
Abstract
A series of 3-aryl coumarin derivatives and 3-phenylazo-4-hydroxycoumarin were evaluated for their monoamine oxidase (MAO) A and B inhibitory activity and selectivity by fluorometric enzymological assays. Among 21 coumarin derivatives, compound 21 (3-phenylazo-4-hydroxycoumarin) displayed a good inhibitory activity (0.12 ± 0.02 µM) and very high selectivity for MAO-B (SI > 833.33). The inhibition was determined as mixed-type and not time-dependent. Docking studies, molecular dynamics and molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) calculations were performed to elucidate in vitro results. Our results reveal that the insertion of an azo linker between coumarin and phenyl rings in 3-arylcoumarins enhances MAO-B selectivity enormously since such a linker leads to the perfect alignment of the coumarin ring in the aromatic cage and the phenyl ring in the entrance cavity of MAO-B active site. Hydrogen bond interactions with Cys172 in the active site entrance of MAO-B also contributes to the remarkably higher inhibitory activity and selectivity for MAO-B.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
It has been verified that coumarin derivatives are strong antioxidants and can prevent the formation of reactive oxygen species (ROS), which play a critical role in the pathogenesis of neurodegenerative diseases. Coumarins such as 3-arylcoumarins possessing methoxy-, hydroxy-, and acetoxy- groups were shown to have antioxidant activities [1,2,3,4,5,6] and coumarins with various substituents have been shown to inhibit monoamine oxidase (MAO) enzymes selectively [7,8,9].
MAO (EC-1.4.3.4) is an enzyme that contains flavin adenine dinucleotide (FAD) and is located outside the mitochondrial membrane and catalyses the oxidative deamination of biological amines. Excessive increase of MAO activity also leads to excessive formation of toxic metabolites such as hydrogen peroxide, ammonia, which are associated with oxidative stress and degeneration of tissues [10]. Due to the role in the oxidation and regulation of intracellular concentration of neurotransmitters, such as serotonin (5-hydroxytryptamine, 5-HT), norepinephrine and dopamine, MAO is an important target in the therapy of neurological disorders such as depression and Parkinson disease [11]. MAO enzyme has two functional isoenzymes, MAO-A and -B, that are differentiated by their distribution in tissues, immunological properties, substrate and inhibitor preferences [12, 13]. MAO inhibitors can be examined in two groups; reversible and irreversible. Clinical use of non-selective and irreversible MAO inhibitors, such as hydrazine, cyclopropylamine, propargylamine and allylamine derivatives, is restricted [14] due to the risk of cheese effect (excessive tyramine accumulation related to extreme consumption of aged cheese), causing sudden elevation of blood pressure and cerebral hemorrhage. Therefore, investigations have focused on selective and reversible inhibitors with different chemical structures which act through non-covalent interactions, enabling the cleavage of the enzyme-inhibitor complex so that the enzyme regains its activity [15].
There are various compounds that are known to selectively and reversibly inhibit MAO isoenzymes. Simoxatone, moclobemide and brofaromine reversibly inhibit MAO-A, whereas safinamide and lazabemide reversibly inhibit MAO-B [16, 17]. Lazabemide was never marketed [18] and adverse effects such as nausea, dizziness, sleeplessness and low blood pressure were reported with use of safinamide in more than 1% in clinical trials [19]. Therefore, discovery of novel selective reversible MAO inhibitors is still essential today.
In the literature, there are various studies emphasizing the high potency of coumarin derivatives for MAO inhibition [20,21,22,23]. It has been found that substitutions at positions 3 or 4 of the main structure of the coumarin modulate the MAO-B inhibitory effect and MAO-A/B selectivity [9, 16, 20, 24]. Among these, 3-arylcoumarins with various substituents in the phenyl rings have been proven to be potent and selective MAO-B inhibitor [7,8,9, 25, 26]. Those 3-arylcoumarins in the literature mostly possess methyl group at the 6th position of the coumarin ring [26,27,28] and/or they are halogenated either on the coumarin ring or the 3-aryl ring [9, 25, 26, 29] or amidated [28].
These studies in the literature prompted us to investigate the MAO inhibitory activities of a series of 3-arylcoumarins with mainly -OCH3, -OH, -OAc substituents at 5, 6, 7, 8 positions of coumarin rings, and p-, m- positions at 3-aryl ring (Scheme 1), which are mostly different than the ones reported in the literature. The MAO inhibitory activities of these compounds which were previously synthesized in our laboratories [30,31,32] are presented for the first time. Moreover, former studies of our group confirmed their strong antioxidant activities [1,2,3,4]. This motivated us to explore their neuroprotective potential as well since dual activity (antioxidant/MAO inhibition) is a valuable strategy for the treatment of neurodegenerative diseases.

Coumarin derivatives used in this study. Compounds 1–20 (left) and 21 (right). See also Table 1 for the description of compounds 1–20
MAO enzymes have been extensively investigated by computational tools owing to the significance of their function and being the target of neurodegenerative disorders, [3, 33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49].
Molecular modeling techniques such as docking are useful tools to predict and analyze interactions between small molecules and their target biomolecules which play an essential role in rational drug design. Many docking studies have been reported for MAO enzymes so far, including various coumarin derivatives [8, 20,21,22, 50,51,52,53,54,55,56,57,58,59,60]. Docking programs are useful tools to evaluate ligands’ binding affinities toward a macromolecule. However, molecular docking is mostly a static approach and cannot properly account for the dynamics of biomolecule-ligand complexes. Molecular dynamics have become a very important tool to obtain structural and chemical data of dynamic protein, nucleic acid and biomolecule-ligand complexes [61, 62]. To accommodate the conformational flexibility of the protein, a newer approach, Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) has been developed which takes into account dynamics of protein–ligand complexes to predict the binding energy that is averaged over a trajectory. Moreover, this method decomposes the total binding energy into components [63].
In this study, twenty-one previously synthesized coumarin derivatives [30,31,32] have been investigated for their in vitro MAO-A and -B inhibitory potencies. To the best of our knowledge, except compounds 7, 10 and 14, MAO inhibitory activities of all other compounds are reported for the first time. In addition, since compound 21 was detected as the best MAO-B inhibitor with high selectivity, in silico studies were also carried out on 21 to provide insight into its inhibitory activity and selectivity.
Experimental
Materials
MAO-A (hMAO-A)/MAO-B (hMAO-B), DMSO, selegiline, rasagiline and iproniazide were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Amplex Red MAO Assay Kit was purchased from Invitrogen (Waltham, MA, USA). All starting chemicals were provided from Sigma-Aldrich (St Louis, MO, USA) or Merck (Darmstadt, Germany).
In vitro MAO-A and -Binhibition fluorescence assay
The synthesized compounds were investigated for their MAO inhibitory activities using recombinant human MAO isoforms by an in vitro continuous fluorometric method. The inhibitory activity was determined by measuring the production of H2O2 from p-tyramine, the common substrate for both hMAO-A and hMAO-B, using the Amplex®-Red MAO assay kit.
3-arylcoumarin derivatives were dissolved in DMSO to prepare a 10 mM stock solution and kept at − 20 °C. 2 μL of the coumarin derivatives prepared at different concentrations and 2 μL of known MAO inhibitors (selegiline, iproniazide and rasagiline) as positive controls in DMSO (not higher than 1% in each well) and as negative controls; 2 μL coumarin samples containing 4.5 μL 100 mM pH 7.4 potassium phosphate buffer instead of hMAO-A/B. 4.5 μL hMAO-A/B were added to a black flat-bottomed 96-well microtest plate. 93.5 μL reaction buffer were added to each well and incubated with stirring for 15 min at 37 °C, in the dark fluorimeter chamber. At the end of the incubation period, the reaction was started by adding 100 μL substrate solution (200 mM Amplex® Red reagent, 1 U/mL horseradish peroxidase and 1 mM p-tyramine). The production of H2O2 and of resorufin was quantified by a microplate reader over a 30 min period at 37 °C, in which the generated fluorescence increased linearly (FLX800™, Bio-Tek® Instruments, Inc., Winooski, VT, USA; excitation/emission: 545/590 nm). Enzymatic activity was calculated by subtracting the baseline fluorescence intensity from the observed fluorescence intensity at the end of 30 min. The reactions carried out with hMAO-A and -B isoenzymes were allowed to proceed at the same time. To observe the effect of DMSO in the coumarin samples, the same experiment was repeated using DMSO instead of distilled water.
Determination of the inhibition mechanism
In order to determine the inhibition mechanism of the coumarin derivative exhibiting the highest inhibitory activity (compound 21), the enzyme activities were determined at 3 different inhibitor concentrations (0.05, 0.1 and 0.2 μM) and at 8 different substrate concentrations (1, 0.75, 0.5, 0.25, 0.15, 0.1, 0.075 and 0.050 mM) as described above. Control experiments were carried out simultaneously by replacing the compounds (coumarin derivatives and reference inhibitors) with appropriate dilutions of DMSO. Km and Vmax values for the MAO-A and -B enzymes were calculated from nonlinear regression analysis of the Michaelis–Menten graphs acquired with GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA. USA). Lineweaver- Burk plots were used to determine the inhibition mechanism of the most potent inhibitor (compound 21) with respect to tyramine as a substrate.
Time-dependent assay for MAO-B inhibitory activity
In studies of the dependence of inhibition on concentration and incubation time of compound 21; 3-phenylazo-4-hydroxycoumarin, which displayed the highest inhibitory activity (Table 1), was evaluated at 0.13 μM. The kinetics of the interaction between compound 21 and MAO-B were further examined by the method of Kitz and Wilson [82]. For further confirmation of the reversibility of enzyme inhibition, four different samples (0.13 μM) of compound 21 were prepared, added to MAO-B solution and incubated at different time periods for 15, 30, 45 and 60 min and the MAO-B activity was determined as described before (Sect. 1.3). Results represent the means ± standard deviation (n = 3). Linear regression analysis was performed using GraphPad Prism 6.0.
Theoretical evaluation of ADME properties
ADME (absorption, distribution, metabolism and excretion) model was applied to coumarin derivatives to confirm the Lipinski’s rule of five (molecular weight, number of hydrogen donors and acceptors, and lipophilicity-expressed as log P). Theoretical prediction of ADME properties was evaluated with Molinspiration software (www.molinspiration.com).
Statistical analysis
IC50 values were determined by nonlinear regression analysis of MAO inhibition versus the logarithm (-log) of the tested compound molar concentration and calculated by mean IC50 value ± SD (standard deviation) using GraphPad Prism 6.0. All the experiments were performed in triplicate and the level of significance was defined as p < 0.05.
Computational studies
Docking studies
Docking simulations were carried out to obtain the most stable complex of compound 21 with MAO-A and MAO-B. Toward this end, the coumarin derivative were subjected to geometry optimization using M062X functional and 6-31G(d,p) basis set using Gaussian 09 [83]. The crystal structures of MAO-A and MAO-B (PDB ID: 2Z5X and 2XFN, respectively) were downloaded from Brookhaven Protein Data Bank. AutoDock4 v.4.2.6 [68] was employed for docking calculation. Grid for docking was described as 60 × 60 × 60 points with a grid spacing of 0.375 Å by using Autogrid4. Center for grid was defined with coordinates (x, y, z) 34.176, 31.342, − 14.785 for MAO-A and 55.199, 150.457, 21.281 for MAO-B, as to center FAD (Flavin Adenine Dinucleotide) cofactor in both enzymes. Conformational space was searched by Lamarckian Genetic Algorithm [84], which was implemented in AutoDock4. Maximum number of evaluations was set to 2,500,000 and 100 runs were performed for both enzymes and each ligand. Prior to docking the coumarin compounds, docking procedure was validated by docking the co-crystalized ligands (harmine for MAO-A and 2-(2-benzofuranyl)-2-imidazoline for MAO-B) and calculating the Root Mean Square Deviation (RMSD) between the docked pose and the co-crystallized structure as 0.7558 Å and 0.1044 Å, respectively. All calculations were performed on Linux platform.
Molecular dynamics simulations
The best MAO-B in vitro selective inhibitor analogue compound 21 complexes with MAO-A and MAO-B were individually subjected to MM/PBSA calculations. Toward this end, MD simulations were carried out with GROMACS 5.0 package [85].
Both enzymes were defined with CHARMM36 force-field and the ligand (compound 21) was parameterized by CGenff. Each complex was solvated in TIP3P water [86, 87] using a periodic cubic box with dimensions of 11.0 nm × 11.0 nm × 11.0 nm to ensure the structure would remain in the box throughout the simulation. A chloride ion was added for a neutralized system. LINCS [88] algorithm was employed for constraining bond lengths. Langevin thermostat was used at 300 K. Particle Mesh Ewald (PME) [89] was employed for long-range electrostatic interactions. Each complex was first minimized by using steepest descent algorithm with GROMACS 5.0 package [85]. Minimization was complete as the maximum force of 10 kJ/mol was reached. Berendsen’s barostat [90] was used to relax the density at a pressure of 1 atm. Both complexes were then subjected to 30 ns MD simulations in explicit water. The simulation time-step was set to 2 fs and data were recorded every 10 ps.
MM/PBSA calculations
The MM/PBSA method, which was originally defined by Kollman et al. [91], employs the equation below to calculate the binding free energy of the protein–ligand complex:
Where Gcomplex refers to the free energy of the complex, Greceptor and Gligand are the free energy of the receptor and the ligand, respectively.
MM/PBSA is a convenient method to evaluate the binding and the energies, since both enzymes (PDB ID: 2Z5X and 2XFN) are mostly located in the cytoplasmic region, except for an alpha helix that is in the transmembrane region of the mitochondria [70, 71]. MM/PBSA calculations were performed using g_mmpbsa method [63]. The grid spacing was set to 0.5 Å. The solute dielectric constant (ε) was set to 2. The ionic strength was set to default value (0.15 M).
Cluster analysis
Cluster analysis was performed to obtain representative structures of MAO-A and MAO-B complexes with compound 21. For this purpose, gmx cluster was employed which is implemented in GROMACS. GROMOS method [92] was selected as the clustering algorithm, which chooses the conformation with the highest number of neighbors as the center of the first cluster within a cut-off range. The cut-off value was kept at the default value of 0.1 nm.
Results and discussion
In vitro MAO-A and -B inhibition fluorescence assay and structure–activity relationship
In vitro studies were carried out to investigate the MAO inhibitory activity of 21 coumarin derivatives as shown in Table 1. Compounds were tested against both human MAO isoforms using a continuous fluorometric assay to determine the inhibitory activities.
As it is shown in Table 1, 3-aryl coumarins display MAO inhibitory activity in the micromolar range. Majority of the compounds show inhibitions below 10 µM IC50 values. The striking feature is that all hydroxy substituted coumarins, except for compound 15, demonstrate the highest MAO inhibitory activities. Hydroxy compounds (1, 3, 5, 7, 9–12, 14–16, 20) exhibit relatively low IC50 values in a range of 1.56 – 3.95 µM for MAO-A and 0.91 – 3.14 µM for MAO-B. Thus, in general, hydroxy substitution enhances the activity, presumably by its hydrogen bond donor feature. On the contrary, acetoxy substitution decreases the inhibitory effect such that compounds 2, 4, 6, 8, 17, 18, 19 exhibit weaker inhibitory effect. For example, replacement of hydroxyl groups in compound 1 (R2 = R3 = OH, R1´ = R2´ = OCH3) with the acetoxy groups in compound 4 (R2 = R3 = OAc, R1´ = R2´ = OCH3) lowered the inhibitory activity fourfold. Similarly, the same fourfold decrease in the activity of compound 8 (R1 = R2 = OAc, R1´ = R2´ = OCH3) is also observed, in regard to compound 7 (R1 = R2 = OH, R1´ = R2´ = OCH3).
Among the twenty 3-aryl coumarins, compound 10, with p- nitro substituent, displays the best inhibitory activity with 0.91 µM value for MAO-B and the highest S.I value, however still lacking the desired selectivity. This is the only 3-aryl coumarin derivative bearing a nitrogen atom at position 3 among the studied compounds. This observation prompted us to test the inhibitory activity with another nitrogen containing coumarin derivative. Therefore, we further determined the MAO inhibitory activity of 3-phenylazo-4-hydroxycoumarin (compound 21), which was previously synthesized in our laboratory [30]. Interestingly, this compound turned out to be the best MAO-B inhibitor (IC50 = 0.13 µM) together with a remarkable selectivity (SI > 833.33). Although synthesis and several applications of 3-phenylazo-4-hydroxycoumarin (21) are documented in the literature [66, 67], to the best of our knowledge, its inhibitory activity to MAO isoforms has not been reported yet. The presence of azo moiety in compound 21 might be partially responsible for the superior inhibitory activity and selectivity toward MAO-B. Further detailed experimental and in silico studies for compound 21 are presented below.
Evaluation of inhibition mechanism and kinetics
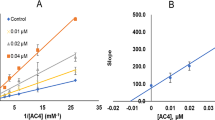
The mechanism of inhibition of the coumarin derivative; compound 21, which displayed the highest MAO-B inhibitory activity and selectivity, was evaluated. Based on dose–response inhibition, three concentrations of the inhibitors were selected, two below and one above IC50 value for the inhibition. The enzyme kinetics data are presented as double reciprocal Lineweaver–Burk plots (Fig. 1). Binding of compound 21 with MAO-B increase the Km value (the Michaelis–Menten constant). As the concentration of the inhibitor increases, Km value apparently increases and Vmax (maximum enzyme activity) decreases, indicating that the inhibitor favors binding to the enzyme substrate complex. The kinetic mechanism results are consistent with mixed-type inhibition mechanism of MAO-B. Ki and Vmax values were determined as 0.0315 µM and 26.58 nM/min/mg of protein.

Kinetic characteristics of inhibition of human MAO-B with compound 21
Analysis of time-dependent enzyme inhibition
The recombinant MAO-B was incubated for different times with compound 21. The activity of enzyme was determined and the percentage of enzyme inhibition was plotted against the pre-incubation time to determine the time dependence of inhibition. The inhibition of MAO-B by compound 21 was not dependent on the pre-incubation time (Fig. 2) and this supports our findings that the inhibition mechanism is reversible.

Time-dependent inhibition of recombinant human MAO-B by compound 21
Computational results
Docking
Since compound 21 exhibited the best selectivity according to the experimental results (Table 1), we focused our computational studies on this compound. Docking studies were carried out to calculate binding energy and explore the binding mechanism for the most promising derivative, compound 21, by using Autodock4 [68]. Docking scores and estimated inhibition constants are displayed in Table 2.
The results are in qualitative agreement with experimental findings such that compound 21 was predicted to be MAO-B selective.
We further examined ligand-enzyme complexes of compound 21 employing MD simulations and calculated the binding energies using MM/PBSA method which is expected to produce more realistic results since the protein dynamics is also taken into account.
MD simulations
Simulations were run to perform MM/PBSA calculations and to obtain time-averaged energy terms. Prior to simulations of MAO-A and MAO-B complexes with compound 21, both enzymes were subjected to 30 ns MD simulations as in holoenzyme form. Later, both complexes obtained from docking were undergone 30 ns simulations. Time dependence of RMSD of both holoenzymes in absence and presence of ligands were plotted (Fig. 3). According to the graphs, it was observed that ligand complexation has increased the stability of MAO-B enzyme, whereas fluctuations have been observed for MAO-A enzyme-ligand complex with remarkably larger RMSD values.

RMSD comparison of proteins obtained from simulations with compound 21 (complex, red) and without ligand (holoenzyme, black) for MAO-A and MAO-B
Moreover, RMSD was calculated for compound 21, with a least square fit to protein backbone for both MAO-A and MAO-B (Fig. 4). RMSD range of MAO-B (0.1–0.2 nm) is smaller than that of MAO-A (0.2–0.35 nm) except for the leap at around 15th ns, which justifies that compound 21 forms a more stable complex with MAO-B than with MAO-A.

Time dependence of RMSD computed for 21 in MAO-A complex (black) and in MAO-B complex (red)
RMSF
The RMSF (root mean square fluctuations) is an assessment of displacement of atoms respective to the reference structure, averaged over the number of atoms [69]. RMSF was calculated to examine the fluctuations in the protein for MAO-A and MAO-B (Fig. 5).

RMSF graphs for MAO-A (left) and MAO-B (right). Black plots represent fluctuations in the absence of ligand. Red plots represent fluctuations in the presence of ligands
Figure 5 shows fluctuations for each residue of MAO-A and MAO-B. For both of the enzymes, the most noticeable thing is the high fluctuation observed for the residues around 480–500 and 460–500 for MAO-A and MAO-B, respectively. These residues correspond to the C-terminal helix buried in the mitochondrial outer membrane [70, 71]. For MAO-A, fluctuations are observed where a helix turn takes place. This is reasonable considering that those residues are in the vicinity of surface of the protein and are more exposed to water, thus more flexible. For MAO-B, more incidents of decrease in fluctuation are observed in the presence of ligand relative to the holoenzyme. This may arise from the ligand binding’s contribution to overall stabilization of the macromolecule. It can be interpreted from Fig. 5 that, presence of ligand in both enzymes cut down the fluctuations. However, displaying more fluctuations in the holo form, MAO-B was stabilized to a greater extent compared to MAO-A upon ligand binding. In general, ligand shows higher conformational stability in MAO-B relative to MAO-A which is revealed by the smaller RMSF values (about 0.1 nm) in MAO-B than in MAO-A (0.2 nm).
MM/PBSA calculations
MM/PBSA calculations were run to examine energy components that constitute the total energy of binding. Table 3 shows the energy terms that contribute to the stability of the protein structures of MAO-A and MAO-B and binding energy of compound 21.
According to Table 3, binding of compound 21 to MAO-A is electrostatically favored over MAO-B. However, polar solvation energy that contributes to binding of compound 21 to MAO-B is lower than that of MAO-A, resulting in a higher affinity (lower binding energy). Polar solvation energy stands for the electrostatic interactions between the continuum solvent and the solute [72]. The difference in polar solvation energy between MAO-A and MAO-B (~ 2.27 kcal/mol), can be due to the binding site of MAO-A, e.g. having less polar groups such as Val210, Cys323, Ile335, Val93 with respect to their corresponding amino acids Thr201, Thr314, Tyr326, Glu84 in MAO-B [73]. Binding energy calculated over simulation is shown in Fig. 6. When different energy components were analyzed in Table 3, it is observed that van der Waals energy (EVDW) is the main energy term that contributes to the total binding energy, and it is substantially more negative than any other energy component. It appears that polar solvation energy (Gpol) is responsible for the difference in total binding energies of compound 21 with MAO-A and MAO-B.

Binding energy (kcal/mol) of compound 21 over time with MAO-A (black) and MAO-B (red)
It is observed that binding energies (∆Gbind) obtained from MM/PBSA (Table 3) are highly negative compared to docking binding energies (Table 2). This discrepancy is a known issue in the literature and there exists plenty of MM/PBSA studies reporting extremely more negative ΔG binding values compared to the docking results [74,75,76], because they are completely different computational approaches related to static (in case of docking) and dynamic (in case of MM/PBSA) aspects of binding process. Therefore, it is not appropriate to make a direct comparison based on the absolute values of ∆Gbind and Ki values. Instead, a qualitative comparison of the SI values can be made. SI obtained from MM/PBSA (25) is notably larger than that from docking (6) revealing that the former method shows better performance in approaching the experimental result. However, it is difficult to make a direct comparison on the absolute value of SI between the calculated and the experimental results because of the complications arising from the mixed-type inhibition kinetics of compound 21. In the literature, results from MM/PBSA analyses have been reported to be correlated with the experimental results [77, 78] although a few contrasting cases have also been presented [72, 79].
Cluster analysis
Cluster analysis was employed by using GROMACS tool gmx cluster. Data obtained from cluster analysis is shown in Table 4. The noticeable difference in total number of clusters between two complexes may account for relatively stable nature of MAO-B complex with compound 21.
We also examined the representative average structures from the most populated clusters of both complexes and their interactions with compound 21 (Fig. 7).

Binding mode and interactions of compound 21 in the active site of MAO-A and MAO-B
Interactions of compound 21 with MAO-A and MAO-B
Interactions of compound 21 with MAO-A and MAO-B were observed by using Discovery Studio 4.5 [80]. Figure 7 displays the interactions in the representative average structures of the most populated cluster for compound 21 in complex with MAO-A and MAO-B. Compound 21 was observed to bind to the active site for both MAO-A and MAO-B based on mainly hydrophobic interactions (π-π stacking and π-alkyl). The striking difference is the two H-bonding type interactions between Cys172 and the hydroxyl group of compound 21 in MAO-B while such interactions are not observed in MAO-A since there is no cysteine residue in its active site. This provides significant insight into selectivity of compound 21 toward MAO-B. It is important to note that this interaction type was not observed in the binding orientation obtained from docking calculations revealing better performance of MD over docking methodology due to dynamic nature of binding. Furthermore, compound 21 makes closer interactions with the surrounding residues in the long and narrow binding site of MAO-B. It is known that MAO-B active site has two cavities; entrance cavity near Ileu199 and the aromatic cage constituting two tyrosines (398 and 435) in the re-face of FAD. The flexibility of the azo linker allows the ligand to fit perfectly the double-pocket active site of MAO-B. As Fig. 7 displays, as coumarin moiety of 21 binds to the aromatic cage, phenyl substituent fits well to the entrance cavity with hydrophobic interactions. This occurrence is supported by a study of Lee et al., who showed a linker between two aromatic rings in 2-aryl-1,3,4-oxadiazin-5(6H)-one, enhanced the binding and selectivity of ligand to MAO-B [81]. Thus, our results verify that, in 3-arylcoumarin skeleton, insertion of a linker azo group between the coumarin and aromatic rings contributes both potency and selectivity remarkably.
Theoretical evaluation of ADME properties
ADME properties of all coumarin derivatives were predicted by using Molinspiration software (www.molinspiration.com). Table 5 shows ADME properties and the number of violations of Lipinski rules. It was observed that none of the compounds violated Lipinski’s rule of five.
Conclusions
In this study, a series of 3-substituted coumarin derivatives were investigated for their MAO inhibitory activities in vitro for the first time. The most potent coumarin derivative has been determined as compound 21 (3-phenylazo-4-hydroxycoumarin) (MAO-A: 25.01 ± 1.35% (inhibition at 100 µM); MAO-B IC50: 0.12 ± 0.02 µM, selectivity index: > 833.33). The inhibition mechanism of this compound has been identified as mixed inhibition. From these results, it may be concluded that compound 21 selectively and reversibly inhibited MAO-B at remarkably low concentrations. Binding affinities calculated by AutoDock4 adequately predicted the MAO-B selectivity of this compound. However, since docking approach cannot deal with dynamical nature of ligand-enzyme complexes, compound 21 was further investigated using MD and MM/PBSA which resulted in higher selectivity toward MAO-B when compared to the Autodock4 results. Our computational results provide mechanistic insight into the underlying aspects of this selectivity. The predominant selectivity of compound 21 toward MAO-B was found to be arising from hydrogen bonding interactions with Cys172 residue which is absent in MAO-A. In addition, the azo group between two aromatic rings served as a linker which provided the molecule a more compatible shape with that of the binding pocket of MAO-B. This might also account for the higher inhibitory activity for MAO-B. Moreover, theoretical ADME predictions showed that compound 21 complies with Lipinski’s rule of five. Thus, in order to increase the MAO-B selectivity and potency of 3-arylcoumarins, insertion of the azo or similar linkers between the coumarin and aromatic rings can be adopted as a new strategy. We propose 3-arylazocoumarin as a new promising scaffold for selective MAO-B inhibition which deserves further investigation to improve its pharmacodynamic and pharmacokinetic properties.
References
Yuce B, Danis O, Ogan A et al (2009) Antioxidative and lipid lowering effects of 7,8-dihydroxy-3-(4-methylphenyl) coumarin in hyperlipidemic rats. ARZNEIMITTEL-FORSCHUNG-DRUG RESEARCH 59:129–134
Danis O, Demir S, Gunduz C et al (2016) Synthesis of selected 3-and 4-arylcoumarin derivatives and evaluation as potent antioxidants. Res Chem Intermed 42:6061–6077
Erzincan P, Saçan MT, Yüce-Dursun B et al (2015) QSAR models for antioxidant activity of new coumarin derivatives. SAR QSAR Environ Res 26:721–737
Ozalp L, Danış Ö, Yuce-Dursun B et al (2020) Investigation of HMG-CoA reductase inhibitory and antioxidant effects of various hydroxycoumarin derivatives. Arch Pharm (Weinheim) n/a:e1900378. https://doi.org/10.1002/ardp.201900378
Roussaki M, Kontogiorgis CA, Hadjipavlou-Litina D et al (2010) A novel synthesis of 3-aryl coumarins and evaluation of their antioxidant and lipoxygenase inhibitory activity. Bioorg Med Chem Lett 20:3889–3892
Rodríguez SA, Nazareno MA, Baumgartner MT (2011) Effect of different C3-aryl substituents on the antioxidant activity of 4-hydroxycoumarin derivatives. Bioorg Med Chem 19:6233–6238
Matos MJ, Teran C, Perez-Castillo Y et al (2011) Synthesis and study of a series of 3-arylcoumarins as potent and selective monoamine oxidase B inhibitors. J Med Chem 54:7127–7137
Viña D, Matos MJ, Ferino G et al (2012) 8-Substituted 3-arylcoumarins as potent and selective MAO-B inhibitors: synthesis, pharmacological evaluation, and docking studies. ChemMedChem 7:464–470. https://doi.org/10.1002/cmdc.201100538
Mellado M, Mella J, González C et al (2020) 3-Arylcoumarins as highly potent and selective monoamine oxidase B inhibitors: which chemical features matter? Bioorg Chem 101:103964
Youdim MBH, Edmondson D, Tipton KF (2006) The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci 7:295–309
van den Berg D, Zoellner KR, Ogunrombi MO et al (2007) Inhibition of monoamine oxidase B by selected benzimidazole and caffeine analogues. Bioorg Med Chem 15:3692–3702
Edmondson DE, Mattevi A, Binda C et al (2004) Structure and mechanism of monoamine oxidase. Curr Med Chem 11:1983–1993
Edmondson DE, Binda C, Wang J et al (2009) Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry 48:4220–4230
Baker GB, Coutts RT, Greenshaw AJ (2000) Neurochemical and metabolic aspects of antidepressants: an overview. J Psychiatry Neurosci 25:481
Chaurasiya ND, Ganesan S, Nanayakkara NPD et al (2012) Inhibition of human monoamine oxidase A and B by 5-phenoxy 8-aminoquinoline analogs. Bioorg Med Chem Lett 22:1701–1704
Chimenti F, Bolasco A, Secci D et al (2010) Investigations on the 2-thiazolylhydrazyne scaffold: synthesis and molecular modeling of selective human monoamine oxidase inhibitors. Bioorg Med Chem 18:5715–5723
Badavath VN, Baysal I, Ucar G et al (2016) Monoamine oxidase inhibitory activity of novel pyrazoline analogues: curcumin based design and synthesis. ACS Med Chem Lett 7:56–61
Cesura AM (2007) Monoamine oxidase B. In: Enna SJ, Bylund DB (eds) xPharm: the Comprehensive Pharmacology Reference. Elsevier, New York, pp 1–10
Cruz MP (2017) Xadago (Safinamide): A monoamine oxidase B inhibitor for the adjunct treatment of motor symptoms in Parkinson’s disease. P T 42:622–637
Pisani L, Muncipinto G, Miscioscia TF et al (2009) Discovery of a novel class of potent coumarin monoamine oxidase B inhibitors: development and biopharmacological profiling of 7-[(3-chlorobenzyl)oxy]-4-[(methylamino)methyl]-2H-chromen-2-one methanesulfonate (NW-1772) as a highly potent, selective, revers. J Med Chem 52:6685–6706. https://doi.org/10.1021/jm9010127
Delogu G, Picciau C, Ferino G et al (2011) Synthesis, human monoamine oxidase inhibitory activity and molecular docking studies of 3-heteroarylcoumarin derivatives. Eur J Med Chem 46:1147–1152. https://doi.org/10.1016/j.ejmech.2011.01.033
Patil PO, Bari SB, Firke SD et al (2013) A comprehensive review on synthesis and designing aspects of coumarin derivatives as monoamine oxidase inhibitors for depression and Alzheimer’s disease. Bioorg Med Chem 21:2434–2450. https://doi.org/10.1016/j.bmc.2013.02.017
Gnerre C, Catto M, Leonetti F et al (2000) Inhibition of monoamine oxidases by functionalized coumarin derivatives: biological activities, QSARs, and 3D-QSARs. J Med Chem 43:4747–4758
Fonseca A, Reis J, Silva T et al (2017) Coumarin versus chromone monoamine oxidase B inhibitors: quo vadis? J Med Chem 60:7206–7212
Matos MJ, Vilar S, García-Morales V et al (2014) Insight into the functional and structural properties of 3-arylcoumarin as an interesting scaffold in monoamine oxidase B inhibition. ChemMedChem 9:1488–1500
Serra S, Ferino G, Matos MJ et al (2012) Hydroxycoumarins as selective MAO-B inhibitors. Bioorg Med Chem Lett 22:258–261
Matos MJ, Viña D, Quezada E et al (2009) A new series of 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg Med Chem Lett 19:3268–3270
Vina D, Matos MJ, Yáñez M et al (2012) 3-Substituted coumarins as dual inhibitors of AChE and MAO for the treatment of Alzheimer’s disease. Medchemcomm 3:213–218
Delogu GL, Serra S, Quezada E et al (2014) Monoamine oxidase (MAO) inhibitory activity: 3-phenylcoumarins versus 4-hydroxy-3-phenylcoumarins. ChemMedChem 9:1672–1676
Danış Ö, Yuce-Dursun B, Gunduz C et al (2010) Synthesis of 3-amino-4-hydroxy coumarin and dihydroxy-phenyl coumarins as novel anticoagulants. Arzneimittelforschung 60:617–620. https://doi.org/10.1055/s-0031-1296335
Gündüz C, Salan Ü, Bulut M (2009) The synthesis of novel 4-(3, 4-dimethoxyphenyl) chromenone-crown ethers and their cation binding, as determined using fluorescence spectra. Supramol Chem 21:724–731
Gündüz C, Salan Ü, Bulut M (2010) The synthesis and fluorescence properties of novel chromenone-crown ethers. Supramol Chem 22:491–498
Erdem SS, Karahan O, Yildiz I, Yelekçi K (2006) A computational study on the amine-oxidation mechanism of monoamine oxidase: insight into the polar nucleophilic mechanism. Org Biomol Chem 4:646–658. https://doi.org/10.1039/b511350d
Abad E, Zenn R, Kästner J (2013) The reaction mechanism of monoamine oxidase from QM/MM calculations. J Phys Chem B 117. https://doi.org/10.1021/jp4061522
Erdem SS, Özpınar GA, Boz Ü (2014) Quantum chemical modeling of the inhibition mechanism of monoamine oxidase by oxazolidinone and analogous heterocyclic compounds. J Enzyme Inhib Med Chem 29:81–86. https://doi.org/10.3109/14756366.2012.753882
Repič M, Vianello R, Purg M et al (2014) Empirical valence bond simulations of the hydride transfer step in the monoamine oxidase B catalyzed metabolism of dopamine. Proteins 82:3347–3355. https://doi.org/10.1002/prot.24690
Zapata-Torres G, Fierro-Huerta A, Barriga-González G et al (2015) Revealing monoamine oxidase B catalytic mechanisms by means of the quantum chemical cluster approach. J Chem Inf Model 55. https://doi.org/10.1021/acs.jcim.5b00140
Oanca G, Purg M, Mavri J et al (2016) Insights into enzyme point mutation effect by molecular simulation: phenylethylamine oxidation catalyzed by monoamine oxidase A. Phys Chem Chem Phys 18:13346–13356. https://doi.org/10.1039/c6cp00098c
Poberžnik M, Purg M, Repič M et al (2016) Empirical valence bond simulations of the hydride-transfer step in the monoamine oxidase A catalyzed metabolism of noradrenaline. J Phys Chem B 120:11419–11427. https://doi.org/10.1021/acs.jpcb.6b09011
Cakir K, Erdem SS, Atalay VE (2016) ONIOM calculations on serotonin degradation by monoamine oxidase B: insight into the oxidation mechanism and covalent reversible inhibition. Org Biomol Chem 14:9239–9252. https://doi.org/10.1039/c6ob01175f
Prah A, Franciskovic E, Mavri J, Stare J (2019) Electrostatics as the driving force behind the catalytic function of the monoamine oxidase A enzyme confirmed by quantum computations. ACS Catal 9. https://doi.org/10.1021/acscatal.8b04045
Akyüz MA, Erdem SS, Edmondson DE (2007) The aromatic cage in the active site of monoamine oxidase B: effect on the structural and electronic properties of bound benzylamine and p-nitrobenzylamine. J Neural Transm (Vienna) 114:693–698. https://doi.org/10.1007/s00702-007-0670-3
Apostolov R, Yonezawa Y, Standley DM et al (2009) Membrane attachment facilitates ligand access to the active site in monoamine oxidase A. Biochemistry 48:5864–5873. https://doi.org/10.1021/bi900493n
Erdem SS, Büyükmenekşe B (2011) Computational investigation on the structure-activity relationship of the biradical mechanism for monoamine oxidase. J Neural Transm (Vienna) 118:1021–1029. https://doi.org/10.1007/s00702-011-0635-4
Borštnar R, Repič M, Kržan M et al (2011) Irreversible inhibition of monoamine oxidase B by the antiparkinsonian medicines rasagiline and selegiline: a computational study. European J Org Chem 2011:6419–6433. https://doi.org/10.1002/ejoc.201100873
Akyüz MA, Erdem SS (2013) Computational modeling of the direct hydride transfer mechanism for the MAO catalyzed oxidation of phenethylamine and benzylamine: ONIOM (QM/QM) calculations. J Neural Transm (Vienna) 120:937–945. https://doi.org/10.1007/s00702-013-1027-8
Atalay V, Erdem S (2013) A comparative computational investigation on the proton and hydride transfer mechanisms of monoamine oxidase using model molecules. Comput Biol Chem 47C:181–191. https://doi.org/10.1016/j.compbiolchem.2013.08.007
Erdem SS, Türkkan S, Yelekçi K, Gökhan-Kelekçi N (2013) Insights into the binding mode of new N-substituted pyrazoline derivatives to MAO-A: docking and quantum chemical calculations. J Neural Transm (Vienna) 120:859–862. https://doi.org/10.1007/s00702-012-0950-4
Pavlin M, Mavri J, Repič M, Vianello R (2013) Quantum-chemical approach to determining the high potency of clorgyline as an irreversible acetylenic monoamine oxidase inhibitor. J Neural Transm 120:875–882. https://doi.org/10.1007/s00702-013-1016-y
Chimenti F, Secci D, Bolasco A et al (2004) Inhibition of monoamine oxidases by coumarin-3-acyl derivatives: biological activity and computational study. Bioorg Med Chem Lett 14:3697–3703. https://doi.org/10.1016/j.bmcl.2004.05.010
Pisani L, Catto M, Nicolotti O et al (2013) Fine molecular tuning at position 4 of 2H-chromen-2-one derivatives in the search of potent and selective monoamine oxidase B inhibitors. Eur J Med Chem 70:723–739. https://doi.org/10.1016/j.ejmech.2013.09.034
Matos MJ, Vilar S, Gonzalez-Franco RM et al (2013) Novel (coumarin-3-yl) carbamates as selective MAO-B inhibitors: synthesis, in vitro and in vivo assays, theoretical evaluation of ADME properties and docking study. Eur J Med Chem 63:151–161
Abdelhafez OM, Amin KM, Ali HI et al (2013) Monoamine oxidase A and B inhibiting effect and molecular modeling of some synthesized coumarin derivatives. Neurochem Int 62:198–209. https://doi.org/10.1016/j.neuint.2012.11.005
Pisani L, Farina R, Nicolotti O et al (2015) In silico design of novel 2H-chromen-2-one derivatives as potent and selective MAO-B inhibitors. Eur J Med Chem 89:98–105. https://doi.org/10.1016/j.ejmech.2014.10.029
Matos MJ, Janeiro P, González Franco RM et al (2014) Synthesis, pharmacological study and docking calculations of new benzo[f]coumarin derivatives as dual inhibitors of enzymatic systems involved in neurodegenerative diseases. Future Med Chem 6:371–383. https://doi.org/10.4155/fmc.14.9
He X, Chen Y-Y, Shi J-B et al (2014) New coumarin derivatives: design, synthesis and use as inhibitors of hMAO. Bioorg Med Chem 22:3732–3738. https://doi.org/10.1016/j.bmc.2014.05.002
Matos MJ, Rodríguez-Enríquez F, Vilar S et al (2015) Potent and selective MAO-B inhibitory activity: amino- versus nitro-3-arylcoumarin derivatives. Bioorg Med Chem Lett 25:642–648. https://doi.org/10.1016/j.bmcl.2014.12.001
Joubert J, Foka GB, Repsold BP et al (2017) Synthesis and evaluation of 7-substituted coumarin derivatives as multimodal monoamine oxidase-B and cholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur J Med Chem 125:853–864. https://doi.org/10.1016/j.ejmech.2016.09.041
Baek SC, Kang M-G, Park J-E et al (2019) Osthenol, a prenylated coumarin, as a monoamine oxidase A inhibitor with high selectivity. Bioorg Med Chem Lett 29:839–843. https://doi.org/10.1016/j.bmcl.2019.01.016
Ferino G, Vilar S, Matos JM et al (2012) Monoamine oxidase inhibitors: ten years of docking studies. Curr Top Med Chem 12:2145–2162
McCammon JA, Gelin BR, Karplus M (1977) Dynamics of folded proteins. Nature 267:585–590
Mackerell AD, Nilsson L (2008) Molecular dynamics simulations of nucleic acid-protein complexes. Curr Opin Struct Biol 18:194–199
Kumari R, Kumar R, Lynn A (2014) g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54:1951–1962. https://doi.org/10.1021/ci500020m
Yang J, Zhang P, Hu Y et al (2019) Synthesis and biological evaluation of 3-arylcoumarins as potential anti-Alzheimer’s disease agents. J Enzyme Inhib Med Chem 34:651–656
Musa MA, Badisa VLD, Aghimien MO et al (2021) Identification of 7, 8-dihydroxy-3-phenylcoumarin as a reversible monoamine oxidase enzyme inhibitor. J Biochem Mol Toxicol 35:e22651
Kappe T, Burdeska K, Ziegler E (1966) Synthesen von Heterocyclen, 80. Mitt.: Über ein durch H-Brücken stabilisiertes 2, 5-Dihydropyrazin. Monatshefte für Chemie und verwandte Teile anderer Wissenschaften 97:77–86
Makhlouf MM, Zeyada HM (2016) Synthesis, structural analysis, spectrophotometric measurements and semiconducting properties of 3-phenyl azo-4-hydroxycoumarin thin films. Synth Met 211:1–13
Morris GM, Huey R, Lindstrom W et al (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. https://doi.org/10.1002/jcc
Martínez L (2015) Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE 10:e0119264. https://doi.org/10.1371/journal.pone.0119264
Son SY, Ma J, Kondou Y et al (2008) Structure of human monoamine oxidase A at 2.2-Å resolution: the control of opening the entry for substrates/inhibitors. Proc Natl Acad Sci U S A 105:5739–5744. https://doi.org/10.1073/pnas.0710626105
Bonivento D, Milczek EM, McDonald GR et al (2010) Potentiation of ligand binding through cooperative effects in monoamine oxidase B. J Biol Chem 285:36849–36856. https://doi.org/10.1074/jbc.M110.169482
Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10:449–461. https://doi.org/10.1517/17460441.2015.1032936
De Colibus L, Li M, Binda C et al (2005) Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc Natl Acad Sci U S A 102:12684–12689. https://doi.org/10.1073/pnas.0505975102
Thirunavukkarasu MK, Karuppasamy R (2021) Drug repurposing combined with MM/PBSA based validation strategies towards MEK inhibitors screening. J Biomol Struct Dyn 1–12
de Gomes I, S, Santana CA, Marcolino LS, et al (2022) Computational prediction of potential inhibitors for SARS-COV-2 main protease based on machine learning, docking, MM-PBSA calculations, and metadynamics. PLoS ONE 17:e0267471
Dankwa B, Broni E, Enninful KS et al (2022) Consensus docking and MM-PBSA computations identify putative furin protease inhibitors for developing potential therapeutics against COVID-19. Struct Chem 1–21
Laurini E, Col VD, Mamolo MG et al (2011) Homology model and docking-based virtual screening for ligands of the σ1 receptor. ACS Med Chem Lett 2:834–839
Burmaoglu S, Kazancioglu EA, Kazancioglu MZ et al (2022) Synthesis, molecular docking and some metabolic enzyme inhibition properties of biphenyl-substituted chalcone derivatives. J Mol Struct 1254:132358
Lee MC, Yang R, Duan Y (2005) Comparison between Generalized-Born and Poisson-Boltzmann methods in physics-based scoring functions for protein structure prediction. J Mol Model 12:101–110
BIOVIA (2016) Discovery studio modeling environment, Release 2016, San Diego: Dassault Systems, 2015
Lee J, Lee Y, Park SJ et al (2017) Discovery of highly selective and potent monoamine oxidase B inhibitors: contribution of additional phenyl rings introduced into 2-aryl-1,3,4-oxadiazin-5(6H)-one. Eur J Med Chem 130:365–378. https://doi.org/10.1016/j.ejmech.2017.02.059
Kitz R, Wilson IB (1962) Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J Biol Chem 237:3245–3249
Frisch M, Trucks GW, Schlegel HB et al (2009) Gaussian 09. Revision A. Gaussian Inc., Wallingford, CT
Morris GM, Goodsell DS, Halliday RS et al (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19:1639–1662
Abraham MJ, Murtola T, Schulz R et al (2015) Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Jorgensen W, Chandrasekhar J, Madura J et al (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. https://doi.org/10.1063/1.445869
MacKerell AD Jr, Bashford D, Bellott M et al (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472
Darden TA, York D, Pedersen LJ (1993) Particle mesh Ewald: an Nṡlog(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Berendsen HJC, Postma JPM, Van Gunsteren WF et al (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690
Kollman PA, Massova I, Reyes C et al (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33:889–897. https://doi.org/10.1021/ar000033j
Daura X, Gademann K, Jaun B et al (1999) Peptide folding: when simulation meets experiment. Angew Chem Int Ed 38:236–240. https://doi.org/10.1002/(SICI)1521-3773(19990115)38:1/2%3c236::AID-ANIE236%3e3.0.CO;2-M
Funding
This work was supported by Marmara University, Commission of Scientific Research Project, FEN-A-110913–0376. L. Ozalp acknowledges Council of Higher Education (YÖK) of Turkey for the PhD scholarship of 100/2000 program.
Author information
Authors and Affiliations
Contributions
Basak Yuce-Dursun: Conceptualization, methodology, experiments. Özkan Danış: Conceptualization, methodology, experiments, data curating, writing, editing Lalehan Özalp: Methodology, data curating, analysis, visualization, writing. Elif Şahin: Experiments, data curating, writing. Serap Demir: Conceptualization, methodology, review. Safiye Sağ Erdem: Analysis, supervision, writing, editing, review. Ayse Ogan: Conceptualization, supervision, project administration, writing, editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yuce-Dursun, B., Danış, Ö., Ozalp, L. et al. In vitro and in silico investigation of inhibitory activities of 3-arylcoumarins and 3-phenylazo-4-hydroxycoumarin on MAO isoenzymes. Struct Chem 34, 1715–1729 (2023). https://doi.org/10.1007/s11224-022-02092-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-022-02092-x