
Abstract
The [3 + 2] cycloaddition (32CA) reactions involving 6-butoxy-5,6-dihydro-4H-1,2-oxazine 2-oxide and dimethyl maleate are examined in this study. Molecular electron density theory (MEDT) is applied at the M06-2X/6-311G(d,p) level, coupled with the D3 dispersion correction. The nitronate 1 species are identified as zwitterionic entities through an analysis of the electron localization function (ELF). This 32CA reaction follows an asynchronous one-step mechanism. Conceptual DFT indices are utilized to classify dimethyl maleate as the electrophilic component and the nitronate as the nucleophilic counterpart. The [3 + 2] cycloaddition processes are predominantly governed by kinetic control, as indicated by activation free energies of − 23.6 and − 11.4 kcal.mol−1 for the exo and endo pathways, respectively, aligning with experimental findings. Despite the nucleophilic and electrophilic character of the reagents, the global electron density transfer at the TSs indicates rather polar 32CA reactions. The formation of a pseudoradical center initiates at carbon atoms C3 and C4. A subsequent docking analysis is conducted on cycloadducts 3 and 4 in relation to the main protease of SARS-CoV-2 (6LU7), alongside the co-crystal ligand. The results of this analysis reveal that cycloadducts 3 exhibit higher binding energy, while cycloadducts 4 display lower binding energy compared to the co-crystal ligand. The results confirm that the presence of isoxazolidine ring increases the affinity of the product 3.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The isoxazolidine ring constitutes a pivotal structural motif found in a multitude of naturally occurring and biologically active compounds, serving as a fundamental scaffold that mimics various natural building blocks and playing a critical role in drug development [1]. Numerous derivatives of this N–O containing five-membered heterocycle have been characterized, exhibiting diverse pharmacological properties, including antiviral [2], antibacterial [3], and anticancer activity [4]. Among the synthetic approaches, the [3 + 2] cycloaddition (32CA) processes have emerged as the most popular and efficient method for the construction of such complex heterocyclic molecules [5]. Specifically, the 32CA reactions involving cyclic nitronate 1 and the double bonds of dimethyl maleate 2 enable the facile synthesis of two diastereomeric isoxazolidines, 3 and 4 (see Scheme 1) [6]. The six-membered cyclic nitronates can be synthesized in the form of individual diastereomers and enantiomers via non-catalyzed [7, 8] and Lewis acid catalyzed [9] hetero-Diels–Alder of nitroalkenes to olefins. Experimental investigations for the reaction of cyclic nitronate 1 dimethyl maleate 2 by Denmark et al. [6] have elucidated the orbital control of these processes, aligning with the principles of Frontier Molecular Orbital (FMO) theory. The experimental results show that an exo orientation is generally preferred. In a complete reversal of the precedent with acyclic nitronates, dimethyl maleate gave a single product resulting from exo approach to nitronates. Accordingly, the associated reaction mechanism is worth investigating in terms of quantum chemical calculations and has not been addressed till date in terms of the MEDT.

32CA reactions of nitronate 1 to dimethyl maleate 2
In the past two decades, the field of computational chemistry has witnessed substantial advancements, leading to the development of specialized software applications tailored for reaction studies [10]. These computational tools empower theoretical chemists to discern intricate mechanistic intricacies. In 2016, Domingo introduced the molecular electron density theory (MEDT) [11], which offers insights into how changes in electron density influence molecular reactivity. Over the past 7 years, the MEDT framework has been employed to analyze various facets of 32CA reactions, encompassing regio- [12], stereo- [12,13,14,15], and chemoselectivity [16, 17], reactivity [18,19,20], substituent effects [21, 22], and other critical aspects [23,24,25]. In this study, a comprehensive MEDT investigation of the 32CA reaction between nitronate 1 and dimethyl maleate 2 (Scheme 1) is presented. Additionally, we explore the role of diastereoselectivity in the interaction of cycloadducts 3 and 4 with the SARS-CoV-2 protein.
This paper is structured into five main sections: (1) An analysis of the electron localization function (ELF) [26] is conducted to elucidate the electronic structure at the ground state (GS) of reagents 1 and 2. (2) Conceptual density functional theory (CDFT) [27, 28] indices are employed to predict electronic flux and polar character in the reagents. (3) The potential energy surface (PES) is systematically examined along the feasible reaction pathways of the investigated 32CA reactions to identify stationary points. (4) ELF topological analysis [26] and QTAIM (Quantum Theory of atoms-in-molecules) [29] parameters are computed to investigate the electronic structure of the located transition states (TSs). (5) Finally, molecular docking studies are conducted to explore the interactions of cycloadducts 3 and 4 with the SARS-CoV-2 main protease.
The global electron density transfer (GEDT) [35] was calculated from the natural bond orbital (NBO) calculations [46, 47] at the TSs using the formula GEDT where q denotes the NBO-derived charges.
Computational methods
The optimization of reagents, transition states (TSs), and cycloadducts entailed the utilization of the Berny analytical gradient optimization approach [30]. Employed for this purpose was the M06-2X [31] functional in tandem with D3 dispersion correction [32] and the 6-311G(d,p) basis set [33]. The M06-2X functional has been endorsed as a suitable computational approach for investigating 32CA reactions in various recent works [16, 34,35,36]. Transition states were ascertained through the observation of a single imaginary frequency, while minima were confirmed by the absence of any imaginary frequencies. The global electron density transfer (GEDT) [37] was calculated from the natural bonding orbitals (NBO) [38] at the TSs using the formula GEDT \(f=\sum q\), where q denotes the NBO-derived charges. The calculation of CDFT indices [28, 39] relied on the B3LYP/6-31G(d) method to delineate the reagents in accordance with conventional reactivity scales [40]. The verification of the minimum energy reaction pathway between reagents, transition states (TSs), and cycloadducts was accomplished via intrinsic reaction coordinate (IRC) computations [41, 42]. The investigation of solvent effects in benzene was carried out employing the polarizable continuum model (PCM) within the self-consistent reaction field (SCRF) framework [43, 44].
The electronic chemical potential (μ) [27], chemical hardness (η) [45], electrophilicity index (ω) [40], and nucleophilicity index (N) [46] were determined using Eqs. (1) to (4) based on the HOMO (EHOMO) and LUMO (ELUMO) energies:
TopMod program [47] and Multiwfn [48] were used to conduct the topological analysis of the ELF and QTAIM [26, 29, 49]. The bonding evolution theory (BET) [50] study of the bonding changes along the experimentally favorable reaction path involving nitronate 1 and dimethyl maleate 2 was also performed. Visualization of the ELF localization domains was performed using Paraview software [51]. All calculations were executed with Gaussian 16 [52].
The compounds 3 and 4, once optimized, were utilized to conduct molecular docking through AutoDock Vina 1.2.0 [53]. The three-dimensional structures of the molecular target were obtained from the Protein Data Bank (PDB) (www.rcsb.org), specifically the SARS-CoV-2 (2019-nCoV) main protease (PDB: 6LU7). Receptor preparation involved the removal of heteroatoms (water and ions), addition of polar hydrogen, and charge assignment. Active sites were defined by grid boxes of suitable dimensions surrounding the bound co-crystal ligands. The docking study was executed with Autodock Vina 1.2.0 [53], and visualization was facilitated by Discovery Studio software [54].
Results and discussion
Analysis of the ELF topology of the reactants nitronate 1 and dimethyl maleate 2
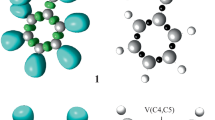
The ELF, initially formulated by Becke and Edgecombe [26] and further developed by Silvi and Savin [49], provides a precise mathematical model for characterizing the electronic structure within a chemical system. The ELF model identifies core, bonding, and non-bonding localization attractors in distinct electronic regions. To classify the three atom components (TACs) involved in 32CA reactions, Domingo’s topological analysis of the ELF is instrumental, categorizing them as pseudodiradical, pseudo(mono)radical, carbenoid, and zwitterionic TACs [11, 55]. Figure 1 illustrates the ELF localization regions and crucial valence basin populations for the optimized reagents nitronate 1 and dimethyl maleate 2 at the M06-2X-D3/6-311G(d,p) level of theory.

M06-2X-D3/6-311G(d,p) basin attractor of nitronate 1 and dimethyl maleate 2 and the ELF localization domains. Protonated basins are shown in blue, monosynaptic basins in red, disynaptic basins in green, and attractor positions in purple (Isovalue = 0.80). Also shown are the suggested Lewis-type structures and the natural atomic charges in terms of the average number of electrons e. Negative charges are shown in red and positive charges in blue
In the ELF analysis of nitronate 1, two monosynaptic basins V(O1) and V′(O1) with a combined population of 5.83e are associated with the non-bonding electron density on the oxygen atom O1. Additionally, two V(C3,N2) disynaptic basins with a total population of 4.20e are linked to the C3-N2 double bond, and a V(N2,O1) disynaptic basin integrating 1.65e is linked to the N2-O1 single bond. Nitronate 1 falls into the category of a zwitterionic TAC, lacking a pseudoradical or carbenoid center. The ELF of dimethyl maleate 2 shows disynaptic basins V(C4,C5) and V′(C4,C5) with a total population of 5.32e related to the C4-C5 double bond. Figure 1 shows the Lewis-like structures of the reactants nitronate 1 and dimethyl maleate 2, as well as the charges from the NBO.
In terms of charges, O1 in nitronate 1 carries a negative charge of 0.402e, while N2 has a positive charge of 0.176e, and C3 has a positive charge of 0.060e. In contrast, dimethyl maleate 2 exhibits negative charges of 0.118 and 0.117 on C4 and C5 carbon atoms, respectively.
Analysis of the CDFT indices
The study of CDFT reactivity indices [27, 28] provides an initial insight into the direction of electronic flow between reactants. CDFT indices were calculated for nitronate 1 and dimethyl maleate 2 at the B3LYP/6-31G(d) level of theory, in line with established reactivity scales [28, 40, 46]. These indices are the electronic chemical potential µ, the chemical hardness η, the electrophilicity ω, and the nucleophilicity N, all expressed in eV. The electronic chemical potential μ of nitronate 1 is found to be − 3.71 eV and exceeds that of dimethyl maleate 2, which is − 4.94 eV (see Table 1). This discrepancy indicates an electronic flux from nitronate 1 to dimethyl maleate 2 during the 32CA reaction. Nitronate 1, with an electrophilicity index (ω) of 1.26 eV, is considered a moderate electrophile, while dimethyl maleate 2, with ω of 2.12 eV, is a strong electrophile. Nitronate 1 is classified as a strong nucleophile (N = 3.04 eV), and dimethyl maleate 2 (N = 1.66 eV) is classified as moderate nucleophiles according to the standard nucleophilicity scale [46].
Analysis of the potential energy surface
The 32CA reaction between nitronate 1 and dimethyl maleate 2 proceeds through two stereoisomeric pathways, exo and endo. Stationary points were identified along these pathways, revealing transition states TS1 and TS2 leading to the formation of cycloadducts 3 and 4. This suggests a one-step mechanism for the 32CA reaction, as shown in Scheme 2.

The stereoisomeric paths for the 32CA reactions of nitronate 1 and dimethyl maleate 2
Taking account that the M06-2X functional was not able to reproduce the experimental selectivity, this 32CA reaction was further studied at the MPWB1K/6‐311G(d,p) computation level. The relative electronic energies, enthalpies, free energies, and entropies of TSs in benzene and their respective products are presented in Table 2. The investigation of the energy profile can yield interesting results. The 32CA reaction exhibits exergonic behavior in benzene, with negative reaction free energies, indicating kinetic control and irreversibility. The above statement is in full agreement with the experimental results, which demonstrate the formation of cycloadducts 3 and 4 [6]. The activation enthalpy of TS1 is 2.2 kcal.mol−1, while TS2 exhibits an activation enthalpy of 4.4 kcal.mol−1 (endo path) using M06-2X functional (see Fig. 2). However, these values slightly increased to 3.9 kcal.mol−1 for TS1 and 5.9 kcal.mol−1 for TS2 when MPWB1K functional was used. The value of relative Gibbs free energies was also increased by 6 and 8.8 kcal.mol−1 for TS1 and TS2, respectively, using MPWB1K functional. Domingo and co-workers [56] have classified the polar reactions into forward and reverse electron density flux (FEDF and REDF) reactions based on the analysis of the GEDT at the TSs. In FEDF cycloaddition reactions, the electron density always fluxes from the TAC or diene towards the ethylene. Non-polar 32CA reactions showing a GEDT lesser than 0.05e are classified as null electron density flux (NEDF). As the analysis of the electron density flux is performed at the actual TSs, this classification is unambiguous. Table 2 also presents the GEDT observed in the TSs, which were used to determine their respective orientations. The analysis of transition states indicates a forward electron density flux (FEDF) [57], signifying a polar reaction. The identified transition states exhibit a GEDT with values of 0.128e and 0.133e.

Relative enthalpies, in kcal.mol−1, for the studied reaction of nitronate 1 and dimethyl maleate 2 in benzene at M06-2X-D3/6–311G(d,p) and MPWB1K/6-311G(d,p) (in parentheses) level of theory
Figure 3 displays the optimized geometry of TS1 and TS2 in the gas phase. Examination of the geometry indicates that the distance between C3 and C4 is shorter than that between O1 and C5, with values of 2.11 and 2.13 Å, respectively, in TS1. However, an opposite trend was found in TS2 with a value of 2.28 Å for the C–C and 2.13 Å for the C-O bond. This finding indicates that TS2 exhibits a somewhat higher degree of asynchrony compared to TS1. The Wiberg bond index [58] between reacted atoms was computed from natural bond orbital analysis and listed in Table 1S. Analysis of the Wiberg bond index reveals that the studied reactive channels are not completely synchronous, because the degree of advancement of the two new bonds between two fragments is not equal in the corresponding transition states.

M06-2X-D3/6–311G(d,p) and MPWB1K/6-311G(d,p) (in parentheses) optimized geometry of TSs and cycloadducts in benzene (bond lengths in Å)
Topological analysis of the ELF at the TSs
The topological characterization of the optimized transition states (TSs) associated with the 32CA reaction between nitronate 1 and dimethyl maleate 2 unveiled their electronic structures. The ELF localization domains and basin attractor locations for the transition states (TSs) involved in the 32CA reaction are depicted in Fig. 4. Analysis of the electron localization function (ELF) of TS1 revealed the presence of monosynaptic basins V(O1) and V′(O1), which together include a population of 5.71e. Similarly, ELF analysis of TS2 indicated the existence of monosynaptic basins V(O1) and V′(O1), with a combined population of 5.74 electrons, associated with the density of nonbonding electrons on the O1 oxygen atom. The ELF of TS1 and TS2 contains V(N2,C3) monosynaptic basins comprising a total population of 2.46e and 2.86e, respectively. These basins are associated with the C3-N2 bonding region. Furthermore, the monosynaptic basin V(N2) integrates 1.62e and 1.68e for TS1 and TS2, respectively. This basin is associated with the density of nonbonding electrons in the nitrogen N2. It is worth noting that there was a decrease in the electron population within the bonding region between C3 and N2. Specifically, the electron population decreased from 4.20e in the case of nitronate 1 to 2.46e in TS1 and 2.86e in TS2. This observation indicates that the double bond between C3 and N2 was cleaved in the transition states, resulting in the generation of nonbonding electron density on the nitrogen atom of N2. The population within disynaptic basin V(O1,N2) shows a decline, decreasing from 1.65e in nitronate 1 to 1.45e in TS1 and further to 1.38e in TS2. The predominant source of electron density in the monosynaptic basin of V(N2) can be attributed to the bonding region between N2 and C3. In TS1, two monosynaptic basins V(C3) and V(C4) with electron densities of 0.46e and 0.36e, respectively, indicate the establishment of pseudoradical centers at C3 and C4. These monosynaptic basins were not observed in TS2.

M06-2X-D3/6-311G(d,p) ELF localization domains and the positions of the TS1 and TS2 attractor basins. Monosynaptic basins are shown in red, protonated basins in blue, disynaptic basins in green, and attractor positions in purple
Additionally, the ELF analysis of TS1 and TS2 revealed the presence of a single disynaptic basin, wherein 2.92e and 3.23e are combined from the C4–C5 bonding area.
Mechanistic implications along the stereoisomeric channels of 32CA reaction from the bonding evolution theory (BET)
The bonding evolution theory (BET), formulated by Krokidis [50] through the utilization of ELF topological analysis and Thom’s catastrophe theory [59], examines the successive alterations in bonding along a given reaction pathway. This provides mechanistic implications for understanding the reaction mechanism. Table 3 presents the populations of ELF basins at the specific sites where the reaction between nitronate 1 and dimethyl maleate 2 occurs during cyclisation, following the stereoisomeric route of TS1. Examination of the ELF basins identified six distinct topological phases belonging to the exo pathway. The ELF structure of the initial point S1 bears a resemblance to the bonding patterns seen in the individual compounds, as indicated in Fig. 1. The ELF studies conducted at the TS1 reveal the presence of a V(N1) monosynaptic basin, which integrates a population of 1.35e at S2, related to the N2 nitrogen lone pair. Pseudoradical centers are observed at carbon atoms C3 and C4 in the S3, leading to the creation of monosynaptic basins V(C3) and V(C4). TS1 belongs to Phase III, as shown in Fig. 5. In this phase, the formation of the single bonds O1–C5 and C3–C4 has not yet occurred. During Phase IV, pseudoradical centers emerge specifically at the carbon atom located at position 5 (C5), resulting in the creation of monosynaptic basins denoted as V(C5). In Phase V, the initial formation of single bonds occurs between carbon atoms C3 and C4. Phase VI witnesses the commencement of forming initial single bonds between atoms O1 and C5, evidenced by the emergence of the disynaptic basin V(C3,C4).

M06-2X-D3/6-311G(d,p) IRC profile of TS1 for the 32CA reaction of nitronate 1 and dimethyl maleate 2
Topological analysis of the AIM at the TSs
The topological analysis of the atoms-in-molecules (AIM) method, as developed by Bader and co-workers [29, 60], is employed to analyze interatomic interactions at the transition states. The values of the total electron density (ρ) and the Laplacian of electron density \({\nabla }^{2}\uprho ({\text{r}}_{\text{c}})\) at the bond critical points (BCP1 and BCP2) corresponding to the formation of O1-C5 and C3-C4 bonds are presented in Table 4. In both instances, the overall electron density is below 0.1 atomic units, in accordance with the topological analysis of ELF. Additionally, the empirical evidence suggests that the interatomic distance required for bond formation exceeds 2.0 Å. This information is further supported by the NCI gradient isosurfaces in Fig. 6 and the reduced density gradient (RDG) plot in Fig. 7. A continuous color-coding scheme based on the second eigenvalue of the Hessian is used, where strong, attractive interactions are represented in blue, weak interactions in green, and strong repulsive interactions in red.

NCI gradient isosurfaces of the transition states TS1 and TS2 for 32CA reaction of nitronate 1 and dimethyl maleate 2

The reduced density gradient and gradient isosurfaces of TS1 and TS2
Molecular docking against SARS-CoV-2
To elucidate the mode of action of the tested cycloadducts (3 and 4), a molecular docking analysis was employed to ascertain their binding modes against the main protease of SARS-CoV-2, a crucial target for the development of anti-SARS-CoV-2 drugs. The selection of these targets was based on their important involvement in viral protein synthesis. Consequently, directing therapeutic efforts towards these proteins offers prospective benefits in terms of virus eradication. The reliability of the docking parameters and procedures was validated by re-docking the co-crystallized ligand, which accurately reproduced the position and orientation observed in the crystal structure (RMSD = 1.34 Å).
The co-crystalized ligand engaged in interactions with the active site of the protein through the formation of five hydrogen bonds, with a corresponding free binding energy of − 7.5 kcal.mol−1, as shown in Table 5. Comparatively, compound 3 exhibited a more favorable binding affinity than the co-crystalized ligand, with a free binding energy of − 7.7 kcal.mol−1, while compound 4 exhibited a lower binding affinity with a value of − 7.2 kcal.mol−1. These findings are visually depicted in Fig. 8.

3D and 2D interactions between tested compounds 3 and 4 with 6LU7 protein
Conclusions
The 32CA reaction of nitronate 1 and dimethyl maleate 2 at the M06-2X-D3/6-311G(d,p) level of theory was investigated. The examination of the CDFT indices predicted the nucleophilic nature of nitronate 1, while dimethyl maleate 2 exhibited electrophilic behavior. The 32CA reaction is found to be polar zwitterionic type and proceeds through one-step mechanism with asynchronous TSs. According to the bonding evolution theory, the pseudoradical center at C3 and C4 formed earlier in the reaction, with the C3-C4 bond forming earlier as well, outweighing the significance of the two-center interaction between the most electrophilic and most nucleophilic centers. The two 32CA reaction variants studied were kinetically controlled and exhibited a one-step mechanism. The activation enthalpies for these reactions in benzene were 2.2 and 4.4 kcal.mol−1 for exo and endo paths, respectively, with activation free energies of 13.5 and 14.9 kcal.mol−1. The initial transition states did not exhibit the creation of C3-C4 and O1-C5 bonds, as indicated by the analysis of geometric parameters and the examination of the ELF and QTAIM. The transition states’ activation free energies derived using M06-2X calculations were in concurrence with the experimental data, suggesting significant diastereoselectivity in the reaction. The analysis conducted by BET indicated that the reaction mechanism followed a one-step mechanism.
Furthermore, the molecular docking simulations provided significant insights into the binding interactions between ligands 3 and 4 and the main protease (6LU7) of the SARS-CoV2 virus. The docking affinity values revealed variations in binding strength, with the cycloadduct 3 (− 7.7 kcal.mol−1) demonstrating the highest affinity, followed by co-crystal ligand (− 7.5 kcal.mol−1), and then cycloadduct 4 (− 7.2 kcal.mol−1). According to the results, the isoxazolidine ring enhances the affinity of the cycloadducts 3 with the SARS-CoV2 compared to the co-crystal ligand.
Data availability
Data and materials are available from the corresponding author on request.
References
Berthet M, Cheviet T, Dujardin G, Parrot I, Martinez J (2016) Isoxazolidine: a privileged scaffold for organic and medicinal chemistry. Chem Rev 116(24):15235–15283
Dmitriev VA, Efremova MM, Novikov AS, Zarubaev VV, Slita AV, Galochkina AV, Starova GL, Ivanov AV, Molchanov AP (2018) Highly efficient and stereoselective cycloaddition of nitrones to indolyl-and pyrrolylacrylates. Tetrahedron Lett 59(24):2327–2331
Yamaguchi M, Matsuda A, Ichikawa S (2015) Synthesis of isoxazolidine-containing uridine derivatives as caprazamycin analogues. Org Biomol Chem 13(4):1187–1197
Khazir J, Singh PP, Reddy DM, Hyder I, Shafi S, Sawant S, Chashoo G, Mahajan A, Alam M, Saxena A (2013) Synthesis and anticancer activity of novel spiro-isoxazoline and spiro-isoxazolidine derivatives of α-santonin. Eur J Med Chem 63:279–289
Jasiński R (2023) Recent progress in the synthesis of nitroisoxazoles and their hydrogenated analogs via [3+2] cycloaddition reactions (microreview). Chem Heterocycl Compd 59(11):730–732
Denmark SE, Seierstad M, Herbert B (1999) Tandem cycloaddition chemistry of nitroalkenes: preparative and theoretical studies on the stereochemical course of [3+ 2] cycloaddition of cyclic nitronates. J Org Chem 64(3):884–901
Woliński P, Kącka-Zych A, Wróblewska A, Wielgus E, Dolot R, Jasiński R (2023) Fully selective synthesis of spirocyclic-1,2-oxazine N-oxides via non-catalysed Hetero Diels-Alder reactions with the participation of cyanofunctionalysed conjugated nitroalkenes. Molecules 28:4586. https://doi.org/10.3390/molecules28124586
Woliński P, Kącka-Zych A, Mirosław B, Wielgus E, Olszewska A, Jasiński R (2022) Green, one-pot synthesis of 1,2-oxazine-type herbicides via non-catalyzed Hetero Diels-Alder reactions comprising (2E)-3-aryl-2-nitroprop-2-enenitriles. J Clean Prod 356:131878
Han H, Li C, Niu X, Wang Y, Zhang W, Wang Q (2022) A combination of polarity reversal, Diels-Alder cycloaddition and skeletal remodeling to access pyridine-fused nitrones. Chem Commun 58(30):4775–4778
Krylov A, Windus TL, Barnes T, Marin-Rimoldi E, Nash JA, Pritchard B, Smith DG, Altarawy D, Saxe P, Clementi C, Crawford TD (2018) Perspective: computational chemistry software and its advancement as illustrated through three grand challenge cases for molecular science. J Chem Physics 149(18).
Domingo LR (2016) Molecular electron density theory: a modern view of reactivity in organic chemistry. Molecules 21(10):1319
Domingo LR, Ríos-Gutiérrez M, Pérez P (2018) A molecular electron density theory study of the reactivity and selectivities in [3+ 2] cycloaddition reactions of C, N-dialkyl nitrones with ethylene derivatives. J Org Chem 83(4):2182–2197
Domingo LR, Ríos-Gutiérrez M (2017) A molecular electron density theory study of the reactivity of azomethine imine in [3+ 2] cycloaddition reactions. Molecules 22(5):750
Acharjee N, Mohammad-Salim HA, Chakraborty M, Rao MP, Ganesh M (2021) Unveiling the high regioselectivity and stereoselectivity within the synthesis of spirooxindolenitropyrrolidine: a molecular electron density theory perspective. J Phys Org Chem 34(6):e4189
Salih SAM, Basheer HA, de Julián-Ortiz JV, Mohammad-Salim HA (2023) Unveiling the stereoselectivity and regioselectivity of the [3+ 2] cycloaddition reaction between N-methyl-C-4-methylphenyl-nitrone and 2-propynamide from a MEDT perspective. Int J Mol Sci 24(10):9102
Aitouna AO, Mohammad-Salim H, Zeroual A, Syed A, Bahkali AH, de Julián-Ortiz JV (2023) Investigation of the molecular mechanism and diastereoselectivity in the [3+ 2] cycloaddition reaction between acetonitrile oxide and Cis-3, 4-dichlorocyclobutene: insights from MEDT and docking study. Comput Theor Chem 1228:114283
Abbiche K, Mohammad-Salim H, Salah M, Mazoir N, Zeroual A, El Alaoui El Abdallaoui H, El Hammadi A, Hilali M, Abdallah H, Hochlaf M (2020) Insights into the mechanism and regiochemistry of the 1, 3-dipolar cycloaddition reaction between benzaldehyde and diazomethane. Theor Chem Acc 139:1–12
Domingo LR, Ríos-Gutiérrez M, Silvi B, Pérez P (2018) The mysticism of pericyclic reactions: a contemporary rationalisation of organic reactivity based on electron density analysis. Eur J Org Chem 2018(9):1107–1120
Domingo LR, Acharjee N, Mohammad-Salim HA (2020) Understanding the reactivity of trimethylsilyldiazoalkanes participating in [3+ 2] cycloaddition reactions towards diethylfumarate with a molecular electron density theory perspective. Organics 1(1):3–18
Mohammad-Salim H, de Julián-OrtizJV, (2023) Theoretical insight into the mechanism and selectivity of the [3+ 2] cycloaddition reaction of N-methyl-1-phenylmethanimine oxide and bicyclopropylidene with a MEDT perspective. Struct Chem 35(2):541–551
Ríos-Gutiérrez M, Domingo LR, Jasiński R (2021) Understanding the different reactivity of (Z)-and (E)-β-nitrostyrenes in [3+ 2] cycloaddition reactions. An MEDT study. RSC Adv 11(16):9698–9708
Domingo LR, Seif A, Mazarei E, Zahedi E, Ahmadi TS (2020) A molecular electron density theory (MEDT) study of the role of halogens (X 2= F 2, Cl 2, Br 2 and I 2) on the aza-Michael-addition reactions. New J Chem 44(44):19002–19012
Mondal A, Domingo LR, Acharjee N (2023) Revealing the influence of tether length on the intramolecular [3+ 2] cycloaddition reactions of nitrones from the molecular electron density theory perspective. J Phys Org Chem 37:e4574
Domingo LR, Aurell MJ, Ríos-Gutiérrez M (2023) A molecular electron density theory study of the domino reaction of N-phenyl iminoboranes with benzaldehyde yielding fused bicyclic compounds. Molecules 28(17):6211
Islam MS, Al-Majid AM, Haukka M, Parveen Z, Ravaiz N, Wadood A, Rehman AU, Ríos-Gutiérrez M, Domingo LR, Barakat A (2023) A novel alpha-amylase inhibitor-based spirooxindole-pyrrolidine-clubbed thiochromene-pyrzaole pharmacophores: unveiling the [3+ 2] cycloaddition reaction by molecular electron density theory. Chem Biol Drug Design 102(5):972–95
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92(9):5397–5403
Parr R, Yang W (1989) Density-functional theory of atoms and molecules. Oxford University Press, New York, Oxford
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21(6):748
Bader RF (1985) Atoms in molecules. Acc Chem Res 18(1):9–15
Schlegel HB (1982) Optimization of equilibrium geometries and transition structures. J Comput Chem 3(2):214–218
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoret Chem Acc 120:215–241
Smith DG, Burns LA, Patkowski K, Sherrill CD (2016) Revised damping parameters for the D3 dispersion correction to density functional theory. The journal of physical chemistry letters 7(12):2197–2203
Hehre WJ (1976) Ab initio molecular orbital theory. Acc Chem Res 9(11):399–406
Shen J, Zhang Y, Xue Y (2022) Theoretical insights into enantioselective [3+ 2] cycloaddition between cinnamaldehyde and cyclic N-sulfonyl trifluoromethylated ketimine catalyzed by N-heterocyclic carbene. J Phys Chem A 126(20):3124–3134
Mohammad-Salim HA, Abdallah HH (2019) Theoretical study of the [4+ 2] cycloaddition reaction of trifluoroethylene with five-membered chalcogens heterocyclic compounds. ARO-The Scientific Journal of Koya University 7(2):69–77
Barrales-Martínez C, Martínez-Araya JI, Jaque P (2021) 1, 3-Dipolar cycloadditions by a unified perspective based on conceptual and thermodynamics models of chemical reactivity. J Phys Chem A 125(3):801–815
Domingo LR (2014) A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4(61):32415–32428
Chocholoušová J, Špirko V, Hobza P (2004) First local minimum of the formic acid dimer exhibits simultaneously red-shifted O-H⋯ O and improper blue-shifted C–H⋯ O hydrogen bonds. Phys Chem Chem Phys 6(1):37–41
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103(5):1793–1874
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58(22):4417–4423
Gonzalez C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94(14):5523–5527
Fukui K (1970) Formulation of the reaction coordinate. J Phys Chem 74(23):4161–4163
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev 94(7):2027–2094
Cances E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107(8):3032–3041
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105(26):7512–7516
Domingo LR, Pérez P (2011) The nucleophilicity N index in organic chemistry. Org Biomol Chem 9(20):7168–7175
Noury S, Krokidis X, Fuster F, Silvi B (1999) Computational tools for the electron localization function topological analysis. Comput Chem 23(6):597–604
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33(5):580–592
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371(6499):683–686
Krokidis X, Silvi B, Alikhani M (1998) Topological characterization of the isomerization mechanisms in XNO (X= H, Cl). Chem Phys Lett 292(1–2):35–45
Ayachit U (2015) The paraview guide: a parallel visualization application. Kitware, Inc
Frisch, ME, Trucks G, Schlegel HB, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Petersson G, Nakatsuji H, Li X (2016) Gaussian 16. Gaussian, Inc. Wallingford, CT
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
BIOVIA DS, (2021) Discovery studio modeling environment. Dassault Systèmes, San Diego, CA, USA
Mohammad-Salim HA, Acharjee N, Domingo LR, Abdallah HH (2021) A molecular electron density theory study for [3+ 2] cycloaddition reactions of 1-pyrroline-1-oxide with disubstituted acetylenes leading to bicyclic 4-isoxazolines. Int J Quantum Chem 121(5):e26503
Domingo LR, Ríos-Gutiérrez M, Pérez P (2020) A molecular electron density theory study of the participation of tetrazines in aza-Diels–Alder reactions. RSC Adv 10(26):15394–15405
Domingo LR, Ríos-Gutiérrez M (2023) A Useful classification of organic reactions based on the flux of the electron density. SciRad 2:1–24
Wiberg KB (1968) Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24(3):1083–1096
Thom R (1977) Stabilité structurelle et morphogénèse: essai d’une théorie générale des modèles. InterÉditions, Paris
Bader RF, Essén H (1984) The characterization of atomic interactions. J Chem Phys 80(5):1943–1960
Funding
This work was funded by the Researchers Supporting Project Number (RSP2024R273), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mohammad-Salim, H., de Julián-Ortiz, J.V., Dahlous, K.A. et al. Elucidating the mechanism and selectivity of [3 + 2] cycloaddition: a DFT and molecular docking investigation of the reaction of 6-butoxy-5,6-dihydro-4H-1,2-oxazine 2-oxide with dimethyl maleate. Struct Chem (2024). https://doi.org/10.1007/s11224-024-02373-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11224-024-02373-7