and bacterial strains used
Breeder seeds of C. arietinum L. cv. ICCV 89314 were obtained from International Crop Research Institute for Semi Arid Tropics (ICRISAT), Patancheru, India. The DH5α strain of E.coli and Agrobacterium AGL-1 cell line were used for cloning and plant transformation purposes, respectively.
Insect strain and rearing
Eggs of H. armigera were supplied by Indian Institute of Pulse Research, Kanpur, India. The 2nd instar larvae/nymphs of H. armigera were used for insect bioassay.
Isolation of genomic DNA from C. arietinum L. leaves
Total genomic DNA was isolated from young green leaves of C. arietinum using Qiagen DNeasy Plant Mini kit (GmbH, Hilden, Germany) following manufacturer’s instructions.
Identification and in silico analysis of promoter region of C. arietinum L. RuBisCO
Small Subunit (rbcS) and Ubiquitin (ubi)
Ubiquitin and RubisCO promoters was isolated by genome walking technique using BD Genome Walker™ Universal Kit (BD Biosciences Clontech, California, USA) according to manufacturer’s instruction. DraI, and EcoRV digested genomic libraries were used for genome walking. Following purification and ligation to the specific adaptor, RAGE-PCR was carried out using primers listed in (Table 1). After two rounds of PCR amplification amplicons were cloned and sequenced. Putative cis-elements were identified using PLACE and MATINSPECTOR. Sequences were submitted to NCBI [rbcS Accsesion no. KC505185, Ubi Accsesion no. KC505186].
Construction of plant transformation vectors
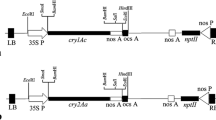
Isolated promoters were primarily cloned in HindIII and XbaI ((Roche, Mannheim, Germany) site of pUC18 vector by replacing CaMV35S promoter. Next, the cry1Ac gene was cloned into BamHI site downstream of CaMV 35S, rbcS and ubi promoters. The 212 bp chloroplast transit sequence was cloned into the XbaI- BamHI sites to generate CaL.rbcS:tp-cry1Ac fusion. Finally, all the four synthetic cassettes were subsequently cloned into the EcoRI and HindIII sites in the MCS region of plant binary expression vector pCAMBIA2301 carrying the nptII gene (Kanamycin) as selectable marker and uidA (gus) gene as reporter system (Fig. 1). For, reporter gene expression study CaL.ubi promoter, CaL.rbcs promoter and CaL.rbcs.tp fusion were cloned in pBI121 binary plant transformation vector in the upstream of gus gene by replacing CaMV35S promoter within HindIII and BamHI sites. Finally, individual binary plant transformation vectors harbouring CaMV35S promoter, CaL.ubi promoter, CaL.rbcS promoter, and CaL.rbcS.tp-cry1Ac fusion were mobilized into competent Agrobacterium tumefaciens AGL-1 cell line.

Schematic diagrams of cry1Ac gene containing chimeric plant expression cassettes. CaMV35S—cauliflower mosaic virus 35S promoter, rbcS—RuBisCO small subunit promoter, Ubi—ubiquitin promoter, ctp—chloroplast transit peptide sequence, nosT—nopaline synthase gene terminator, RB—right border, LB—left border
Determination of transcription start site (TSS) by 5′ rapid amplification of c-DNA ends
Total RNA was prepared from 16 day old mature chickpea leaves using Trizol reagent (Himedia, India) as described in the manufacturer’s protocol. mRNA isolation and 5’-Rapid amplification of cDNA ends were carried out to determine the TSS of the isolated promoters according to the user manual given in the RACE kit (Invitrogen, USA). PCR was performed using a forward primer [AAP, supplied with the kit] and a reverse primer specific for the target genes i.e rbcS and Ubi (R3 and R4, respectively). Primers used for RACE were listed in (Table 1). PCR amplified fragments were purified using Qiagen Gel extraction kit (Qiagen, Germany) and subsequently cloned into the pGEMT- Easy vector (Promega, USA) and sequenced.
Chickpea transformation
Agrobacterium-mediated chickpea transformation, was carried out as described by (Chakraborti et al. 2006; Atif et al. 2013) with a modified rooting protocol. The regenerated shoots were transferred to shoot elongation medium supplemented with 0.25 mg/l IAA for additional 10 days and then 3–4 cm elongated shoots were transferred to rooting medium (1/2MSsalts, B5 vitamins, 1 mg/l IBA and 20 g/l sucrose) (Murashige and Skoog 1962). After 10 days the rooted shoots were transferred to pots for hardening and finally transferred to the glasshouse. All cultures were maintained in culture room 24–25 °C under a cool white light of 60 µmol/m2/s for 16 h photoperiod.
Assay for β-glucuronidase (GUS) activity
The histochemical gus assay was performed according to the method of Jefferson (1987), using 5-bromo-4-chloro-3-indoxyl-beta-d-glucuronide (X-gluc) (Duchefa Biochemie, The Netherlands) as a substrate.
PCR analyses for monitoring transgene insertion
Genomic DNA was isolated from young green leaves of both the transformed and control chickpea plants followed by CTAB extraction method (Saghai-Maroof et al. 1984). PCR analysis was carried out using cry1Ac gene specific primers CF1 and CR1 (Table 1). Amplicon obtained from plasmid served as positive control.
Southern blot analysis
Southern blot analysis was carried out using extracted genomic DNA of control and PCR +ve transgenic events according to (Sambrook and Russell 2001) with some modifications. 25 μg of EcoRI (Roche, Mannheim, Germany) digested genomic DNA was separated by electrophoresis on 0.8 % (w/v) agarose gel. The gels were subsequently blotted onto a positively charged nylon membranes (Hybond-N+; Amersham™ Biosciences, Buckinghamshire, UK). A 0.6 kb cry1Ac amplicon was radiolabelled with [α32P] dCTP using Rediprime II™ Random Prime Labeling System, (Amersham™ Biosciences, Buckinghamshire, UK) and used as a probe. Following hybridization and washing, the membranes were wrapped and exposed to Kodak X-ray film for seven days at −80°C and subsequently developed.
Segregation analysis of the transgene
T1 seeds, obtained from primary transformants were screened for antibiotic sensitivity on 100 mg/l kanamycin supplemented media. Two weeks old seedlings were scored for resistance or sensitivity. PCR analysis was further carried out using both kanamycin resistance and sensitive lines with primer pairs CF1 and CR1. Segregation pattern of cry1Ac gene in T2 progeny plants was calculated and validated by comparing the data with the expected ratio by χ2-test.
Protein extraction from chloroplast enriched fraction
Chloroplast isolation and chloroplastidic protein extraction from the plant harbouring rbcS:tp construct were performed according to the Bio-protocol (Klinkenberg 2014) with minor modifications. Briefly, 5 g of leaf tissue was homogenized with 20 ml of ice-cold isolation buffer and filtered through a nylon mesh and homogenate containing chloroplast fraction was poured carefully on top of the 40 % percoll layer and centrifuged. The supernatant was discarded and the chloroplasts were re-suspended gently in 750 μl of isolation buffer. Finally, centrifugation was done to pellet the chloroplasts. The entire procedure was essentially maintained in dark and chilled condition.
For protein extraction, the freeze-fractured chloroplasts were gently dissolved in 1 ml of ice-cold protein extraction buffer and centrifuged to obtain the supernatant containing soluble chloroplastidic proteins. The pellet, enriched with non-soluble chloroplastidic proteins, was re-suspended in 1 ml of ice-cold protein extraction buffer. All the centrifugations were carried out at 4°C. Protein concentration was determined using Bradford assay (Bradford 1976). 20–25 μg of protein sample from both the fraction (soluble and non-soluble) were used for subsequent western blot analysis.
Western blot analysis of transgenic plants
Total soluble protein was extracted from mature green leaves of 1-month-old T1 transgenics and wild type plants. The extraction buffer contained 20 mM TrisCl pH 7.5, 0.02 M phenylmethylsulphonyl fluoride (PMSF). The amount of protein was quantified using Bradford method (Bradford 1976). One microgram purified Cry1Ac protein was used as positive control. Approximately 40 µg of total protein from individual lines was separated on 15 % SDS-PAGE and blotted onto a positively charged Hybond C membrane (Amersham Biosciences, Buckinghamshire, UK) using a semidry transfer apparatus (Applied Biosystems, USA). After blocking, the membrane was probed with anti-Cry1Ac polyclonal primary antibody. Substrate (ECL reagents GE Healthcare, Germany) based detection was done using anti-rabbit IgG-horse radish peroxidase (HRP) conjugated (Sigma-Aldrich, USA) secondary antibody.
Immunohistoflourescence analysis
Transverse leaf sections from both transgenic and untransformed plants were primarily incubated in 10 % (v/v) trichloroacetic acid (Sigma-Aldrich, USA) at 4 °C for 1 h followed by ethanol: acetic acid (3:1, v/v) wash with three to four changes until complete removal of chlorophyll. The tissue sections were then passed successively through a series of graded ethanol to water (90, 70, 50, 30 % (v/v), respectively, each of 10-15 min duration) and blocked with 3 % (w/v) bovine serum albumin (BSA, Merck, Germany) in 1× phosphate buffered saline (PBS) at room temperature for 2 h. The tissue samples were then subjected to incubation with an anti-Cry1Ac antibody (1:5000) dissolved in BSA for overnight at room temperature. After washing with 1x PBS, the sections were treated with an anti-rabbit IgG-FITC conjugated (1:20,000) (Sigma-Aldrich, USA) secondary antibody for 1 h at room temperature. Finally, the slides were examined under an Axioscope Carl Zeiss inverted fluorescent microscope using excitation filter of 450–490 nm for FITC. Images were captured with the AxioCam ICc3 digital camera and the AxioVision imaging software system (Carl Zeiss Micro Imaging, GmbH, Germany).
ELISA of soluble protein extracts
Quantitative estimation of Cry1Ac protein expression was determined by indirect ELISA as described by Dutta et al. (2005). Crude protein extract (90 µg) from T0 and T1 transgenic leaves and purified native Cry1Ac (serially diluted from 5 µg to 10 ng in coating buffer) were stored at 4 °C overnight. Wells of a microtitre plate (Immunomaxi, Trassadingen, Switzerland) were coated with coating buffer (15 mM Sodium carbonate, 35 mM Sodium bicarbonate, 3 mM Sodium azide; pH 9.6). Next day after washing the plates, it was blocked with 3 % (w/v) non-fat milk (Merck) in PBS for 2 h at 37 °C. The wells were incubated with anti-Cry1Ac primary antibody with 1:5000 dilutions in PBS for 1 h at 37 °C and subsequently incubated with HRP conjugated anti-rabbit secondary antibody, (Sigma, St. louis, USA) diluted at 1:10,000 in PBS for 1 h at 37 °C. All the washing steps were eventually performed according to the method described by (Dutta et al. 2005). After addition of substrate, O-phenylenediamine hydrochloride (OPD, Sigma), dissolved in citrate buffer, the colour was developed and the reading of the microtitre plate was recorded at 415 nm in a plate reader (ELx 800, Bio-Tek Instruments Inc., Winooski, VT, USA).
Insect bioassay of transgenic plant
The entomocidal activity of Cry1Ac toxins expressed in transgenic chickpea plants were assayed by larval feeding bioassay using H. armigera second instar larvae. All the larvae were reared on an artificial diet rich in gram flour. About 200–250 mg fresh chickpea twigs and healthy green pods were placed in 35 mm petridish containing 3 % water-agar poured in slant. Ten neonate larvae were used for each petridish, and three replicates were made for each individual toxin expressing transgenic line. Plates were sealed with parafilm to prevent desiccation and incubated at 25-26oc for 16 h photoperiod and 70 % relative humidity.Larval mortality rate was recorded at 24, 48 and 72 h time point (Fig. 9).
Statistical analysis
The data were analysed using Graphpad prism 5 software (GraphPad Software, La Jolla, CA, USA). One-way analyses of variance (ANOVAs) were executed to compare the differences between the untransformed control and the transgenic plants with all four constructs. P < 0.05 was considered to be statistically significant.
Results
Agrobacterium-mediated transformation and regeneration of transgenic chickpea plants
A total 3367 explants were transformed with Agrobacterium tumefaciens AGL-1 strain harbouring different promoter-cry1Ac fusion cassettes under similar conditions. A total 58 putative chickpea transformants were recovered with multiple shoot branches after three-four rounds of kanamycin selection (100 mg/l). 46 plants out of 58 showed normal phenotype with no alteration in flowering and pod setting (Fig. 2). The total number of 46 transformants generated using different constructs and transformation frequency was documented in (Table 2).

Different in vitro developmental stages of genetically transformed chickpea explants, a single cotyledon with half embryo as explants for transformation, b–e multiple shoot initiation after 2 weeks co-cultivation, f elongation of regenerated shoots in shoot elongation medium, g in vitro rooting of the elongated shoots, h hardening of the rooted plantlets in artificial soil, i putative transformants growing under greenhouse condition
Histochemical GUS assay
The isolated promoters (CaL.Ubi, CaL.rbcS and CaL.rbcS:tp) driven gus expression was evaluated in selected independent events. The T1 seedling obtained from the pUbi-GUS primary transformants showed constitutive GUS expression. The prbcS-GUS exhibited a less intense GUS activity compared to prbcS:tp-GUS construct in transgenic chickpea leaves and stems. CaMV35S promoter driven GUS expression was found to be much weaker than any of our isolated promoters. pUbi-GUS and prbcS:tp-GUS executed a very intense GUS activity in pod walls of transgenic chickpea. Thus, GUS expression pattern of pUbi-GUS and prbcS:tp-GUS clearly demonstrates a uniform and high level of GUS expression in different plant parts throughout plant growth and development compared to the CaMV35S. However, the untransformed plant did not show any endogenous GUS activity (Fig. 3).

Monitoring the promoter activity, a schematic representation of promoter-gus fusion cassettes, b T1 seedling of pUbi-gus line showing constitutive gus expression, c histochemical gus staining of mature leaves from transgenic chickpea plants using different promoter-gus fusion cassettes, d shoot tissues of different transgenic gus lines showing differential GUS expression, e GUS activity in the pod wall of mature transgenic plants
Molecular characterization of putative transformants
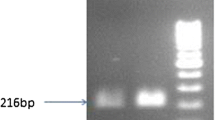
Preliminary molecular screening was determined by PCR analysis and Southern hybridization. Genomic DNA was isolated from a total 40 transgenics (10 from each construct) along with untransformed plant to perform PCR with gene specific primers. PCR analyses identified an amplification of 621 bp amplicon, which was alike to the plasmid DNA taken as positive controls. Whereas, the untransformed plants did not show any amplification under identical assay condition (Fig. 4).

PCR analysis of T1 generation transgenic chickpea plants using cry1Ac gene specific primers, lane (+ve)—621 bp PCR amplicon using plasmid DNA of cry1Ac positive clone; lane (−ve) DNA from untransformed control plant, a T1 generation of different 35S promoter carrying transgenic lines (35S-5,7,9,11and 29) showing 621 bp amplification of cry1Ac gene, b PCR amplification of rbcS:tp-cry1Ac transgenic lines (rTP-8,10,11,14,16,17 and 20), c PCR amplification for the presence of cry1Ac coding sequence in7 T1 progenies of rbcS-cry1Ac (R-4,7,12,15,16,23 and 31), d 621 bp amplification of Ubi- cry1Ac (U-1,2,4,7,13,15 and 34) transgenic lines using gene specific primers
Southern hybridization revealed stable integration of all the synthetic cassettes along with their T-DNA constituents in the plant nuclei and number of copies inserted in individual transgenic events. The hybridization signal of T1 transgenic plants unveiled single copy insertion ranges between 2.3 to 9.5 kb, while untransformed plant did not show any hybridization signal (Fig. 5a). Amongst the lines, 35S9, R7 and U4 showed double copy transgene insertion. Finally, four T2 plants from rbcS:tp-11 and three from Ubi-13 were further subjected to hybridization with αP32dCTP labeled cry1Ac probe. Results confirmed efficient transmission of cry1Ac gene to T2 progeny (Fig. 5b, c).

Southern blot analysis of EcoRI digested genomic DNA from transgenic chickpea leaves. Lane (+ve) 0.62 kb cry1Ac gene was used as positive control and lane (–ve) genomic DNA from wild type plants used as negative control. EcoRI/HindIII digested λ DNA markers are indicated on the left. a lane (1–4) 35S-cry1Ac harboring transgenic chickpea lines 35S5,7,9 and11; lane (5–8) rbcS:tp-cry1Ac carrying transgenic lines rTP8,11,16 and 20; lane (9–13) rbcS-cry1Ac transformed transgenic lines R4,16,23, 31and a double copy bearing R7; lane (14–18) U2,U13, double copy bearing U4, U15 and U34, b and c analysis of T2 progeny transgenic lines obtained from rTPT1L-11 and UT1L-13 respectively
Inheritance of selectable marker gene and cry1Ac
The inheritance pattern of cry1Ac gene in T1 seeds harvested from primary transformants was assessed by germinating seeds on 100 mg l−1 kanamycin supplemented medium. Antibiotic screening followed by PCR analysis attributed to prepare the segregation chart (Table 3). Segregation data was analyzed by Chi square test. The results indicate that both the genes segregate according to Mendelian ratio 3:1 (resistant: susceptible p ≤ 0.05, \(\chi^{2}=3.841\)) for kanamycin tolerance.
Expression analysis of Cry1Ac protein in T1 transgenic lines
Expression of synthetic Cry1Ac toxin in transgenic chickpea lines was validated by western blotting using anti Cry1Ac antibody. Three independent selected transgenic lines from 35S constructs and four events each from the other three constructs were subjected to immunoblot assay. Results displayed a sharp band of ~68 kDa identical to purified native Cry1Ac protein band (Fig. 6). The green tissue specific expression of Cry1Ac toxin using rbcS-cry1Ac and rbcS:tp-cry1Ac systems was further verified by the immunoblotting using both chloroplast enriched fraction as well as total cell extract. The level of Cry1Ac expression was found to be ~2.0 fold higher in green tissues of transgenic chickpea in the case of rbcS:tp-cry1Ac compared to rbcS-cry1Ac system. Moreover, Cry1Ac expression in rbcS:tp-cry1Ac transgenic chickpea is found to be restricted in the chloroplast enriched fractions than the corresponding cytsolic fractions (Fig. 6b). However, untransformed plants did not show any band when subjected to immunoassay in either case.

Western blot using total protein extract from transgenic chickpea lines expressing Cry1Ac. (+ve)—2 µg purified Cry1Ac of ~68 kDa as positive control, (−ve)—total protein isolated from untransformed chickpea plant as negative control. a western blot analysis of 35S-Bt chickpea lines, b chloroplast enriched (CP) as well as total soluble protein (TSP) extracts from transgenic chickpea leaves of rbcS:tp-cry1Ac (lines 8, 11,16 and 20) were probed with anti-Cry1Ac antibody, c western blot analysis of crude protein extracts from leaves of four independent T1transgenic chickpea lines (4,16,23 and 31)of rbcS-cry1Ac construct, d four T1 progeny plants of Ubi-cry1Ac lines showing a sharp ~68 kDa band . Total cellular protein extracted from mature leaves of 30 days field grown chickpea plants and 40 µg of total protein of each line was used for western blot analysis
A quantitative assessment of Cry1Ac protein accumulation in transgenic chickpea was performed by indirect ELISA. Among the two constitutive promoters (35S and Ubq), the Bt-Cry1Ac protein expression level varied between 10–15 ng mg−1 of TSP and 15–25 ng mg−1 of TSP for CaMV35S promoter and CaL.Ubq promoter driven constructs respectively. Cry1Ac accumulation under green tissue specific promoter (rbcS:tp-cry1Ac) ranges between 25 and 40 ng mg−1 of TSP which is ~1.5 fold higher in comparison to rbcS-cry1Ac event (18–24 ng mg−1 of TSP) (Fig. 7). While no endotoxin accumulation was traced in case of untransformed control plant.

Immunohistofluorescence assay of transgenic chickpea leaves. Transverse leaf sections were incubated with anti-Cry1Ac anti-serum as primary antibody and anti-rabbit IgG-FITC conjugated secondary antibody. The accumulation of Cry1Ac is indicated by the green fluorescence. a the transverse sections from untransformed control leaf showed no green fluorescence, b the leaf section of p35S-cry1Ac plants showing detection of faint green fluorescence of constitutive Cry1Ac expression in all the cells types, c cross sections of leaf blade of chickpea plants transformed with prbcS-cry1Ac showing localization of Cry1Ac in the mesophyll cells, d the prbcS:tp-cry1Ac showing more accumulation of Cry1Ac protein in the chloroplast containing green tissues of transgenic chickpea. The expression of Cry protein is indicated by deep green fluorescence, e the pUbi-cry1Ac showed a bright fluorescence signal compared to the p35S-cry1Ac, indicating a higher amount of Bt protein accumulation. Bars represent 10 µm
Expression analysis by western blot and ELISA was found to be well coordinated with the outcome from Immunohistofluorescence study. Significant variation in green fluorescence pattern of FITC was detected in transgenic leaf samples, established the distinct regulation pattern of individual promoters. Moreover, the toxin expression mediated by rbcS:tp promoter was profoundly intense in leaf tissue than the rest of the three (Fig. 8). WT untransformed plant did not show any fluorescence activity when probed with FITC conjugated secondary antibody.

Quantitative estimation of accumulated Cry1Ac protein in different T1 generation transgenic plants. Average quantity of Cry1Ac toxin in transgenic plants is shown as ng mg−1 of TSP ± standard deviation on the top of histogram bars and also indicated by horizontal mark. ELISA reading represented after subtracting the background readings from untransformed plants
In planta insect bioassay of transgenic plants
The entomocidal activity of T2 transgenic chickpea plants expressing Bt-Cry1Ac protein was assayed via detached leaf feeding bioassay against H. armigera 2nd instar larvae. Thirty days old chickpea twigs of individual transgenic events were incubated with larvae of H. armigera. Three replicates for the individual event each were prepared with 10 larvae. A comparative account of insect mortality was represented (Fig. 9c, d) for pUbi-cry1Ac, prbcS-cry1Ac, and prbcS:tp-cry1Ac. All the plants derived from above mentioned three constructs showed a significant (p ≥ 0.05) fold enhanced mortality when compared to CaMV35S-cry1Ac. U34, U13, R31, R12, R16 and rTP11, with significant amount of toxin accumulation, served as the most promising lines, which accounted for ≥90 % insect mortality. Survived larvae fed on transgenic twigs showed less weight gain when compared to those fed on the untransformed plant. A little or no weight gain was recorded for survived larvae fed on transgenic twigs between 48 and 72 h; whereas WT untransformed twigs displayed a huge weight gain of the neonate larvae with massive chewing of entire twigs (Fig. 9a). Pods from untransformed control plant were found to be severely bored by larvae along with complete damage of growing embryo. Interestingly, transgenic pods did not show such pod borer infestation even after 72 h of incubation (Fig. 9b).

Insect bioassay performed on T2 generation Bt transgenic chickpea lines with Helicoverpa armigera 2nd instar larvae. a detached leaf bioassay performed with T2 progenies of 35S7, U13, R16 and rTP11 lines against H armigera 2nd instar larvae, b infestation of WT and transgenic chickpea pods by H armigera 2nd instar larvae recorded after 72 h time point, c comparison of survived larval bodyweight fed on WT and transgenic twigs at 0 h.24 h,48 h and 72 h time interval, d comparison of mean larval mortality fed on different transgenic lines harboring CaMV35S-Bt, Ubi-Bt, rbcS-Bt and rbcS:tp- Bt. The values were represented as a percentage of mean larval mortality and each bar diagram represents three independent transgenic events from each construct and each with three technical replicates. Error bar represent standard errors. Statistical significance was analyzed using one way ANOVA followed by tukey’s multiple-range test (p value < 0.05)
Discussion
Transgenic crop plants expressing Cry proteins from Bacillus thuringiensis had shown significant resistance against insect pests. It also mitigates the use of synthetic pesticides (Koziel et al. 1993; Sardana et al. 1996; Perlak et al. 2001). One of the major constrains in heterologous gene expression is the instability of transcript due to the lack of optimized codon usage, absence of proper AT: GC ratio, premature degradation of mRNA resulting lower protein production etc. Several modifications have been implicated in the native cry1Ac gene to restore the transcript stability by elimination of polyadenylation sites, exploitation of plant preferred codons and balancing GC content (Perlak et al. 1990; De Rocher et al. 1998). Native cry genes do not express well in plants due to different codon usage from bacterial origin. Previous reports suggested that proper codon optimization results in 10–100 fold enhancement of Cry1Ab translational efficiency for plant expression (Perlak et al. 1991). Eventually, synthetic cry genes with plant preferred codon have been introduced into several crops for insect tolerance (Nayak et al. 1997; Bao et al. 2009; Kim et al. 2009; Tohidfar et al. 2013). Previous studies reported that high expression of cry1Ac gene often negatively affect plant regeneration (Rawat et al. 2011). The present study has convincingly illustrated that, moderate to high level of toxin expression in transgenic chickpeas using homologous promoters do not cause any perturbation with normal plant growth and development.
Three different vector chimera were developed to increase the transcriptional efficacy of synthetic cry1Ac in transgenic events. Homologous ubiquitin promoter was used for constitutive cry1Ac expression. Tissue specific expression was achieved using RuBisCO small subunit promoter and its chloroplast transit sequence. CaL.Ubi promoter showed constitutive GUS expression in T1 progeny of pUbi-GUS line. Green tissue specific GUS expression was monitored both in prbcS-GUS and prbcS:tp-GUS lines. The GUS expression in either cases was found to be more intense compared to CaMV35S-GUS line. Images clearly indicate that pUbi and prbcS:tp are able to strongly induce the gus expression in the pod walls of transgenic chickpea (Fig. 3).
Detailed molecular screening of transgenic plants established stable cry1Ac integration. PCR positive transgenic plants harboring four different constructs were chosen for Southern blot analysis. Southern hybridization showed single copy insertion in most of the lines, confirming stable T-DNA integration at a single locus in the plant genome. T1 plants from ubi-13 and rbcS:tp-11 successfully transmitted the single copy transgene in T2 progeny. Antibiotic screening of T1 progeny seeds obtained from primary transformants segregated more or less in Mendellian fashion (3:1).
The toxin expression in transgenic chickpea leaves was validated by western blotting and immunohistofluorescence assay. Quantitative estimation of CaL.ubi promoter derived Cry1Ac expression in transgenic chickpea ranges between 15 and 19 ng mg−1 TSP. CaL.ubi promoter showed ~2.0 fold higher toxin accumulation compared to CaMV35S promoter. In case of pActin-Bt, the protein expression ranges from 4 to 18 ng mg−1 TSP in transgenic chickpea plants (Ganguly et al. 2014). Also, a ZmUbi1 promoter accounted for an increased expression of foreign gene in native maize tissues, transgenic rice callus, protoplasts (Sullivan et al. 1989; Cornejo et al. 1993; Streatfield et al. 2004), and also in sugarcane (Wei et al. 2003). Present investigation has detected 2.5 fold enrichment of Cry1Ac protein in rbcS:tp-cry1Ac plant compared to that exclusive of transit peptide (rbcS-cry1Ac) which was even ~6–8 fold higher than CaMV35S promoter. Our current finding was well substantiated with previously reported Arabidopsis rbcS:tp system accounting a 10–20 fold increment in both cry1Ac mRNA and protein levels compared to CaMV35S promoter (Wong et al.1992). However, a higher amount of toxin accumulation does not always correlate with the tolerance level. A moderately expressing (<0.01 % TSP) Cry1Ab protein was found to be sufficient for exhibiting 100 % mortality of stem borer in rice (Bregitzer et al. 1998). In our study transgenic lines expressing toxin (0.02–0.03 % of TSP) demonstrated ≥90 % insect mortality. Detached leaf bioassay with all the transgenic chickpea plants expressing Cry1Ac exhibited enhanced resistance to H. armigera 2nd instar larvae. The bioassay result indicated a significant growth reduction of the insect larvae. Survived larvae fed on WT control twigs displayed a much higher weight gain, but those who fed on transgenic twigs ended up with severe growth retardation between 24 and 72 h. Larvae showed a different feeding pattern on control and transgenic twigs as reported by Chakrabarti et al. (2000). The mortality percentage was found to be highest in rbcS:tp-cry1Ac line (>90 %). Kim et al. (2009) also reported rbcS:tp-cry1Ac plant showed high level of protection against rice leaf folder. In the present experiment bioassay was also conducted on pod tissues using WT and transgenic plants at 24, 48, and 72 h time interval. In case of WT pod the insect larvae severely invaded the pod wall within 24 h time period. But no such severe infestation was found in the pod tissues of transgenic plants even after 72 h. A slight damage was noticed in T2 progenies of 35S7 at 72 h. The larvae completely engulfed growing embryo of WT pod at 48 h time point, whereas the larval entry was completely inhibited in transgenic pods. Similar results were reported in chickpea plants transformed using cry1Ac downstream of soybean pod specific msg promoter (Ganguly et al. 2014). Bioassay results clearly indicated the toxin expression in transgenic plants limits the insect damage significantly. To summarize, the present study was an attempt to delineate the function of these homologous promoters in expressing the insecticidal gene through a more systemic manner. Attributing to all the revelations and explanations, it may be confidently concluded that Cry1Ac expression under these homologous promoters provides a potent resource to combat the challenges against the threatening pest. Effective use of such homologous promoters will be a safe and reliable arena to boost up host resistance and sustainable agricultural practices.
References
Abu-Salem FM, Abou EA (2011) Physico-chemical properties of tempeh produced from chickpea seeds. Arab J Am Sci 7:107–118
Acharjee S, Sarmah BK, Ananda Kumar P, Olsen K, Mahon R, Moar WJ, Moore A, Higgins TVJ (2010) Transgenic chickpeas (Cicer arietinum L.) expressing a sequence-modified cry2Aa gene. Plant Sci 178:333–339
Alvarez JM, Dotseth E, Nolte P (2005) Potato tuberworm: a threat for Idaho potatoes. Univ Idaho Ext Bull CIS1125, p4. http://info.ag.uidaho.edu/pdf/CIS/CIS1125.pdf
Arvinth S, Arun S, Selvakesavan RK, Srikanth J, Mukunthan N, Kumar PA, Premachandran MN, Subramonian N (2010) Genetic transformation and pyramiding of aprotinin-expressing sugarcane with cry1Ab for shoot borer (Chilo infuscatellus) resistance. Plant Cell Rep 29:383–395
Asharani BM, Ganeshaiah KN, Kumar ARV, Makarla U (2011) Transformation of chickpea lines with Cry1X using in planta transformation and characterization of putative transformants T1 lines for molecular and biochemical characters. J Plant Breed Crop Sci 3:16413–16423
Atif RM, Patat-Ochatt EM, Svabova L, Ondrej V, Klenoticova H, Jacas L, Griga M, Ochatt SJ (2013) Gene transfer in legumes. Prog Bot 74:37–100
Bao JH, Chin DP, Fukami M, Ugaki M, Nomura M, Mii M (2009) Agrobacterium-mediated transformation of spinach (Spinacia oleracea) with Bacillus thuringiensis cry1Ac gene for resistance against two common vegetable pests. Plant Biotechnol 26:249–254
Bates SL, Zhao J, Roush RT, Shelton AM (2005) Insect resistance management in GM crops: past, present and future. Nat Biotechnol 23:57–62
Bradford MM (1976) A rapid and sensitive method for the quantitation of proteins using the principle of protein-dye binding. Anal Biochem 72:248–254
Bregitzer P, Halbert SE, Lemaux PG (1998) Somaclonal variation in the progeny of transgenic barley. Theor Appl Genet 96:421–425
Canedo V, Benavides J, Golmirzaie A, Cisneros F, Ghislain M, Lagnaoui A (1999) Assessing Bt-transformed potatoes for potato tuber moth, Phthorimaea operculella (Zeller), management. Program report 1997–98, Impact on a Changing World International Potato Centre (CIP), Lima, pp 161–170
Chakrabarti SK, Mandaokar AD, Pattanayak D, Chandla VK, Ananada Kumar P, Naik PS, Sharma RP (2000) Transgenic potato lines expressing a synthetic cry1Ab gene acquired tolerance to both potato tuber moth and defoliating caterpillar. In: Paul Khurana SM, Shekhawat GS, Singh BP, Pandey SK (eds) Potato global research and development. Indian Potato Association, Shimla, pp 249–255
Chakraborti D, Sarkar A, Das S (2006) Efficient and rapid in vitro plant regeneration system for Indian cultivars of chickpea (Cicer arietinum L.). Plant Cell Tissue Organ Cult 86:117–123. doi:10.1007/s11240-005-9072-0
Cornejo M-J, Luth D, Blankenship KM, Anderson OD, Blechl AE (1993) Activity of a maize ubiquitin promoter in transgenic rice. Plant Mol Biol 23(3):567–581
Datta K, Vasquez A, Tu J, Torrizo L, Alam MF, Oliva N, Abrigo E, Khush GS, Datta SK (1998) Constitutive and tissue specific differential expression of cryIA(b) gene in transgenic rice plants conferring resistance to rice insect pest. Theor Appl Genet 97:20–30
Davidson MM, Jacobs JME, Reader JK, Butler RC, Frater CM, Markwick NP, Wratten SD, Conner AJ (2002) Development and evaluation of potatoes transgenic for a cry1Ac9 gene conferring resistance to potato tuber moth. J Am Soc Hortic Sci 127:590–596
Davidson MM, Takla MFG, Jacobs JME, Butler RC, Wratten SD, Conner AJ (2004) Transformation of potato (Solanum tuberosum) cultivars with a cry1Ac9 gene confers resistance to potato tuber moth (Phthorimaea operculella). NZ J Crop Hortic 32:39–50
De Rocher EJ, Vargo-Gogola TC, Diehn SH, Green PJ (1998) Direct evidence for rapid degradation of Bacillus thuringiensis toxin mRNA as a cause of poor expression in plants. Plant Physiol 117:1445–1461
Douches DS, Li W, Zarka K, Coombs J, Pett W, Grafius E, Ei-Nasr T (2002) Development of Bt-cry5 insect-resistant potato lines ‘Spunta-G2’ and ‘Spunta-G3’. HortScience 37:1103–1107
Dutta I, Majumder P, Saha P, Ray K, Das S (2005) Constitutive and phloem specific expression of Allium sativum leaf agglutinin (ASAL) to engineer aphid (Lipaphis erysimi) resistance in transgenic Indian mustard (Brassica juncea). Plant Sci 169:996–1007
Dutton A, Romeis J, Bigler F (2005) Effects of Bt maize expressing cry1Ab and Bt spray on Spodoptera littoralis. Entomol Exp Appl 114:161–169
Ganguly M, Molla K, Karmakar S, Datta K, Datta S (2014) Development of pod borer resistant transgenic chickpea using apod specific and a constitutive promoter driven fused cry1Ab/Acgene. Theor Appl Genet 127:2555–2565
Gatehouse JA (2008) Biotechnological prospects for engineering insect-resistant plants. Plant Physiol 146:881–887
Gaur PM, Tripathi S, Gowda CLL, Ranga Rao GV, Sharma HC, Pande S, Sharma M (2010) Chickpea seed production manual. ICRISAT 502:1–28
Jain M, Misra G, Patel RK, Priya P et al (2013) A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. doi:10.1111/tpj.12173
Jefferson RA (1987) Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol Biol Rep 5:387–405
Jouanin L, Bonadé Bottino M, Girard C, Morrot G, Giband M (1998) Transgenic plants for insect resistance. Plant Sci 131:1–11
Jurat-Fuentes JL, Adang MJ (2006) Cry toxin mode of action in susceptible and resistant Heliothis virescens larvae. J Invertebr Pathol 92:166–171
Khan GA, Baksh A, Ghazanfar M, Riazuddin S, Husnain T (2013) Development of transgenic cotton lines harbouring a pesticidal gene (cry1Ab). Emir J Food Agric 25(6):434–442
Kim EH, Suh SC, Park BS, Shin KS, Kweon SJ, Han EJ, Park S-H, Kim YS, Kim J-K (2009) Chloroplast-targeted expression of synthetic cry1Ac in transgenic rice as an alternative strategy for increased pest protection. Planta 230:397–405
Klinkenberg J (2014) Extraction of chloroplast proteins from transiently transformed Nicotiana benthamiana leaves. Bio-protocol 4(18): e1238. http://www.bio-protocol.org/e1238
Kota M, Daniell H, Varma S, Garczynski SF, Gould F, Moar WJ (1999) Overexpression of the (Bt) Cry2Aa2 protein in chloroplasts confers resistance to plants against susceptible and Bt-resistant insects. Proc Natl Acad Sci USA 96:840–845
Koziel MG, Beland GL, Bowman C, Carozzi NB, Crenshaw R, Crossland L, Dawson J, Desai N, Hill M, Kadwell S, Launis K, Lewis K, Maddox D, McPherson K, Meghji MR, Merlin E, Rhodes R, Warren GW, Wright M, Evola SV (1993) Field performance of elite transgenic maize plants expressing an insecticidal protein derived from Bacillus thuringiensis. Biotechnology 11:194–200
Kumar H, Kumar V (2004) Tomato expressing Cry1A(b) insecticidal protein from Bacillus thuringiensis protected against tomato fruit borer, Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae) damage in the laboratory, greenhouse and field. Crop Prot 23:135–139
Maiti RK (2001) The chickpea crop. In: Maiti R, Wesche-Ebeling P (eds) Advances in chickpea science. Science Publishers Inc, Enfield, pp 1–31
Maqbool SB, Riazuddin S, Nyguyen TL, Gatehouse AMR, Gatehouse JA, Christou P (2001) Expression of multiple insecticidal genes confers broad resistance against a range of different rice pests. Mol Breed 7:85–93
Mehrotra M, Singh AK, Sanyal I, Altosaar I, Amla DV (2011) Pyramiding of modified cry1Ab and cry1Ac genes of Bacillus thuringiensis in transgenic chickpea (Cicer arietinum L.) for improved resistance to pod borer insect Helicoverpa armigera. Euphytica 182:87–102
Meiyalaghan S, Davidson MM, Takla MGF, Wratten SD, Conner AJ (2004) Effectiveness of four cry genes in transgenic potato for conferring resistance to potato tuber moth. New directions for a diverse planet. In: Proceedings of the 4th international crop science congress Risbane, Australia. ISBN 1 920842 20 9
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissues cultures. Physiol Plantarum 15:473–479
Narváez-Vásquez J, Orozco-Cárdenas ML, Ryan CA (1992) Differential expression of a chimeric CaMV-tomato proteinase Inhibitor I gene in leaves of transformed nightshade, tobacco and alfalfa plants. Plant Mol Biol 20:1149–1157
Nayak P, Basu D, Das S, Basu A, Ghosh D, Ramakrishnan NA, Ghosh M, Sen SK (1997) Transgenic elite indica rice plants expressing cry1Ac-endotoxin of Bacillus thuringiensis are 24 resistant against yellow stem borer (Scirpophaga incertulas). Proc Natl Acad Sci USA 94:2111–2116
Perera OP, Willis JD, Adang MJ, Jurat-Fuentes JL (2009) Cloning and characterization of the Cry1Ac-binding alkaline phosphatase (HvALP) from Heliothis virescens. Insect Biochem Mol Biol 39:294–302. doi:10.1016/j.ibmb.2009.01.006
Perlak FJ, Deaton RW, Armstrong TA, Fuchs RL, Sims SR, Greenplate JT, Fischhoff DA (1990) Insect-resistant cotton plants. Bio/Technology 8:939–943
Perlak FJ, Fuchs RL, Dean DA, McPherson SL, Fischhoff DA (1991) Modification of the coding sequence enhances plant expression of insect control protein genes. Proc Natl Acad Sci USA 88:3324–3328
Perlak FJ, Oppenhuizen M, Gustafson K, Voth R, Sivasupramaniam S, Herring D, Carey B, Ihring RA, Roberts JK (2001) Development and commercial use of Bollgard® cotton in the USA-early promises versus today’s reality. Plant J 27:489–501
Rawat P, Singh AK, Ray K, Chaudhary B, Kumar S, Gautam T, Kanoria S, Kaur G, Kumar P, Pental D, Burma PK (2011) Detrimental effect of expression of Bt endotoxin Cry1Ac on in vitro regeneration, in vivo growth and development of tobacco and cotton transgenics. J Biosci 36:363–376
Rondon SI, DeBano SJ, Clough GH, Hamm PB, Jensen A, Schreiber A, Alvarez JM, Thornton M, Barbour J, Dogramaci M (2007) Biology and management of the potato tuberworm in the Pacific Northwest. PNW.594
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York (PCR)
Sardana R, Dukiandjiev S, Giband M, Cheng X, Cowan K, SauderC Altosaar I (1996) Construction and rapid testing of synthetic and modified toxin gene sequences CryIA (b&c) by expression in maize endosperm culture. Plant Cell Rep 15:677–681
Sarkar A, Hess D, Mondal HA, Banerjee S, Sharma HC, Das S (2009) Homodimeric Alkaline Phosphatase located at Helicoverpa armigera midgut, a putative receptor of Cry1Ac Ccntains α-GalNAc in terminal glycan structure as interactive epitope. J Proteome Res 8:1838–1848. doi:10.1021/pr8006528
Sengupta A, Sarkar A, Priya P, Ghosh Dastidar S, Das S (2013) New insight to structure-function relationship of GalNAc mediated primary interaction between insecticidal Cry1AcToxin and HaALP receptor of Helicoverpa armigera. PLoS One 8(10):e78249
Somers DA, Samac DA, Olhoft PM (2003) Recent advances inlegume transformation. Plant Physiol 131:892–899
Streatfield S, Magallanes-Lundback M, Beifuss K, Brooks C, Harkey R, Love R, Bray J, Howard J, Jilka J, Hood E (2004) Analysis of the maize polyubiquitin-1 promoter heat shock elements and generation of promoter variants with modified expression characteristics. Trans Res 13(4):299–312
Sullivan TD, Christensen A, Quail P (1989) Isolation and characterization of a maize chlorophyll a/b binding protein gene that produces high levels of mRNA in the dark. Mol Gen Genet 215:431–440
Tabashnik BE, Huang F, Ghimire MN, Leonard BR, Siegfried BD, Rangasamy M, Yang Y, Wu Y, Gahan LJ, Heckel DG, Bravo A, Soberón M (2011) Efficacy of genetically modified Bt toxins against insects with different genetic mechanisms of resistance. Nat Biotechnol 29:1128–1131
Tohidfar M, Zare N, Jouzani GS, Eftekhari SM (2013) Agrobacterium-mediated transformation (Medicago sativa) using a synthetic cry3a gene to enhance resistance against alfalfa weevil. Plant Cell Tissue Organ Cult 113:227–235
Van Rheenen HR, Pundir RPS, Miranda JH (1993) How to accelerate the genetic improvement of a recalcitrant crop species such as chickpea. Curr Sci 654:414–417
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Taŕan B, Millan T, Zhang X, Ramsay LD, Iwata A, Wang Y, Nelson W, Farmer AD, Gaur PM, Soderlund C, Penmetsa RV, Xu C, Bharti AK, He W, Winter P, Zhao S, Hane JK, Carrasquilla-Garcia N, Condie JA, Upadhyaya HD, Luo MC et al (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 3:240–246
Wei H, Wang M-L, Moore PH, Albert HH (2003) Comparative expression analysis of two sugarcane polyubiquitin promoters and flanking sequence in transgenic plants. J Plant Physiol 160:1241–1251
Wong EY, Hironaka CM, FischhoV DA (1992) Arabidopsis thaliana small subunit leader and transit peptide enhance the expression of Bacillus thuringiensis proteins in transgenic plants. Plant Mol Biol 20:81–93
Wunn J, Kloti A, Burkhardt PK, Biswas GC, Ghosh K, Iglesias VA, Potrykus I (1996) Transgenic indica rice breeding lineIR58 expressing a synthetic CryIA(b) gene from Bacillus thuringiensis provides effective insect pest control. Biotechnology 14:171–176
Ye G, Tu J, Hu C, Datta K, Datta SK (2001) Transgenic IR72 with fused Bt gene cry1Ab/cry1Ac from Bacillus thuringiensis is resistant against four lepidopteran species under field conditions. Plant Biotechnol 8(2):125–133
Acknowledgments
JC and SS are indebted to Indian Council of Agricultural Research (No. NFBSFARA/AB-2010(2010-11 dt. 24.01.2011) for providing financial support. PG and AS are thankful to Bose Institute for financial assistance. Dr. Dipankar Chakraborty, St. Xavier’s college, Kolkata is acknowledged for his intellectual contribution. Dr. Sujayanand G.K. of Indian Institute of Pulse Research is duly acknowledged for providing H. armigera strain. All the authors are grateful to Bose Institute for infrastructural facilities. Special thanks are extended to Mr. Sudipta Basu, Mr. Swarnava Das and Mr. Surajit Maity for their sincere efforts in the field and necessary laboratory assistance.
Author contributions
JC, SS, DB and SD conceived and designed the experiments. JC, SS and PG carried out Southern analysis, Immunohistofluorescence, ELISA and statistical analysis. JC and SS conducted all the other experiments. JC, SS, PG, AS and SD analyzed the data. JC, SS and PG drafted the manuscript. DB and SD edited the manuscript and supervised the work. SD contributed all the reagents, materials and analysis tools. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Joydeep Chakraborty and Senjuti Sen contributed equally to this work.
Rights and permissions
About this article

Cite this article
Chakraborty, J., Sen, S., Ghosh, P. et al. Homologous promoter derived constitutive and chloroplast targeted expression of synthetic cry1Ac in transgenic chickpea confers resistance against Helicoverpa armigera . Plant Cell Tiss Organ Cult 125, 521–535 (2016). https://doi.org/10.1007/s11240-016-0968-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-016-0968-7