
Abstract
The interaction of three potent antifilarial compounds (4C, 4F, and 3F) with filarial proteins thioredoxin, glutathione s-transferase and cyclophilin were investigated using molecular docking and density functional theory (DFT) studies. Molecular docking was performed using YASARA tool, Hex 8.0.0 Cuda tool and PatchDock server and docked complex were visualized by Discovery Studio 3.0. The predicted binding energy of antifilarial compounds 4C (−247.6, −243.8, −256.8 kcal mol−1), 4F (−242.6, −246.4, −232.4 kcal mol−1) and 3F (−272.4, −248.5, −277.7 kcal mol−1) with filarial protein 4FYU, 5D73, and 1A33, respectively. Docking results were strongly supported by molecular dynamics data and molecular mechanics-generalized born surface area (MM-GBSA) calculations. The optimized geometries of all three compounds were used for calculating the energies of the frontier molecular orbitals highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The lowest HOMO–LUMO energy gap in compound 3F suggested that it is the most bioactive molecule among all these three compounds, which is in accordance with the docking results of these compounds. The interaction energies between ligand and protein are mainly due to hydrogen bonds, hydrophobic interactions, and van der Waals interactions which give the stability to the complex. The structural information and docking studies of different filarial proteins with antifilarials obtained from this study could aid in screening and designing new antifilarial or selective inhibitors for chemotherapy against filariasis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Molecular docking is a computational technique providing important information for protein–ligand docking and structure-based rational drug design [1,2,3]. Moreover, the effective screening of potential drug candidates by docking studies at an early stage provides large cost savings at a later stage of the overall drug discovery process [4]. In the finishing decade of 1970, molecular docking was introduced which made straightforward method for protein–ligand docking (PLD) and structure-based drug design (SBDD) [5]. It has been used in drug designing since 1980s [6]. Docking programs are generally based on different algorithms and are developed to study the protein–ligand and protein–protein interaction. In structural molecular biology and pharmaceutical research, molecular docking plays an important role. Various reports and research papers on protein–protein and protein–ligand molecular docking have been published in the past years [7,8,9,10,11]. Molecular docking is used to study the different types of interaction between various small molecules such as drugs, novel active compounds, and protein at the atomic level. This virtual screening also helps in explicating elementary biochemical processes and comportment portrayal of many small molecules within the active site of target protein [12]. For an efficient docking it is necessary to find out the position of active binding site in the target protein while for the unknown active binding sites, cavity detection programs or online servers such as GRID [13, 14], POCKET [15], Surf Net [16, 17], PASS [18] and MMC [19] is utilized to identify putative active binding sites within target proteins. Emil Fischer in 1894 [20], proposed lock and key theory, where ligand acts as key which fits into the active cavity of proteins behaving as a lock. Daniel Koshland in 1958, proposed a step further theory called as induced-fit theory [21, 22]. As per this hypothesis, a conformational change by the dynamic site stash is accomplished when a substrate come in the region of the objective protein and an arrangement or regulation of the dynamic site is produced.
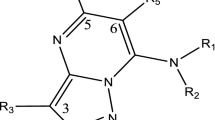
Computer-based molecular docking can facilitate the early stages of drug discovery through systematic prescreening of ligand (i.e., small molecules) for shape and energetic compatibility with a receptor (i.e., protein) prior to test assessment. The structure of the intermolecular buildings framed between at least two atoms were predicted by molecular docking studies [23]. It is a key device in basic atomic science and drug design. Later on computational density functional theory (DFT) has become an effective tool in the investigation of molecular structure and also used to enumerate different chemical reactivity descriptors of the reported complexes. The aim of the present work is to describe and characterize the molecular structure, and theoretical properties of methyl-1(4-methyl-phenyl)-9H-pyrido(3,4-b) indole-3-carboxylate (4C), methyl-1(2-chloro-phenyl)-9H-pyrido(3,4-b) indole-3-carboxylate (4F) and methyl-1(2-chloro-phenyl)-1,2,3,4-tetrahydro-9H-pyrido(3,4-b) indole-3-carboxylate (3F). Numerous reports are available in the literature concerning the structures and DFT studies [24, 25]. On the basis of above reports and characterization of molecular interaction in silico, the present work was completed to ponder the biochemical nature and the binding of different filarial proteins i.e., 4FYU, 5D73, and 1A33 with potent antifilarial compounds 4C, 4F, and 3F. The investigation was also supported by analyzing the drug likeness potential of 4C, 4F, and 3F compounds.
Wuchereria bancrofti (filarial parasite) contains thioredoxin glutathione reductase (TGR) which plays a chief role in the maintenance of cellular redox status. It is a homo dimeric flavor protein. The prosthetic group is flavin adenine dinucleotide (FAD) and the binding domain is nicotinamide adenine dinucleotide phosphate (NADPH) in each dimeric subunit. Each subunit of TGR contains a glutaredoxin (Grx) domain fused to thioredoxin reductase (TR) domain. The glutathione reductase (GR) domain and thioredoxin reductase (TR) domain are analogous. Both the TR and GF enzyme belong to the same family. The filarial worm Wuchereria bancrofti has lost the genes encoding of TR and GR enzymes. By RNA interference silencing of TGR gene, parasites are killed. This confirms that TGR act as potential drug target for the treatment of elephantiasis [26].
Cyclophilins are ubiquitous proteins, found in various organisms such as bacteria, mammals, and plants [27]. They possess peptidylprolylisomerase (PPIase) activity and act as catalysts in many protein-folding events, predominantly in the folding of proline-rich proteins [28, 29]. Cyclophilins has also strong affinity to bind with immunosuppressive drug cyclosporin A (CsA) [30]. Sequence of different amino acid and structural comparisons of the cyclophilins allow grouping of different isoforms depending on their cellular location. Cyclophilin A being the copious one is present in cytosol while Cyclophilin B is present in endoplasmic reticulum [31] and other isoform of cyclophilins are present in the mitochondrion [32]. Cyclophilins are found in human filarial nematode Brugia malayi and are also present in other filarial parasites i.e., Onchocerca volvulus. B. malayi is a medically important digenetic endoparasite found in tropical countries. The primary and secondary hosts are mosquitoes and humans, respectively, and infection mostly occurs in lower lymphatic vessels which is transmitted via mosquito vectors during larval stage (i.e., worms are not mature). Chronic debilitating filarial disease symptoms may be the next consequence, including elephantiasis [33]. Benzodiazepine and γ–amino butyric acid (GABA) receptors interact with centrally acting agents. They are potent anthelmintic agents [34,35,36,37]. It is well reported that 3-carboxy-β-carbolines exhibit a high order of affinity for benzodiazepine receptors [38, 39]. On the basis of above reported literature, we have designed and synthesized a small library of 45 compounds and evaluated them for micro and macrofilaricidal activities against L. cariniiin cotton rats (Sigmodonhispidus) and, A. viteae and B. Malayi in Mastomyscoucha [40, 41]. From the library of compounds, we choose three most active compounds i.e., methyl-1(4-methyl-phenyl)-9H-pyrido(3,4-b) indole -3-carboxylate (4C), methyl-1(2-chloro- phenyl)-9H-pyrido(3,4-b) indole-3-carboxylate (4F) and methyl-1(2-chloro-phenyl)-1,2,3,4-tetrahydro-9H-pyrido(3,4-b) indole-3-carboxylate (3F), for study. These compounds flourished as potent macrofilaricidal activity against A. viteae (Fig. 1). In order to understand the interaction of active antifilarial compounds 4C, 4F, and 3F with filarial proteins viz. thioredoxin, glutathione s-transferase and cyclophilin, we performed the docking studies using YASARA (Yet Another Scientific Artificial Reality Application) tool, HexCuda 8.0.0, and PatchDock server.

Structures of ligands
In the present paper, molecular docking and quantum chemistry (DFT) are employed to discuss the charge distribution and frontier orbital energy of these three potent antifilarial compounds (4C, 4F, and 3F). This study may provide the theoretical information for designing and synthesis of novel antifilarial compounds in future.
Materials and methods
Structure retrieval and verification
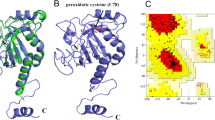
The three dimensional (3D) structures of target proteins were taken from Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) and ligands were designed using ChemDraw Ultra 7.0. The geometries and energies of these ligands were optimized using the Gaussian09W software, and Discovery Studio 3.0 was used for SDF to PDB format conversion. The latter was also used to retrieve and optimize 3D structures of thioredoxin (PDB ID: 4FYU), glutathione s-transferase (PDB ID: 5D73) and cyclophilin (PDB ID: 1A33). Thioredoxin (Resolution: 2.00 Å, R-Value: 0.218), glutathione s-transferase (Resolution: 2.33 Å, R-Value: 0.258) and cyclophilin (Resolution: 2.15 Å, R-Value: 0.211) proteins of filarial worms were obtained from PDB server and were recognized as targets (Fig. 2). PDB advance BLAST analysis was used for selecting proteins and their assembly. BLAST facilitates the utilization of the protein assemblies by query coverage and scoring to maximum. Protein validation was done by RAMPAGE and PDBSum server [42].

Secondary structures of targeted filarial proteins 4FYU, 5D73, and 1A33
Binding site identification and active site residues analysis
MetaPocket 2.0 server was used to search noticeable active binding sites of thioredoxin, glutathione s-transferase and cyclophilin proteins [43]. To compare docking results and to scrutinize active binding lees best 5 key binding pockets were salvaged [42]. MetaPocket, server was used to identify active binding sites of selected receptors. http://projects.biotec.tu-dresden.de/metapocket/ is the free link which facilitates MetaPocket 2.0. The small molecules/ligands bind with the protein surface voids and receptacles. Thus, the starting point for PLD and SBDD is to identify these nooks. Therefore, to find out drug candidates in PLD/SBDD or high-throughput screening (HTS) processes, it is very important to identify the appropriate ligand binding site residues [44].
Ligand collection and optimization
To extend our research, we chose 3 compounds from the pool of 45 compounds (Fig. 1) showing the best micro and macrofilaricidal activities and studied their interaction with 4FYU, 5D73 and 1A33. The structures of 4C, 4F, and 3F ligands were collected based on earlier published research paper [45]. Further the collected ligands were designed using ChemDraw Ultra 7.0 tool and optimized in terms of geometry and energy using the Gaussian software and finally converted in PDB format using Discovery Studio 3.0 (Fig. 3). Discovery Studio 3.0 is a suite of software for simulating small molecule and macromolecule system. Discovery Studio 3.0 provides software application covering the following area, ligand design, pharmacophore modeling, structure-based design, macromolecule design and validation. Discovery Studio 3.0 (Accelrys Software, Inc.) was used for visualization of protein/ligand or docked complexes.

Retrieved ligands structure (4C, 4F, and 3F)
Ligand verification
All active compounds (4C, 4F, 3F) with potent macrofilaricidal activity (90-100%) against Acanthochelinema vitae at 50 mg/kg × 5days by interaperitoneal route from our earlier publication were selected for molecular docking studies [45].
Drug likeness, adsorption, distribution, metabolism, excretion, and toxicity prediction
Lipinski filter (http://www.scfbio-iitd.res.in/software/drugdesign/lipinski.jsp), was used for prediction of drug likeness of thioredoxin, glutathione s-transferase and cyclophilin proteins. According to Lipinski filter, an orally active drug should comply with minimum of four of the five laid down criteria for drug likeness. These four factors are molecular mass, cLogP, hydrogen donor and acceptor and molar refractive index [46]. The link http://lmmd.ecust.edu.cn:8000/ provides admet SAR facility, was used to test the ligands 4C, 4F, and 3F for evaluating their absorption, distribution, metabolism, excretion and toxicity (ADMET) [47]. The link http://fafdrugs4.mti.univ-paris-diderot.fr/ provides FAF-Drugs 4 facility, was used to predict extra ADMET properties of 4C, 4F, and 3F.
Molecular docking study and envisioning
YASARA, an AutoDock-based tool for molecular docking and virtual screening was used for analyzing dissociation constant (Kd) and comparative binding energy of the docked molecular complexes [48, 49]. Hex 8.0.0 Cuda and PatchDock servers were used for molecular docking. HexServer (http://hexserver.loria.fr/) is a handy and speedy online tool which generates a graded record of up to 1000 docking calculations. It also offers an expedient way to perform comprehensive graphics processor unit hastened fast Fourier transform-based rigid body docking calculations with no necessity of operator [50].
PatchDock server is a geometry built molecular docking algorithm [51], which was used for docking analysis of thioredoxin, glutathione s-transferase and cyclophilin proteins. It was used to upload the PDB files of target proteins and ligand for docking evaluation. The same was also used to analyze approximate interface area (AI area) and geometric shape complementarity score (GSC score). It was operated by managing the RMSD to 4.0. [42]. PatchDock is laced with flexible constraints for the ease of docking of variety of binding entities [52].
Chemistry
Three different ligands were selected for docking analysis. The structure of ligands (Fig. 1), i.e.,methyl-1(4-methyl- phenyl)-9H-pyrido(3,4-b) indole -3-carboxylate (4C) and methyl-1(2- chloro- phenyl)-9H-pyrido(3,4-b) indole -3-carboxylate (4F) and methyl-1(2- chloro- phenyl)-1,2,3,4-tetrahydro-9H-pyrido(3,4-b) indole -3-carboxylate (3F) were drawn by using ChemDraw Ultra 7.0, Cambridge Soft Corp. (http://www.cambridgesoft.Com), USA. Discovery Studio 3.0, Accelrys Inc. (www.accelrys.com), USA was used to convert the 2-D structures to enhanced 3D structures. All the three compounds have been reported as new lead molecules in antifilarial chemotherapy [45].
Molecular docking
The crystal structures of thioredoxin, glutathione s-transferase and cyclophilin were obtained from the protein data bank (PDB ID: 4FYU, 5D73, 1A33). Molecular docking was conducted in order to determine the interaction of 4C, 4F, and 3F compounds with target proteins viz. thioredoxin, glutathione s-transferase and cyclophilin [53]. Molecular docking was performed by using YASARA tool, Hex 8.0.0 with Cuda version tool and PatchDock server. The Hex 8.0.0 is a simple and easy protein docking and molecular superposition program for docking calculations. It improves the quality of analysis and reduces the docking time of the predicted docked complexes [54]. Prediction of protein–ligand and protein–protein docked complexes was done by PatchDock server [51]. Random starting positions, orientations and torsions were employed for all ligands. Protein–ligand docking results were clustered into groups with root-mean-square deviation <2.0 Å. Visualization and analysis of molecular structures and different protein–ligand and protein–protein interfaces were performed by using Discovery Studio 3.0, AccelrysInc [26].
Molecular dynamics simulations
For authenticating whether the results obtained by docking (YASARA) are robust or coincidental. The docked structures of 5D73 receptor with compounds 4C, 4F, and 3F were subjected to the molecular dynamics (MD) simulations for 50 ns in aqueous environment and Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA), molecular mechanics generalized born surface area (MM-GBSA) can effectively deal with the conformational change upon ligand binding. The 4C, 4F, and 3F compounds were prepared by AMBER’s Xleap module and the partial charges to the compounds were derived by generation of the electrostatic potentials employing the restrained electrostatic potential fitting method (RESP) with 6-31G(d) basis set utilizing the Hartree-Fock level [55,56,57] in GAUSSIAN [58]. MD simulations performed with periodic boundary conditions employing the AMBER suite (AMBER 14 version) with parm99SB along with gaff.dat force field [59, 60]. The complexes were enclosed in a periodic water box with chloride counter ions, and the resulting trajectory is then post processed by removing the solvent and the periodicity, and calculating the average free energy over a series of static frames. About 200 frames of complex, extracted from the last 20 ns of the stable molecular dynamics simulation trajectories for compounds 4C, 4F, and 3F, were used to analyze the binding free energy of the complex using molecular mechanics-generalized born surface area methodology [61,62,63]. The structures after MD were generated and analyzed using LigPlot. To our best knowledge, this work represents one of the most extensive studies of MM/PBSA or MM/GBSA for molecular docking.
Density functional theory
In the present study, the Gaussian09 software was used to optimize the structure and energy of 4C, 4F, and 3F molecules. Computational calculations were done at the B3LYP/6-31+G (d) basis set level using the Gaussian software. The optimized geometry was then used as input file for calculating the energies of the frontier molecular orbitals highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).
Results and discussion
The PDB Sum server was used to study target protein molecule stereochemistry. The complete physical configuration and structural patterns of proteins were determined by various parameters considered by the server. The server also search and evaluate suitable voids in protein for better binding. It showed compliant regions of filarial proteins exposed for docking with ligands [64].
Docking properties of thioredoxin, glutathione s-transferase and cyclophilin were characterized with the claimed enzymes. The result showed formation of H-bond between thioredoxin, glutathione s-transferase, cyclophilin and amino acids residues situated in the major binding sites 1, 2, 3, 4, and 5 of protein. Protein–ligand communication was found boosted up by ionic interactions, hydrophobic interactions and van der Waals forces along with hydrogen bonds [65].
Docking analysis of 4C, 4F, and 3F compounds
The ligand structures (4C, 4F, and 3F) were salvaged from PubChem database and then YASARA tool, PatchDock server, and Hex 8.0.0 Cuda tool were used to examine docking. Docking of 4C compound with the filarial proteins 4FYU discovered the absence of H-bonding between 4C and amino acid residues traced in the noticeable binding sites 1, 2, 3, 4, and 5 (Table 1). Conformational shift of active sites in the target proteins was observed when ligand 4C was docked and indicating diminished catalytic activity. Pro41, Arg126, Pro134, Ala133, Gly131, Lys132, Ser130, Gln44, Ile48, and Phe45 were found as the most accepted amino acid residues existing at the outstanding binding sites 1 and 4. May be the detection and alignment of 4C molecules was supported by the favorable arena provided by all preceding amino acid residues. Leu13, Glu157, Val153, Pro16, Ser63, His98, Gly64, Gln62, Arg95, and Thr99 were detected as commonly appeared amino acid residues of filarial protein 5D73 existing at the outstanding binding site 1 while Gln122, Gly85, Gly86, Gly120, Thr79, Ser53, Glu87, Gly92, Met93, Thr118, and Lys114 were found as frequently occurring amino acid residues of filarial proteins 1A33 located at the binding site 1. All these amino acid residues including alanine are believed to be capable in the formation of H-bonds, ionic bonds, van der Walls forces, charged interactions and hydrophobic interactions. GSC score and AI area of the docked complexes made easy to analyze their binding potency. Complying docking results for ligand–protein complexes were interpreted from the energetics data obtained. The range of binding energy was found to be −232.4 kcal mol−1 to −277.7 kcal mol−1 (Tables 2, 3, 4, 5) of the docked complexes, which signify the robust docking of the considered active antifilarial compounds with good potency against filarial proteins. After then, Discovery Studio 3.0 was used to envision docked complexes (Figs. 4 and 5) and to expose different interactions ligand–protein docking comprising herein.





Docked complexes visualized by Discovery Studio 3.0 showing interactions of 4C, 4F, and 3F with the target proteins, 4FYU, 5D73 and 1A33





2D model of docked complexes as visualized by Discovery Studio 3.0 showing interactions of 4C, 4F, and 3F with the target proteins, 4FYU, 5D73, and 1A33
Out of these target proteins, 5D73 and 1A33 were the best targets of selected compounds 4C, 4F, and 3F. From Table 5, it is clear that most of the interacting amino acid residues of filarial proteins were present prominent active site 1 residues. The residues His98, Leu13 of 5D73, and Lys114 of 1A33 were common amino acids in all results obtained from all three docking software, Hex, PatchDock, and YASARA tool.
MD simulation
In the practice of virtual screening to estimate the quality of molecular dynamics simulations, energetic and structural properties were examined during the whole MD simulation of every complex. The average RMSD value for the 5D73 system is 2.75 Å. indicating no dynamical collapse. For systems with more than 4000 atoms, low potential energy and RMSD values indicate that the MD simulation process is stable and reliable. The binding free energy values of compounds 4C, 4F, and 3F with 5D73 were found to be 7.6 kcal/mol, 7.3 kcal/mol, and 7.0 kcal/mol, respectively. The formation of more stable complex of 5D73 with studied antifilarial compounds may be inferred due to presence of their hydrophobic interactions with amino acid residues of protein. The amino acid residues involved in hydrophobic interactions with 4C (Tyr7, Phe8, Ile10, Gly12, Leu13, Tyr106, and Gly204), with 4F (Tyr7, Phe8, Ile10, Gly12, Leu13, Tyr106, and Gly204) and with 3F (Tyr7, Phe8, Ile10, Leu13, Trp38, Leu50, and Gly204) are shown in Fig. 6. Additionally, Tyr7 and Trp38 were found to provide further stability to 4F, 3F-5D73 complex by formation of hydrogen bonds (Fig. 6). Binding affinities of receptor with all the ligands (4C, 4F, and 3F) were calculated by post processing the ensembles of structures extracted from MD trajectories using MM-GBSA and MM-PBSA calculations (implemented in the AMBER 14 program). The observed binding energy for 4C using MM-GBSA was −29.3 ± 1.5 kcal/mol, and using the MM-PBSA method, the binding energy was −7.1 ± 0.3 kcal/mol. For compounds 4F and 3F, the binding energies were −37.1 ± 2.1 kcal/mol (MM-GBSA) and −7.2 ± 0.2 kcal/mol (MM-PBSA), −43.1 ± 1.6 kcal/mol (MM-GBSA), and −9.1± 0.7 kcal/mol (MM-PBSA), respectively. Thus, it is evident from the MM-PB/GBSA studies that the compound 3F shows better binding affinity as compared to compounds 4C and 4F.

5D73 active site residues interacting with 4C, 4F, and 3F after MD simulations, red residues showing H-bonding between residue and molecule docked, red are the hydrophobic interaction between the hydrophobic residues of the active site amino acid of the 5D73
DFT calculation
DFT studies of these three ligands were carried out using the hybrid functional DFT/B3LYP/6-31G(d) method using the Gaussian software for better precision and calculation speed [66, 67]. The geometries of these ligands were optimized in the ground electronic state, and their energy minimization was done. The optimized geometries were then used for calculating the energies of the frontier molecular orbitals (HOMO and LUMO). Owing to the importance of frontier molecular orbitals (HOMO and LUMO) and their properties, in order to predict the reactivity and charge delocalization within these molecules, Gauss-View 5.0 was employed for visualization and constructing the shape of their frontier molecular orbitals (Fig. 7) [68, 69]. It is very well documented that molecules having small and large HOMO–LUMO gaps correspond to more and less reactive species [69, 70]. The HOMO–LUMO gaps of these present compounds are shown in Fig. 7. The lowest HOMO–LUMO gap obtained in the case of 3F suggested that it is the most bioactive molecule among all these three compounds, which is in accordance with the docking results of these compounds. Also, the electron density in the case of compound 3F is localized over one side of ring in the case of HOMO (differently from 4C and 4F), while in the case of LUMO, it is localized mostly over all the molecule, revealing delocalization of electrons from HOMO to LUMO.

Frontier molecular orbital diagrams for HOMO and LUMO for a 4C, b 4F, and c 3F calculated at the B3LYP/6-31G(d) level
According to DFT studies, order of activity 3F>4C>4F, which matches with the docking results (3F>4C>4F).
Testing of drug likeness of 4C, 4F, and 3F
Lipinski filter revealed for the drug likeness of 4C, 4F, and 3F ligands. AdmetSAR was employed to predict pharmacokinetic properties of the ligands (Table 6). Together Lipinski filter and admetSAR play crucial role to drug design and prediction for their binding ability. The evaluated data obtained for 4C, 4F, and 3F compounds supports for their implementation as antifilarial drugs in living systems. cLogP value calculated for 4C, 4F, and 3F compounds were 1.9111460, 1.697250, and 1.144460, respectively. Thus, all these 3 compounds passed drug ability test because negative cLogP value favors hydrophilicity and bioavailability of a compound. Akin score were obtained for all the 3 compounds on the basis of the Lipinski filter and admetSAR parameters. FAF-Drugs4 was used to evaluate pharmacokinetic properties (ADMET) of 4C, 4F, and 3F compounds. The results were in compliance with Lipinski's rule of five.
Conclusions
A successful study of the interaction of potent antifilarial compounds 4C, 4F, and 3F with filarial proteins were carried out using molecular docking and DFT calculations. In overall docking scores, compound 3F was found to be the most superior to compounds 4F and 4C. The compound 3F binds with amino acids Gly85, Gly86, and Gln120 of proteins 1A33 through H-bond interactions. The predicted binding energy of antifilarial compounds 4C (−247.6, −243.8, −256.8 kcal mol−1), 4F (−242.6, −246.4, −232.4 kcal mol−1) and 3F (−272.4, −248.5, −277.7 kcal mol−1) with filarial protein 4FYU, 5D73, and 1A33, respectively. His98, Leu13, and Lys114 are common amino acids present at prominent binding site 1. MD simulations have been proved to be a remarkable approach for identifying protein–ligand stability, structural transformations, binding energies change in complexes, etc. MM-PB/GBSA studies confirmed that the compound 3F shows better binding affinity as compared to compounds 4C and 4F. The lower HOMO–LUMO energy gap obtained in the case of 3F suggested that it is the most bioactive molecule among all these three compounds. The interaction energies are mainly due to hydrogen bonds, hydrophobic interactions, and van der Waals interactions which provide stability to the complex. Hence, it could be found that compounds having identical parent structure with different functional groups could modulate the biological activity of filarial protein and could be considered better candidates as antifilarial agents.
Data availability
Not applicable.
Code availability
ChemDraw Ultra 7.0, Discovery Studio 3.0, GaussView 5.0.
References
Kellenberger E, Rodrigo J, Muller P, Rognan D (2004) Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 57:225–242
Meng XY, Zhang HX, Mezei M, Cui M (2011) Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des 7:146–157
Zhu H, Pisabarro MT (2010) MSPocket: an orientation-independent algorithm for the detection of ligand binding pockets. Bioinformatics 27:351–358
Nagamine N, Sakakibara Y (2007) Statistical prediction of protein–chemical interactions based on chemical structure and mass spectrometry data. Bioinformatics 23:2004–2012
Ruyck JD, Brysbaert G, Blossey R, Lensink MF (2016) Molecular docking as a popular tool in drug design, an in silico travel. Adv Appl Bioinforma Chem 9:1–11
Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE (1982) A geometric approach to macromolecule-ligand interaction. J Mol Biol 161:269–288
Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3:935–949
Halperin I, Ma B, Wolfson H, Nussinov R (2002) Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins 47:409–443
Coupez B, Lewis RA (2006) Docking and scoring—theoretically easy, practically impossible. Curr Med Chem 13:2995–3003
Kontoyianni M, Madhav P, Suchanek E, Seibel W (2008) Theoretical and practical considerations in virtual screening: a beaten field. Curr Med Chem 15:107–116
Brooijmans N, Kuntz ID (2003) Molecular recognition and docking algorithms. Annu Rev Biophys Biomol Struct 32:335–373
McConkey BJ, Sobolev V, Edelman M (2002) The performance of current methods in ligand-protein docking. Curr Sci 83:845–856
Goodford PJ (1985) A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem 28:849–857
Kastenholz MA, Pastor M, Cruciani G, Haaksma EE, Fox T (2000) GRID/CPCA: a new computational tool to design selective ligands. J Med Chem 43:3033–3044
Levitt DG, Banaszak LJ (1992) POCKET: a computer graphics method for identifying and displaying protein cavities and their surrounding amino acids. J Mol Graph 10:229–234
Laskowski RA (1995) SURFNET: a program for visualizing molecular surfaces, cavities, and intermolecular interactions. J Mol Graph 13:323–330
Glaser F, Morris RJ, Najmanovich RJ, Laskowski RA, Thornton JM (2006) A method for localizing ligand binding pockets in protein structures. Proteins 62:479–488
Brady Jr GP, Stouten PF (2000) Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des 14:383–401
Mezei M (2003) A new method for mapping macromolecular topography. J Mol Graph Mode 121:463–472
Fischer E (1894) Einfluss der Configuration auf die Wirkung der Enzyme. BerDtChemGes 27:2985–2993
Koshland Jr DE (1963) Correlation of structure and function in enzyme action. Science 142:1533–1541
Hammes GG (2002) Multiple conformational changes in enzyme catalysis. Biochemistry 41:8221–8228
Sergio FS, Pedro AF, Maria JR (2006) Protein–ligand docking: current status and future challenges. Proteins Struct Funct Bioinf 65:15–26
Arunagiri C, Anitha AG, Subashini A, Selvakumar S (2018) Synthesis, X-ray crystal structure, vibrational spectroscopy, DFT calculations, electronic properties and Hirshfeld analysis of (E)-4-Bromo-N'-(2, 4-dihydroxy-benzylidene) benzohydrazide. J Mol Struct 1163:368–378
Bharti S, Choudhary M, Mohan B, Sharma SR, Ahmad K (2018) Syntheses, crystal structures, DFT, molecular docking and inhibition studies of jack been urease by nickel (II) and copper (II) Schiff base complexes. Inorgan Nano-Metal Chem 48:211–224
Huang J, Hua W, Li J, Hua Z (2015) Molecular docking to explore the possible binding mode of potential inhibitors of thioredoxin glutathione reductase. Mol Med Rep 12:5787–5795
Galat A, Metcalfe SM (1995) Peptidylproline cis/trans isomerases. Prog Biophys Mol Biol l63:67–118
Hsu VL, Heald SL, Harding MW, Handschumacher RE, Armitage IM (1990) Structural elements pertinent to the interaction of cyclosporin a with its specific receptor protein, cyclophilin. Biochem Pharmacol l40:131–140
Davis JM, Boswell BA, Bachinger HP (1989) Thermal stability and folding of type IV procollagen and effect of peptidyl-prolylcis-trans-isomerase on the folding of the triple helix. J Biol Chem 264:8956–8962
Braun W, Kallen J, Mikol V, Walkinshaw MD, Wuthrich K (1995) Three-dimensional structure and actions of immunosuppressants and their immunophilins. FASEB J 9:63–72
Mikol V, Kallen J, Walkinshaw MD (1994) X-ray structure of a cyclophilin B/cyclosporin complex: comparison with cyclophilin A and delineation of its calcineurin-binding domain. Proc Natl Acad Sci U S A 91:5183–5186
Taylor P, Page AP, Kontopidis G, Husi H, Walkinshaw MD (1998) The X-ray structure of a divergent cyclophilin from the nematode parasite Brugiamalayi. FEBS Lett 425:361–366
Page AP, Landry D, Wilson GG, Carlow CKS (1995) Molecular characterization of a cyclosporin A-insensitive cyclophilin from the parasitic nematode Brugiamalayi. Biochemistry 34:11545–11550
Moore EC, Zedeck MS, Agrawal KC, Sartorelli AC (1970) Inhibition of ribonucleoside diphosphate reductase by 1-formylisoquinoline thiosemicarbazone and related compounds. Biochemistry 23:4492–4498
Campbell WC, Fisher MH, Stapley EO, Albers-Schonberg G, Jacob TA (1983) Ivermectin: a potent new antiparasitic agent. Science 221:823–828
Johnston GAR (1976) Physiologic pharmacology of GABA and its antagonists in the vertebrate nervous system. In: Roberts E Chase TN, Tower DB (1976) editors. GABA in Nervous System Function. New York: Raven Press 394-411.
Wang CC (1984) Parasitic enzymes as potential targets for antiparasitic chemotherapy. J Med Chem 27:1–9
Braestrup C, Nielson M, Olsen CE (1980) Urinary and brain β-carboline-3-carboxylates as potent inhibitors of brain benzodiazepine receptors. Proc Natl Acad Sci USA 77:2288–2292
Dodd HR, Ouannes C, Robert-Gero M, Potier P (1989) Hybrid molecules: growth inhibition of Leishmania donovani promastigotes by thiosemicarbazones of 3-carboxy-β-carbolines. J Med Chem 32:1272–1276
Kumar S, Seth M, Bhaduri AP, Visen PKS, Misra A, Gupta S, Fatima N, Katiyar JC, Chatterjee RK, Sen AB (1984) Syntheses and anthelmintic activity of alkyl 5(6)-(substituted carbamoyl)- and 5(6)-(Disubstitutedcarbamoyl)benzimidazole-2-carbamates and related compounds. J Med Chem 27:1083–1089
Murthy PK, Tyagi K, Chowdhury TKR, Sen AB (1983) Susceptibility of Mastomysnatalensis (GRA strain) to a subperiodic strain of human Brugiamalayi. Indian J Med Res 77:623–630
Yadav S, Pandey SK, Singh VK, Goel Y, Kumar A, Singh SM (2017) Molecular docking studies of 3-bromopyruvate and its derivatives to metabolic regulatory enzymes: implication in designing of novel anticancer therapeutic strategies. PLoS ONE 12:e0176403
Huang B (2009) MetaPocket: a meta approach to improve protein ligand binding site prediction. OMICS 13:325–330
Zhang Z, Li Y, Lin B, Schroeder M, Huang B (2011) Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics 27:2083–2088
Srivastava SK, Agarwal A, Chauhan PMS, Agarwal SK, Bhaduri AP, Singh SN, Fatima N, Chatterjee RK (1999) Potent 1,3-disubstituted-9H-pyrido[3,4-b]indoles as new lead compounds in anti-filarial chemotherapy. Bioorg Med Chem 7:1223–1236
Lipinski CA (2004) Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 11:337–341
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y (2012) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099–3105
Krieger E, Vriend G (2014) YASARA View-molecular graphics for all devices-from smartphones to workstations. Bioinformatics 30:2981–2982
Trott O, Olson AJ (2010) AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie DW (2010) Hex Server: an FFT-based protein docking server powered by graphics processor. Nucleic Acids Res 38(Web Server issue):W445–W449
Schneidman-Duhovny D, Inbar Y, Nussinov R, &Wolfson HJ (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 33(Web Server issue):W363-W367.
Duhovny D, Nussinov R, Wolfson HJ (2002) Efficient unbound docking of rigid molecules. 185-200. In: Guigo R, Gusfield D (2002) editors. Algorithms in Bioinformatics. WABI 2002. Lecture Notes in Computer Science, Berlin, Heidelberg: Springer, 2002,Vol 2452.
Mphahlele MJ, Gildenhuys S, Parbhoo N (2017) Synthesis, cytotoxicity and molecular docking studies of the 9-substituted 5-styryltetrazolo [1,5-c]quinazoline Derivatives. Molecules 22:1719
Ritchie DW, Kozakov D, Vajda S (2008) Accelerating and focusing protein-protein docking correlations using multi-dimensional rotational FFT generating functions. Bioinformatics 24:1865–1873
Cornell WD, Cieplak P, Bayly CI, Kollmann PA (1993) Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J Am Chem Soc 115:9620–9631
Bayly CCI, Cieplak P, Cornell WD, Kollman PA (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem 97:10269–10280
Gordon MS, Schmidt M W Chapter 41 (2005) Advances in electronic structure theory: GAMESS a decade later. In Theory and applications of computational chemistry. The First Forty Years; Dykstra C E, Frenking G, Kim K S, Scuseria G E, Eds, Elsevier: Amsterdam 1167−1189.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd J, Brothers EN, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09. In. Gaussian, Inc., Wallingford, CT, USA
Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossv PA (2014). Amber:14
Wang J, Wolf RM, Caldwell JW, Kollamn PA, Case DA (2004) Development and testing of a general Amber force field. J Comput Chem 25:1157–1174
Hou T, Zhang W, Case DA, Wang W (2008) Characterization of domain–peptide interaction interface: a case study on the amphiphysin-1 SH3 domain. J Mol Biol 376:1201–1214
Hou T, Xu Z, Zhang W, McLaughlin WA, Case DA, Xu Y, Wang W (2009) Characterization of domain-peptide interaction interface: a generic structure-based model to decipher the binding specificity of SH3 domains. Mol Cell Proteomics 8:639–649
Gohlke H, Kiel C, Case DA (2003) Insights into protein–protein binding by binding free energy calculation and free energy decomposition for the Ras–Raf and Ras–Ral GDS complexes. J Mol Biol 330:891–913
Schmidt T, Bergner A, Schwede T (2014) Modelling three-dimensional protein structures for applications in drug design. Drug Discov Today 19:890–897
Zhao H, Huang D (2011) Hydrogen bonding penalty upon ligand binding. PLoS ONE 6:e19923
Kumar GSS, Prabhu AAM, Bhuvanesh N, Ronica XAV, Kumaresan S (2014) Molecular structure investigation of organic cocrystals of 1,10-phenanthroline-5,6-dione with aryloxyacetic acid: a combined experimental and theoretical study. Spectrochim Acta A: Molecular and Biomolecular Spectroscopy 132:465–476
Yadav P, Sharma B, Sharma C, Singh P, Awasthi SK (2020 Mar 18) Interaction between the antimalarial drug dispiro-tetraoxanes and human serum albumin: a combined study with spectroscopic methods and computational studies. ACS omega 5(12):6472–6480
Yadav P, Yadav JK, Dixit AK, Agarwal A, Awasthi SK (2019) Insight into the interaction of benzothiazole tethered triazole analogues with human serum albumin: Spectroscopy and molecular docking approaches. Luminescence 34:812–822
Yadav P, Yadav JK, Agarwal A, Awasthi SK (2019) Insights into the interaction of potent antimicrobial chalcone triazole analogs with human serum albumin: spectroscopy and molecular docking approaches. RSC Adv 9:31969–31978
Ali A, Asif M, Alam P, Alam MJ, Sherwani MA, Khan RH, Ahmad S (2017) DFT/B3LYP calculations, in vitro cytotoxicity and antioxidant activities of steroidal pyrimidines and their interaction with HSA using molecular docking and multispectroscopic techniques. Bioorg Chem 73:83–99
Funding
JKY, VKS, and AA are grateful to Banaras Hindu University, Varanasi, India, for the financial assistance. PY is thankful to UGC, Delhi, India, for providing SRF.
Author information
Authors and Affiliations
Contributions
Jitendra Kumar Yadav: conceptualization, data curation, formal analysis, investigation, methodology, project administration, visualization, writing—original draft, reviewing and editing.
Priyanka Yadav: conceptualization, data curation, investigation, methodology, writing—original draft, reviewing and editing.
Vinay Kumar Singh: conceptualization, investigation, methodology, writing—original draft, reviewing and editing.
Alka Agarwal: conceptualization, formal analysis, methodology, project administration, resources, supervision, writing—review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declares no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yadav, J.K., Yadav, P., Singh, V.K. et al. Molecular docking and density functional theory studies of potent 1,3-disubstituted-9H-pyrido[3,4-b]indoles antifilarial compounds. Struct Chem 32, 1925–1947 (2021). https://doi.org/10.1007/s11224-021-01772-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-021-01772-4