
Abstract
Lymphatic filariasis (LF) remains a significant health challenge for populations in developing countries. LF is a parasitic disease transmitted by mosquitoes, mainly caused by the filarial nematode, Wuchereria bancrofti, prevalent in tropical and subtropical regions. Since the present drugs develop complications, including adverse side effects, lack of specificity, and development of drug resistance, the present study focused on developing the potential anti-filariasis drugs targeting crucial proteins for the nematode life cycle. We have identified the therapeutic compounds by targeting the enzyme thioredoxin peroxidase 1 (WbTPx1), which facilitates the conversion of hydrogen peroxide into water, an essential mechanism by which the nematode survives against oxidative stress in the host. This approach might resolve treatment efficacy and activity difficulties at various stages of filarial parasitic worms. We modeled the structure of WbTPx1 and employed the structure-based virtual screening approach, focusing on the dimer interface region of the protein. ADMET prediction profiles of the non-toxic, top-ranked hits with higher docking scores demonstrate higher affinity to the nematode protein than its human homolog. The molecular dynamic simulation studies show WbTPx1-hit complexes’ stability and the intactness of hits in the binding site. Further, in vitro validation of identified hits using Setaria digitata, a cattle nematode, showed better IC50 and higher inhibition than the standard drug ivermectin, indicating the potential to inhibit enzyme activity and the development of drug candidates for controlling LF.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lymphatic filariasis (LF), is a disease transmitted to humans by infected mosquitoes. It is also known as elephantiasis, and this disfiguring disease is caused by three different species of parasitic nematodes: Brugia malayi, Brugia timori, and Wuchereria bancrofti and more than 90% of cases were reported by W. bancrofti [1, 2]. Early report states that, the number of people affected due to LF stands at around 120 million across 76 countries throughout the tropical regions of Africa, Asia, and other sub-tropical regions [3, 4]. Another insight is that India and Sub-Saharan parts of Africa are the major regions affected by LF. As per the 2021 World Health Organization (WHO) statistics, LF continues to concern 882 million people in 44 countries worldwide. According to the global baseline estimate, over 15 million people were affected by lymphedema and 25 million males from hydrocele. If we consider the filarial infection in Asia, it accounts for nearly 63% global burden, mostly in the region of Southeast Asia, India alone harbors 40% of the world filarial burden, despite the existence of the National Filaria Control Programme (NFCP) since 1955 [5]. This vector-borne disease is transmitted to humans, especially through the members of the Aedes, Anopheles, Culex, Mansonia, and Ochlerotatus mosquitoes that serve as carriers, transmitting them based on the unique biological characteristics and geographic locations [6]. Eventually, they grow and affect the lymphatic system, causing permanent damage to humans and leading to disfiguration and disability of the limbs. Being a digenetic parasite, W. bancrofti has its lifecycle in two hosts. The humans support the adult form, and the mosquitoes support the larval form. During a blood meal, the mosquito injects infectious filarial larvae into a healthy human, where they developed in the lymphatic system into adult worms. Over a period, these adult undergo sexual reproduction giving rise to larval offspring referred to as microfilariae. Subsequently, these microfilariae migrate from lymphatic system into bloodstream and perpetuating the cycle when mosquito ingest the blood from infected person [7, 8]. WHO formed GPELF (Global Programme to Eliminate Lymphatic Filariasis) in 1997, where Mass Drug Administration (MDA) and morbidity management activities were initiated to educate people on personal hygiene management and the health impact of preventing LF. Ivermectin, albendazole (ALB), and diethylcarbamazine (DEC) are generally used to treat filarial worms. However, their mechanism has yet to be deciphered clearly, and these drugs works well for larval stage of the worms and not against adult and larval parasites [9, 10]. Currently the filarial treatment switching from two-drug regimen to a three-drug regimen with the combination of ivermectin, diethylcarbamazine, and albendazole (IDA) in addition, chemotherapy was also given as medication. The major problem in the current treatment for LF is that the exact mechanism of the anti-filarial are not known clearly and development of drug resistance to its molecular targets. Due to the prolonged use of these drugs, the nematode developed resistance due to the single nucleotide polymorphisms in its molecular targets. These drugs have developed resistance due to SNPs, mutations and lack of specificity with the filarial targets. Ivermectin exhibit varying therapeutic efficacy due to the polymorphisms in cytokine genes IL-10, TGF-β, IFN-γ, IL-4 and IL-5 that hinder the success rate of the treatment of LF [11]. Because of the existing mutations in the gene that encodes β-tubulin in the filarial worm, drug resistance has been developed in albendazole. It was reported that DEC alters the host arachidonic acid and nitric oxide metabolic pathways and it showed extreme adverse side effects and is not recommended for treating LF [12]. Mutations that confer resistance to anthelmintic medications can emerge on their own or become more prevalent as a result of pharmacological therapy. There is no conclusive evidence establishing a clear relationship between these genes and the development of resistance [13].
Upon W. bancrofti infection, the host’s initial immune system reaction activates macrophages, neutrophils, and eosinophils. This response hits to the production of ROI “Reactive Oxygen Intermediates” and RNI “Reactive Nitrogen Intermediates”, which include the superoxide anion radical (O2–), hydrogen peroxide (H2O2), peroxyl (RO2.) and hydroxyl radicals (OH–). Collectively, referred to as ROS (reactive oxygen species) generated by the host to manage the nematode invasion [14, 15]. Nevertheless, the primary characteristics of a lymphatic filariasis infection involve the nematode’s ability to survive within the host for an extended period (years). Their effective neutralization of ROS facilitates this resilience through well-adapted antioxidant enzymes. The crucial survival tactic employed by filarial nematodes revolves around their antioxidant enzymes, which include catalase (CAT), glutathione peroxidase (GPx), glutathione S transferases (GST), peroxiredoxins (Prxs) and superoxide dismutase (SOD). These enzymes are essential in neutralizing the ROS activity of the host, thereby securing the ongoing viability of the nematodes within the host [16, 17]. So, focusing on these antioxidant enzymes might serve as a viable and effective approach to target the worms at all phases of their life cycle. Peroxiredoxins (Prxs) display remarkable efficiency as cysteine-dependent peroxidases actively participating in antioxidant, regulatory, and signaling systems with remarkable efficiency. Prxs play a crucial role in defending against peroxynitrite (ONOO−) and ROS that arise from both the host and endogenous sources [18]. A widespread family of peroxidases found in most of the eukaryotes, Prxs play a critical role in reducing hydrogen peroxide (H2O2), peroxynitrite, and alkyl hydroperoxides of various affinities. The active site, catalytic cycle, and fold of prxs were conserved and they interact with the peroxide substrate through a conserved cysteine residue known as the peroxidatic Cys (CP) [19, 20]. Peroxiredoxins may be categorized into five subfamilies: BCP, Prx1, Prx5, Prx6 and Tpx based on sequence similarity. Thioredoxin peroxidase-1 (TPx-1) is a member of the thiol-specific antioxidant protein family that is mostly found in the cytoplasm and they effectively reduced and detoxified the H2O2 using the redox-active cysteine. Three varieties of TPxs are distinguished by the location and number of cysteine (Cys) residues: 1-Cys, typical 2-Cys, and atypical 2-Cys type [21, 22]. They follow a uniform reaction mechanism in which the catalytic cysteine, referred to as the peroxidatic cysteine, undergoes oxidation by hydroperoxides, resulting in hyper oxidation and forming sulfenic (–SOH), sulfinic (–SO2H), or sulfonic acid (–SO3H) states of the cysteine. Subsequently, the cysteine is regenerated to its reduced form by various molecules, such as thioredoxin (TRX), glutaredoxin (GRX) or glutathione [23]. Because the filarial parasite is susceptible to disruption of its antioxidant system, it is an ideal potential target for developing new drug candidates [24].
In the present work, we used the in silico methods to modeled and study the dynamic structural stability of WbTPx1 using homology modeling and simulation studies. To identify promising hits that could bind with WbTPx1 we performed structure-based virtual screening. Moreover, the potent hit molecules undergo complex dynamics using molecular dynamics (MD) simulations to evaluate their dynamic intactness with the enzymes. The quantum mechanical method DFT (Density Functional Theory) examines the reactivity of identified hits. The hit compounds identified in the present study were further substantiated through in vitro validation using Setaria digitata. Our study’s findings and associated observations could pave the way for designing and developing innovative therapeutic drug compounds that could effectively target the filarial worm throughout its whole life cycle, with the ultimate goal of eradicating LF.
Materials and methods
Modeling of thioredoxin peroxidase 1 (TPx1) of Wuchereria bancrofti (WbTPx1)
Homology modeling techniques have been employed to predict a reliable 3D structure as target protein since experimental 3D structure of WbTPx1 is not been reported. The WbTPx1 (UniProt ID: B8YNX5) has a 228 amino acid protein sequence that was obtained from the UniProt sequence database. To find the best template, BLASTp was utilized to search against the Protein Databank (PDB) and the determination of the optimal template relied on factors such as E-value, query coverage, percentage identity, and resolution. The Schrödinger Prime suite [25] was used to model the 3D coordinates of WbTPx1 using the best template structure and the structural deviation was calculated for the selected template and target using PyMol. The PROCHECK module present in SAVES server was used to examine the stereochemistry for the modeled structure.
Molecular dynamics simulations of WbTPx1
The GROMACS 5.1.2 suite [26] was utilized to carried out MD simulation to study the dynamicity of WbTPx1 using GROMOS96 53a6 force field [27] to create the protein topology. The WbTPx1 system was parameterized with a 1.2 nm dimension in every direction at the center of the cubic box. Using an SPC water model, the system was solvated with about 22,958 water molecules [28]. The system’s total charge was neutralized by introducing the proper quantity of 0.1 M ionic strength sodium (Na+) and chloride (Cl–) ions. To eliminate the steric conflicts and poor geometry in the solvated system, the energy minimization was implemented using steepest descent method [29] for maximum of 50,000 steps, then the system was brought to equilibrium for 500 ps under the NVT ensemble (isothermal-isochoric) with constant particles, volume and temperature using the Berendsen external bath [30]. Further, Parrinello-Rahman barostat [31] was used to equilibrate for 2000 ps with constant particles, pressure and temperature under NPT ensemble (isothermal-isobaric) at 310 K. LINCS algorithms [32] were implement to constraint the bond and bond length and the geometry of the solvent were constraints by implementing the SETTLE algorithm [33]. Applying the Particle Mesh Ewald (PME) method, the long-range electrostatic interactions were computed [34]. Finally, production MD simulation was applied to the well-equilibrated protein system for 200 ns time scale, storing structure trajectories every 10 ps. The dynamic stability profile of the WbTPx1was examined by analyzing the fluctuations in the RMSF, RMSD, Rg and SASA by GROMACS utilities and VMD [35] was used to analyze and visualize the trajectories.
Preparing the WbTPx1 for structure-guided virtual screening
The Schrödinger suites VSW (Virtual Screening Workflow) module was utilized to screen for potential inhibitors. Ligand Preparation, Filtering, and Docking are the three main process in VSW. First, the Ligprep module was used to prepare the small molecules dataset [36], which involves the conversion of 2D into 3D conforms using the OPLS-3e force field [37] that generates up to 32 conformations for each molecule. The ligand molecules prepared were allowed to pass through the filters based on the “Lipinski rule of five”, ADME properties, and the elimination of compounds with special reactive groups, such as alkali metals and halogens. Next, we prepared the WbTPx1 protein using Schrödinger protein preparation wizard, which refines and corrects the charges, bond order, bond angle, and side chains of the atoms in proteins. By utilizing the OPLS-3e force field, the intra-molecular hydrogen bonds were optimized, and the modeled structure underwent energy minimization. Identifying and selecting suitable binding pockets have become an essential aspect of computational screening. By utilizing the Schrödinger SiteMap module [38], we predict the binding site of the WbTPx1. It calculates the properties of the residue includes acceptor, donor, and hydrophilic regions in WbTPx1 and ranks the possible binding pocket based on the SiteScore function. Finally, the best-ranked binding site was chosen for docking, the grid box was created with 15 Å from all three sides that cover all the residues. Glide module of Schrödinger [39], uses the three-stage docking method to identify the best binding small molecules which covers HTVS (High Throughput Virtual Screening), SP (Standard Precision) and XP (Extra Precision). In each docking stage, only top 10% of the molecules were allowed for the subsequent level of virtual screening. Further, Prime MM-GBSA modules which use the OPLS-AA force field to perform the ΔGbind (binding free energies) calculation for the WbTPx1-ligand bound complexes with default parameters.
Density functional theory (DFT) studies
Top five identified hits compounds were taken for the DFT calculation using the Gaussian 09 program [40]. The electronic structure is a fundamental component in understanding the thermodynamic properties of molecular interactions and the electronic, chemical, and molecular dynamics nature of small molecules. To identify the energy levels of the “Highest Occupied Molecular Orbital” (HOMO) and “Lowest Unoccupied Molecular Orbital” (LUMO) for the identified hits using the hybrid function B3LYP-D3 with the basis set 6-311G** + + (2d, 2p). Calculating the energy gap (HOMO–LUMO) enables the determination of various electronic properties, including chemical potential, chemical stability, electron affinity and hardness [41]. This information is critical in comprehensively assessing the chemical potential of the hits and helps in understanding their electronic properties.
Stability analysis of protein–ligand complexes
The MD simulations assess the dynamic stability of the best five non-toxic hits complex with the WbTPx1. The hit topologies were parameterized using the online PRODRG server [42]. The WbTPx1-hit complexes system was constructed similarly implemented in native WbTPx1, which includes replicating the protein topology, setting up the system using the periodic boundary condition, solvation, neutralization and equilibration. The dynamicity of the complexes was investigated by performing 50 ns production MD simulation.
Essential dynamics (PCA) for the native and hits bound WbTPx1
To analyze the conformational changes and atomic movements of WbTPx1 and hit-bound WbTPx1 complexes, the PCA (Principal Component Analysis) was employed to the their respective trajectory structure obtained from MD simulations [43]. PCA is a popular statistical technique used to investigate the protein movements that are crucial to understanding the biological roles [44]. The unsupervised multivariate statistical method that explains the greatest variability across the datasets is often used to reduce the dimensionality of large datasets [45]. The initial step in PCA construction is to prepare the trajectory in cartesian coordinates, that involves eliminating the solvents and periodicity that establish the necessary initial parameters, then the next crucial metrics is the construction of a covariance matrix with dimensions of 3N × 3N, where N represent the overall number of atoms in trajectory obtained from simulation studies for the WbTPx1 and hit-bound systems. The eigenvectors and eigenvalues that describe the motion and magnitude of the WbTPx1 are obtained through the diagonalization of the covariance matrix [46]. Most often, the highest variability in the data can be attributed to the first three eigenvalues, which correspond to the principal components PC1, PC2, and PC3. Using the bio3D package in R software, the PCA analysis was carried out on the WbTPx1 protein in two distinct states: the unbound-native and hit-bound WbTPx1 [47].
Anti-filarial activity using in vitro studies
Setaria digitata, a filarial worm that infects cattle and is homologous to human parasites W. bancrofti, was used in in vitro tests to evaluate the hit compounds identified through in silico approaches [48, 49]. S. digitata is a well-known and extensively studied in vitro model organism popularly used for testing the macrofilaricidal activity [50,51,52]. Adult motile worms are collected from the peritoneal cavity of a calf that had been newly slaughtered, and they were subsequently washed multiple times using normal physiological saline (0.95%). Without delay, the adult worms were expeditiously transferred into DMEM (Dulbecco’s modified eagle’s medium), with 0.01% Pen-Strep antibiotic and 10% heat-inactivated fetal bovine serum (FBS) and the container was brought to the lab less than an hour. The chemical compounds were acquired from San Diego, California’s ChemBridge Corporation. To evaluate the viability of S. digitata, the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reduction assay was performed. The worms were exposed to different concentrations (0.625, 1.250, 2.500, 5.000, 10.000, 15.000, and 20.000 µM), for a duration of 24 h [53]. Pen-Strep (0.01%) and 10% heat-inactivated FBS were added to DMEM throughout testing [54]. Each worm was exposed for 24 h before being incubated for 30 min with.
0.25 mg mL−1 MTT on phosphate-buffered saline (pH 7.4). After the incubation period was over, each worms were moved into microplate plate with 400 µL dimethyl sulphoxide (DMSO) and allowed to gently shake for an hour at room temperature. Using a multimode plate reader (Synergy H1, BioTek, USA), the absorbance of formazan produced by treated–T, control–C (untreated), and negative control –H (heat-killed worms) was measured at 492 nm.
The positive control group consisted of adult worms that were exposed to media but not treated with hits. The negative control (H) were heat-killed adult worms for 30 min at 56 °C and then incubated with MTT. The concentration of the inhibitor and the percentage of inhibition from the triplicate tests and controls were used to generate the inhibition profile [54, 55]. Using GraphPad PRISM 8.0.1, the IC50 values using dose–response are determined and matched with reference drug, ivermectin.
Results and discussion
Modeling and validation of WbTPx1
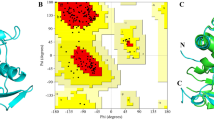
The sequence similarity search was carried out against non-redundant RCSB-PDB using NCBI-BLASTp that identified the crystallographic structure of peroxiredoxin-1 (PDB ID: 4FH8) from Ancylostoma ceylanicum (AcePrx-1) as the best template with the sequence identity of 64.71% along with the query coverage of 82% and e-value of 1e-89. Thus, the 3D structure of WbTPx1 was modeled using the template (PDB ID: 4FH8) and the structural alignment showed an RMSD deviation of 0.68 Å. The quality of the modeled WbTPx1 3D structure (Fig. 1) is demonstrated by the Ramachandran plot with respect to φ and ψ angles, which shows that 75.3% of amino acid residues are present in the most favored regions, 22.2% are in additional allowed region, 2.5% are in the generously allowed region, and none (0.0%) of the amino acids were observed in the disallowed regions, the detail statistics of the Ramachandran plot is given in Supplementary Fig. 1. The WbTPx1 possesses an amino acid of 228 residues that forms a compact and globular structure featuring the characteristic fold of the Prxs (peroxiredoxins) family that is organized around a twisted sheet composed of seven β-strands, encircled by five α-helices, particularly it forms thioredoxin fold-a unique structural motif consists of four-stranded β-sheet and three flanking α-helices [56]. The existence of conserved motif ‘‘GGLG’’ (Gly-Gly-Leu-Gly) and ‘‘YF’’ (Tyr-Phe) that are the invariant features of Prxs family, the sequence of WbTPx1 also found to have conserved motif as like in other parasites [18]. The amino acid sequence of WbTPx1 has two peroxidative cysteine residues (conserved cysteine) at positions 78 and 199 that are essential for the reduction and detoxification of hydrogen peroxides as well as aiding in head-to-tail dimer interface formation composed of residues 166 to 192 as remain conserved across Prxs family [19].

A Superimposition between target (WbTPx1) and the template (Ancylostoma ceylanicum, Peroxiredoxin-1 B Cartoon illustration of a three-dimensional structure of WbTPx1 that characterized with thioredoxin fold consisting of five α-helices and seven β-strands and the catalytic important residues peroxidatic cysteine (C78) are represented as red sticks, and C Ramachandran Plot for the WbTPx1 model
Dynamic stability studies of WbTPx1 through MD simulations
The MD simulation were performed for 200 ns at 310 K in the SPC water model to examine the dynamicity of WbTPx1. The processed trajectories (20,000 structures) were used to study the dynamic structural changes in the WbTPx1. After 20 ns, it was observed that the systems had achieved a stable equilibrium. The RMSD of WbTPx1 was plotted, which showed a deviation between 0.38 and 0.49 nm with the average RMSD and standard deviation of 0.44 nm and 0.015 nm, respectively. A lower deviation in the RMSD value signifies dynamic stability and equilibrium in the modeled structure, making it suitable for further studies. The RMSF was calculated to assess and evaluate the each amino acid residue flexibility of in WbTPx1, the residue-specific flexibility along the backbone of the WbTPx1 exhibits RMSF between 0.04 and 0.51 nm with average RMSF and SD of 0.15 nm and 0.09 nm, respectively.
It was noted that the region of the N-terminal loop of WbTPx1, starting from Met1 to Phe42, showed higher fluctuations. In contrast, the residue Ser23 exhibited the highest N-terminal fluctuation with the RMSF value of 0.46 nm. The loop-III which spans from Met57 to Lys63, which connects the β-sheet III (β-III) with the α-helix I (α-I), exhibits a higher backbone fluctuation between 0.08 nm (Met57) and 0.12 nm (Lys61). The longer loop-V that starts from Thr116 to Pro129 showed fluctuation between 0.05 (Pro129) and 0.17 nm (Glu120). Similarly, the loop-VI (143–154 residues) that connect the α-III with β-V showed higher residual fluctuation that ranged from 0.10 to 0.23 nm; among them, the residue Glu148 had the maximum fluctuation. Additionally, the C-terminal region, ranging from residues 195 to 228 and encompassing loop-IX and α-V, exhibited higher fluctuations, mainly because of its inherent flexibility in terminal.
The RMSF analysis revealed that the elevated fluctuations noted in the amino acid residues align in the loops of C- and N-terminals of the WbTPx1. The dynamic compactness of the WbTPx1 was monitored based on changes in the Rg, that fluctuate between 1.78 nm and 1.89 nm, averaging 1.82 nm. This observation indicates the structural compactness and stability of the intact architecture of WbTPx1 during the simulation. Further, the SASA was computed to evaluate the folding profile and the protein’s interaction to solvent molecules over time, the SASA value for WbTPx1 was found between 113 and 130 nm2, with an average of 120 nm2 (Fig. 2). These overall MD simulation results revealed that the modeled WbTPx1 is stable with intact structural architecture during the timescale of 200 ns MD simulation.

The data indicate that the WbTPx1 is stable and preserves its structural architecture throughout the simulation: (A) RMSD, (B) RMSF, (C) Rg and (D) SASA calculated from 200 ns simulations
Prediction of binding site and structure-guided virtual screening of WbTPx1 inhibitors
The Schrödinger SiteMap module, we identify the regions of the binding sites in WbTPx1, we utilized the potentially energy minimized structure (− 968439.19 kJ/mol) obtained from the 200 ns trajectories structure for binding site predictions and molecular docking studies, the detailed of all the predicted sites of WbTPx1 are shown in supplementary Fig. 2 and the best site is selected based on the site score, Dscore (druggability score) and volume. The predicted site revealed a highest site score of 0.929 and a druggability score of 0.957 with the cavity volume of 315.217 Å3 through diverse descriptors such as enclosure, size, cavity, volume, solvent exposure degree, interaction tightness, and hydrogen bond donor and acceptor characteristics. The predicted binding site comprises the following residues: Ser8, Leu27, Ala28, Ile30, Pro35, Ile165, Arg166, His167, Ser168, Leu169, Val170, Glu181, Arg184, Thr185, Ala188, Phe189, Val192, Glu193, Glu197, Val198, Cys199, Pro200, Ala201, Asn202 and Trp203, respectively. Interestingly, the predicted binding site lies in the reported region involved in the dimer interface (166 to 192) [57], and targeting the interface region was reported to be a successful approach in inhibiting the activity of proteins and enzymes [58,59,60]. The grid for docking was created to cover all the binding site residues of WbTPx1 using the grid size of X = 53.05, Y = 47.62, and Z = 47.15 with the inner grid box of 10 × 10 × 10 Å3 and the outer grid dimensions of 32.41 × 32.41 × 32.41 Å3 to perform molecular docking within the specified grid space boundaries. To screen against WbTPx1, we used small molecule compounds from the in-house ChemBridge database, the prefiltering first eliminates molecules with reactive functional groups and those that violate the five rules of Lipinski. Approximately 698,709 small compounds from the ChemBridge database were chosen following the filtering procedure. The final dataset, which consisted of 2,086,614 structures, was carefully prepared and then subjected to a three-phase molecular docking steps (HTVS, SP, and XP) for screening against WbTPx1, finally a total of 601 molecules in with docking scores ranging from − 7.84 to − 5.46 kcal/mol were obtained based on XP results following screening using HTVS and SP docking. The top 25 hit compounds (Supplementary Table 1) were tested for toxicity profile prediction using the Mclue server, based on their binding affinity for WbTPx1 [61].
The prediction revealed five non-toxic hit molecules (ChemBridge ID: 26319496, 58522350, 24425757, 38651207 & 29025530) and ADME calculations indicate that the molecules are in the acceptable range (Table 1). The highest drug-likeliness attribute is represented by the #star descriptor value of zero across the top five non-toxic hits, signifying a higher probability of drug-like quality. All the top five hits exhibit favorable aqueous solubility, cell permeability, absorption, and zero violation in Lipinski’s rule of five, which clearly depict the identified hits have higher oral bio-availability. The extra Precision (XP) docking results (Fig. 3) for the five hits of WbTPx1 are given in Table 2 where the glide score ranges from − 7.41 to − 6.88 kcal/mol and the glide energy lies from − 48.73 to − 36.22 kcal/mol. The 2D representation interaction map of the hits with the WbTPx1, are shown in Fig. 4. The first hit, N-benzyl-N-(2-hydroxyethyl)-3-[5-(1H-indol-3-ylmethyl)-1,3,4-oxadiazol-2-yl] propenamide (ID-26319496) formed three hydrogen bonds with residues Ser168, Arg184 and Pro200 with the Glide score of − 7.41 kcal/mol.

A Structural alignment of top five hits which showed higher affinity with better satisfied the ADMET profile that bind in the binding site of WbTPx1 were represented in black, red, blue, green, and purple (ID: 26319496, 58522350, 24425757, 38651207 & 29025530) sticks, respectively. B Surface representation rendered transparently and integrated with a blue cartoon shows the hits bound in the WbTPx1 binding site. C Cartoon representation of the WbTPx1 and the sticks represent the binding site residues that are labeled in single letter

The 2D representation illustrating the ligand interaction map of the five hit molecules in the binding site of WbTPx1 A 26319496, B 58522350, C 24425757, D 38651207 and E 38651207. The interaction profile indicates that hydrogen bonds and salt bridge interactions contribute to the stabilization of the hits with WbTPx1. Additionally, the hits (26319496, 38651207 and 38651207) showed Pi–Pi interaction. F Surface representation rendered transparently and integrated with a blue cartoon shows the hits in the WbTPx1 binding site
The hit also formed three pi-pi stacking interactions with the His167 moiety. The second hit, 1-(3-{[(S*)-(cis-3-hydroxycyclo butyl) (phenyl)methyl]amino}-3-oxopropyl) piperidine-4-carboxamid (ID: 58522350) forms four hydrogen bonds with residues Ala28, Ser168, Arg184 and Trp203 with the Glide score of − 7.37 kcal/mol whereas the thirds hit, 4-[(1-{[3-(4-hydroxyphenyl)-1H-pyrazol-5-yl]carbonyl}pyrrolidin-3-yl)methyl]benzoic acid (ID: 24425757) exhibit three hydrogen bonds and a salt bridge with the residues Leu27, His167, Trp203 and Arg184 having the Glide score of − 7.13 kcal/mol.
The fourth hit, 1-{[5-(hydroxymethyl)-2-furyl]methyl}-N-[4-(1,3 -thiazol-4-yl)phenyl]-3-piperidine carboxamide (ID: 38651207) showed three hydrogen bonds and two pi-pi stacking with residues Leu27, Arg166, Ser168, His167, and Phe189 with the Glide score of − 6.93 kcal/mol whereas the fifth hit, N-[(cis-3-hydroxycyclobutyl)(2-thienyl)methyl]-2-(2-oxoazepan-1-yl)acetamide (ID: 29025530) two hydrogen bonds and one pi-pi stacking with the residues Cys199, Trp203 and His167 having the Glide score of − 6.88 kcal/mol, respectively. The distance of H-bond occurred between the hit molecule and the protein is also one factor that decides the stability and strength of protein-hit complex formation. The H-bond is considered stronger if the distance is less than 2.5 Å, and it is considered moderate if it is between 2.5 and 3.5 Å [55, 62]. All five hits exhibited a strong H-bond with a distance lesser than 2.5 Å. Besides the bonded interactions, the stability and binding of potential hits are influenced by non-bonded contributions, including charged, hydrophobic and polar groups of WbTPx1. The Prime MMGBSA method uses the molecular mechanics and continuum solvation models to calculate the ΔG Bind (binding free energy) analysis between the docked complex structure of the selected five hit molecules of WbTPx1. It showed the ΔGbind values between − 47.15 and − 63.06 kcal/mol, which lies in the order of hit ID: 58522350 > 38651207 > 24425757 > 26319496 > 29025530. Further, the cross-docking study was performed with human TPx1 (PDB ID: 5UCX) to evaluate the drug specificity. Hs and WbTPx1 show an identity of 56.19% with a structural RMSD value of 0.8 Å. Both organism exhibits similar thioredoxin structural folds, with five α-helices and seven β-strands which are the invariant features of members peroxiredoxins family. We have carried out cross-docking with the top five identified potent hits at the same dimer interface area of HsTPx1 to confirm the selectivity of the identified hits towards the WbTPx1. The extra Precision (XP) docking results showed the Glide score of − 3.14, − 3.33, − 4.51, − 1.75 and − 3.14 kcal/mol for the hits IDs: 26319496, 58522350, 24425757, 38651207, and 29025530, respectively. Additionally, we observed differences in the volume of the binding site cavity and the lack of bonded interaction between the hits and HsTpx1, which may account for the lower docking scores and affinity, and these results revealed that hits exhibit higher and strong affinity towards Wb. Thus, studies were carried out for these selected hit molecules to analyze binding stability and complex integrity.
Density functional theory (DFT) analysis of potent hits WbTPx1
In quantum calculations, the HOMO and LUMO play a significant influence in reactivity of the hits. HOMO signifies an inclination to donate electrons, whereas the LUMO accepts electrons, and its energy aligns with the electron affinity. The energy gap (Egap) is utilized to determine the chemical reactivity of the hits. The calculated energies of the LUMO orbital for five hits lie between − 0.02558 and − 0.06691 eV, whereas the HOMO orbital energies lies between − 0.20061 and − 0.24194 eV.
Additionally, the five hits energy gaps (Egap) ranged from 0.12 to 0.29 eV (Table 3). The HOMO and LUMO energy of the five hits, along with the molecular orbitals diagram, are shown in Fig. 5. The distribution of energy in the hit molecules is shown by the colors green and red, which indicate the positive and negative charged moities. Based on Egap value, the chemically reactive of the hits lies in the order of ChemBridge ID: (29025530 (0.20966 eV) > 58522350 (0.20094 eV) > 26,319,496 (0.18668 eV) > 38651207 (0.16548 eV) > 24425757 (0.15936 eV), respectively.

HOMO and LUMO for the five potential hits A 26319496, B 58522350, C 24425757, D 38651207 and E 38651207 are shown with orbital contour map. The noticeable difference in energy gap between the LUMO and HOMO signifies the stability and chemical reactivity of the hits, revealing their strong binding affinity with WbTpx1
MD simulation: dynamic stability of hit-bound WbTpx1 complexes
MD simulations were carried out for all five hits bound to WbTpx1 individually to monitor the dynamicity of the complexes. All the five WbTpx1 complex systems (WbTpx1 + ChemBridge ID: 26319496, 58522350, 24425757, 38651207 & 29025530) attain equilibrium after 5 ns. The RMSD deviation of complex 1 (WbTpx1 + ChemBridge ID: 26319496) ranged from 0.30 to 0.44 nm, with average RMSD and SD value of 0.37 nm and 0.02 nm, whereas the complex 2 (WbTpx1 + ChemBridge ID: 58522350) and complex 3 (WbTpx1 + ChemBridge ID: 24425757) exhibited the RMSD deviation within 0.31 to 0.47 nm and 0.30 to 0.46 nm had the average of 0.38 and 0.39 nm and the standard deviation of 0.02 and 0.01 nm. Complex 4 (WbTpx1 + ChemBridge ID: 38,651,207) and 5 (WbTpx1 + ChemBridge ID: 29025530) showed similar trends in the RMSD values, that fluctuated between 0.33 and 0.52 nm and 0.41 to 0.55 nm showed the average of 0.42 and 0.45 nm with the standard deviation of 0.03 and 0.01 nm, respectively. To investigate the level fluctuations of residues upon hit binding, the backbone RMSF of WbTpx1 was examined. As observed in the native WbTPx1 all the five hit bound complexes exhibited higher reside-wise RMSF fluctuations in both the terminal regions due to the terminal flexibility. The N-terminal loop that spans from Met1 to Phe42 showed a higher RMSF value and similarly, the C- C-terminal from His195 to Thr228 has the highest RMSF value due to terminal flexibility. The loop-V spanning from the residues Thr116 to Pro129 and loop-VI from Glu143 to Arg154 was observed to exhibit higher RMSF value in all the five complex systems of WbTPx1. The RMSF of the complex1 fluctuated between 0.05 and 0.69 nm with an average of 0.17 nm and the residue Ser119 of loop-V showed the highest backbone fluctuation of 0.29 nm whereas in loop-VI the residue His147 exhibit maximum fluctuation of 0.20 nm. In complexes 2 and 3, the average RMSF fluctuation was found to be 0.16 and 0.15 nm, exhibiting a similar range between 0.05 and 0.71 nm. In both complexes, the loop-V residue Ser119 displays higher fluctuations of 0.22 and 0.15 nm and the residue Glu148 of loop-VI shows larger backbone fluctuations of 0.17 and 0.21 nm, respectively. The average RMSF of complex 4 and 5 were found to be 0.16 and 0.17 nm with a range from 0.05 to 0.49 nm and 0.06 to 0.80 nm, respectively, where the loop-V residue Glu120 and Ser119 account for more fluctuation of 0.17 and 0.22 nm while the loop-VI the residue Glu148 was observed to have more fluctuation of 0.22 and 0.17 nm among other counterparts residues of loop-VI. The Rg (radius of gyrations) measures the compactness of the hit-bound state of WbTPx1, the five WbTpx1 complex systems exhibit intact Rg with the average value of 1.95, 1.93, 1.91, 1.93 and 1.92 nm with the SD of 0.02, 0.01, 0.01, 0.02 and 0.02, respectively. The SASA was also used to evaluate the complexes surface area that interacts with the nearby water molecules. The average SASA for WbTPx1-hit complexes was determined to be 134.65, 135.07, 137.54, 131.70, and 132.69 nm2, with corresponding standard deviations of 2.74, 3.17, 2.51, 3.16, and 2.78 nm2, as seen in (Fig. 6).

The stability profiles: A RMSD, B RMSF, C Rg and D SASA are calculated from simulations for the WbTpx1—hits complexes: complex 1–5 (26319496, 58522350, 24425757, 38651207 and 38651207) are showed in black, red, blue, green and purple, respectively, these analyses clearly indicate the intact and consistent bounding of the hits with WbTpx1 throughout complex simulations
The complex MD simulation based on the profiles of RMSF, RMSD, SASA and Rg revealed that throughout the simulation period, all the five hits (ChemBridge ID: 26319496, 58522350, 24425757, 38651207& 29025530) bind intact inside the binding site of WbTPx1. The simulated trajectories were analyzed by calculating Coulomb and L–J energies to examine non-bonded energy contributions in the interactions between WbTPx1 and hit molecules. Table 4 signifies that the Lennard–Jones potential energy was more dominant than coulomb energy that accounts for the stability of the five complexes. Based on the calculation of binding energy, it is evident that non-bonded energies were also crucial in maintaining the intact and stable binding of hits within the WbTPx1 throughout complex dynamics. Supplementary Fig. 3 showcases ensembles of WbTPx1 complexes with hit molecules, captured every 10 ns throughout the simulation. Its evident from these snapshots that the hits remained securely bound within WbTPx1 binding site throughout the entire simulation period.
Comparative PCA and FEL analysis of native and hit-bound dynamics
PCA projects the molecular trajectories generated in the MD simulation onto a 2D plane, with the first three principal components (PC1 to 3) elucidating the maximum variability in the data (Table 5). A comparison between the hits bound and native WbTPx1 revealed that the native WbTPx1 PC1 magnitude was smaller, whereas the magnitude of PC2 of the WbTPx1 native is observed to be higher than the hits bound WbTPx1. In the native system, the first two PCs account for 49.9% variability and the hit bound five systems (WbTpx1 + ChemBridge ID: 26319496, 58522350, 24425757, 38651207 & 29025530), the first two PCs contribute 49.61%, 50.02%, 48.47%, 51.93% and 60.85% as total variability of the systems. Therefore, the initial two eigenvectors, PC1 and PC2, encompassed the maximum principal variances in the original distribution of the conformational space within the simulation systems. This indicates that PC1 and PC2 predominantly reflect the shifts in the conformational state. PCA scatter plots comparing the native and hit-bound states of the WbTpx1 simulation systems were generated by projecting along the directions of the first two principal components, PC1 and PC2, where the red and blue dots indicate the conformationally stable and unstable, respectively. The scatter plot distribution of native clearly showed irregular and messy. However, in the case of five hits complex system the scatter plot was found to be well organized where the red and blue spots in clusters were equally spaced parallel to each other on both sides of the diagonal with respect to PC1, PC2 and PC3. This PCA clearly showed the intact and stable binding of the hits in the binding site of the WbTpx1 that remained consistent with the RMSD, RMSF, Rg and SASA analysis results.
Although the initial two principal components were adequate to explain the fluctuations in conformational changes and motion throughout the simulation, we also considered the variability explained by the PC3, which is clearly shown in supplementary Figs. 4, 5, 6, 7, 8, 9.
Further, the correlated motions at the residue level were also determined by analyzing the cross-correlation map of residues for the atoms in the backbone of both native WbTPx1 and hit bound (WbTpx1 + ChemBridge ID: 26319496, 58522350, 24425757, 38651207 & 29025530). The maximum negative or anticorrelated backbone motion was represented by the magenta region that range from 0 to − 1, indicating that the residues shift in the opposite direction and the backbone motion with positive correlation of WbTPx1 was showed by the cyan regions that range from 0 to 1, implying that the residues move in the same direction. The stronger the correlation, the darker the displayed color. The figure demonstrated that overall, the correlated motion of the two systems displayed a similar motion trend, but it was clear that these two systems correlated motion had altered upon binding of the hit molecules with the WbTpx1. Especially in key areas related to binding site regions (represented as black box in Fig. 7). It was generally believed that the reduction of negatively correlated motion would reduce the flexibility of the protein. Therefore, the five hits binding to the WbTpx1 binding site would hinder the complex systems correlated motion, making it more difficult for WbTpx1 to be exposed to forming dimers and thereby accomplishing the purpose of inhibiting protein activity. The cross-correlation map revealed that both native and hits bound WbTPx1 exhibit a similar trend in the correlated motion among the backbone atoms of the residues. We observed a minute change in the motion of the residue’s backbone located in N– C– terminals and in the dimer interface region where the hits bind with the WbTPx1, as shown in Fig. 7. The porcupine plotting was generated for the PC1 which accounts for the greatest variability of the native and hit-bound states to visualize the direction of movement of the backbone atoms. Observations indicated that the WbTPx1 bound to hits exhibits reduced backbone atom movement compared to the native WbTPx1 (Fig. 8) that in accordance with the results of PCA and cross-correlation analysis.

Cross-correlation map for A native WbTPx1 and B–F hit bound WbTPx1 (WbTPx1- ChemBridge ID-26319496, 58522350, 24425757, 38651207 and 29025530 with positive correlation in cyan and negative (anticorrelation) in magenta colors, respectively. The black box highlighted the key areas where the correlated motions of the native and hit bound systems of WbTPx1 were found to be significantly different

Porcupine plot illustrating the backbone atom movement for A native WbTPx1 and B–F hit bound WbTPx1 (WbTPx1- ChemBridge ID-26319496, 58522350, 24425757, 38651207 and 29025530 from the trajectories. The directional arrows in the projection depict the movement of the WbTPx1 backbone atoms
To explore the conformational dynamics of native and hits bound WbTPx1 the FEL (free energy landscape) was calculated. This enabled the observation of various states like local and global minima that are sampled throughout the entire time scale period of the MD simulation. The Gibbs free energy ranges from 0 to 11.50 kJ/mol for the native WbTPx1 while for the complexes-WbTPx1 + ChemBridge ID: 26319496, 58522350, 24425757, 38651207 & 29025530, it ranges from 0 to 11.50,11.50,11.60, 13.40, and 11.60 kJ/mol and FEL is represented as contour diagram is shown in Fig. 9. The dark blue area indicates energetic minima and conformationally stable states, while the red and yellow sections show higher energy states.

The FEL comparison: A native WbTPx1 and with five complexes B–F ((WbTPx1- ChemBridge ID-26319496, 58522350, 24425757, 38651207 and 29025530)) The contour profile constructed based on first two principal components (PC1 & PC2) along the x, y axis and the z-axis represented by Gibbs free energy
The native WbTPx1 exhibits a single global lowest energy conformation with a steep, narrow valley single basin. The FEL analysis of complexes 1 and 2 exhibit a similar trend in energy state with a broad basin that converges into numerous energy states from multiple local minimum states and confined to a single global energy state. The Complexes 3, 4, and 5 adopted a similar energy spread that of native, containing multiple local energy states but confined into a single sharp global minima energy state. Thus, FEL analysis offers atomic-level resolution of a protein–ligand bound system, identifies potential binding transition states and metastable states, and can be useful for designing inhibitors. The results of the essential dynamics, cross-correlation profile, porcupine and FEL analyses provide precise insights into the changes in the molecular conformational that occurred during MD simulation period as hits bound to the dimer interface site of WbTPx1.
Viability evaluation of filarial worms through MTT-formazan colorimetric assay
The MTT assay was performed at concentrations spanning from 0.625 to 20 µM over a 24-h duration. At the maximum dosage of 20 mM concentration, three hits exhibited inhibition efficacy higher than 92% (94.50 ± 1.56, 93.83 ± 0.96 and 92.78 ± 0.91).
In contrast, at lower concentrations (0.625 µM), it showed the inhibition percentages of 32.70 ± 0.74%, 38.79 ± 0.85% and 34.60 ± 1.27%, respectively, for the hits- ChemBridge ID: 58522350, 24425757 and 26319496. Interestingly, all three hits had greater inhibition percentages at concentrations between 0.625 and 20 µM, that is found be to higher than the standard anti-filarial drug Ivermectin, as shown in Table 6. The IC50 values were calculated by the Michalis Menten equation and were matched with the standard drug ivermectin, IC50 value of hits of WbTPx1 58522350, 24425757 and 26319496 lies between the range of 1.069 ± 0.204 µM, 1.373 ± 0.259 µM and 1.429 ± 0.246 µM. Interestingly, we noted that the IC50 value of two hits (24425757 & 26319496) was lesser than the IC50 value of Ivermectin with 1.374 ± 0.152 µM, shown in Fig. 10. The hits found using the in silico technique have the ability to eliminate adult worms, as revealed by the MTT reduction assay which showed better IC50 and higher inhibition than Ivermectin. Further, we employed molecular docking in the same binding site of WbTPx1 to examine the interacting profile of Ivermectin. Interestingly, Ivermectin showed a glide score of − 3.352 kcal/mol, suggesting a lower binding affinity than the identified potent hits of WbTPx1, which ranged from − 7.410 to − 6.887 kcal/mol, the interaction profile of the Ivermectin with the WbTPx1 is shown in Fig. 10. We observed two moderate hydrogen bonds with the distance of 2.9 Å and 3.0 Å between His 167 and Glu 181 of WbTPx1 with Ivermectin. In the case of the identified five hit compounds, we observed the complex is stabilized by the hydrogen bonds, salt bridge, Pi–Pi interaction and hydrophobic interactions that accounts for higher binding affinity and these interaction profiles were found to be less prevalent in Ivermectin- WbTPx1 complex.

A In vitro validation using the adult S. digitata worms, for top three hits of WbTpx1 along with the standard drug Ivermectin (ChemBridge ID-58522350, 24425757and 26319496) at different concentrations 0.625 to 20 µM, B two dimensional interaction profile of ivermectin interactions with the binding site residues of WbTpx1, where H-bonds were represented by solid arrows and C transparent surface representation embedded with blue cartoon representation clearly showing the ivermectin bind in the binding site of WbTpx1
Conclusion
Our current research is primarily on employing structure-guided drug design to identify potent inhibitors targeting the dimer interface region of WbTPx1. The WbTPx1 exhibited higher binding affinity with the five best non-toxic hits obtained from ChemBridge database (ChemBridge ID-26319496, 58522350, 24425757, 38651207 & 29025530). The reactive of the hits were found using the DFT analysis, which also revealed important information for further hit enrichment and enhanced activity. Furthermore, the MD simulation studies of these WbTPx1-hit complexes demonstrated that all five hits were found to be intact and bound inside the WbTPx1 binding region. Its interesting to note that these five hits showed more binding selectivity for Wb than for Hs. The in vitro validation suggested that, in comparison to the standard medicine Ivermectin, the two discovered hits (ID: 24425757 & 26319496) showed a better percentage of inhibition and an improved IC50. In future these identified hit molecules are the starting point for improvement of innovative small molecular chemical scaffolds and fragments that can be enriched using in vitro and in vivo models that likely could delivered strong drug-like characteristics features which might helpful towards addressing LF infections.
References
Simonsen PE, Mwakitalu ME (2013) Urban lymphatic filariasis. Parasitol Res 112:35–44. https://doi.org/10.1007/s00436-012-3226-x
Routh HB, Bhowmik KR (1993) History of elephantiasis. Int J Dermatol 32:913–916. https://doi.org/10.1111/j.1365-4362.1993.tb01418.x
Michael E, Bundy DAP, Grenfell BT (1996) Re-assessing the global prevalence and distribution of lymphatic filariasis. Parasitology 112:409–428. https://doi.org/10.1017/s0031182000066646
Ramaiah KD, Das PK, Michael E, Guyatt HL (2000) The economic burden of lymphatic filariasis in India. Parasitol Today 16:251–253. https://doi.org/10.1016/s0169-4758(00)01643-4
Bizhani N, Hashemi Hafshejani S, Mohammadi N et al (2021) Lymphatic filariasis in Asia: a systematic review and meta-analysis. Parasitol Res 120:411–422. https://doi.org/10.1007/s00436-020-06991-y
Famakinde DO (2018) Mosquitoes and the lymphatic filarial parasites: research trends and budding roadmaps to future disease eradication. Trop Med Infect Dis 3:4. https://doi.org/10.3390/tropicalmed3010004
Freedman DO (1998) Immune dynamics in the pathogenesis of human lymphatic filariasis. Parasitol Today 14:229–234. https://doi.org/10.1016/S0169-4758(98)01244-7
Paily KP, Hoti SL, Das PK (2009) A review of the complexity of biology of lymphatic filarial parasites. J Parasit Dis 33:3–12. https://doi.org/10.1007/s12639-009-0005-4
Rao UR, Chandrashekar R, Subrahmanyam D (1987) Effect of ivermectin on serum dependent cellular interactions to Dipetalonema viteae microfilariae. Trop Med Parasitol 38:123–127
Critchley J, Addiss D, Gamble C et al (2005) Albendazole for lymphatic filariasis. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.cd003753.pub3
Reaves BJ, Wallis C, McCoy CJ et al (2018) Recognition and killing of Brugia malayi microfilariae by human immune cells is dependent on the parasite sample and is not altered by ivermectin treatment. Int J Parasitol Drugs drug Resist 8:587–595. https://doi.org/10.1016/j.ijpddr.2018.09.002
Kwarteng A, Ahuno ST, Akoto FO (2016) Killing filarial nematode parasites: role of treatment options and host immune response. Infect Dis poverty 5:86. https://doi.org/10.1186/s40249-016-0183-0
Cobo F (2016) Determinants of parasite drug resistance in human lymphatic filariasis. Rev Esp Quimioter 29:288–295
Weiss SJ, Test ST, Eckmann CM et al (1986) Brominating oxidants generated by human eosinophils. Science 234:200–203. https://doi.org/10.1126/science.3018933
Cross AR, Jones OTG (1991) Enzymic mechanisms of superoxide production. BBA: Bioenerg 1057:281–298. https://doi.org/10.1016/s0005-2728(05)80140-9
Callahan HL, Crouch RK, James ER (1988) Helminth anti-oxidant enzymes: a protective mechanism against host oxidants? Parasitol Today 4:218–225. https://doi.org/10.1016/0169-4758(88)90162-7
Selkirk ME, Smith VP, Thomas GR, Gounaris K (1998) Resistance of filarial nematode parasites to oxidative stress. Int J Parasitol 28:1315–1332. https://doi.org/10.1016/s0020-7519(98)00107-6
Gretes MC, Poole LB, Karplus PA (2012) Peroxiredoxins in parasites. Antioxid Redox Signal 17:608–633. https://doi.org/10.1089/ars.2011.4404
Wood ZA, Schröder E, Robin Harris J, Poole LB (2003) Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci 28:32–40. https://doi.org/10.1016/s0968-0004(02)00003-8
Flohé L (2010) Changing paradigms in thiology from antioxidant defense toward redox regulation. Methods Enzymol 473:1–39. https://doi.org/10.1016/s0076-6879(10)73001-9
Knoops B, Loumaye E, Van Der Eecken V (2007) Evolution of the peroxiredoxins. Subcell Biochem 44:27–40. https://doi.org/10.1007/978-1-4020-6051-9_2
Hakimi H, Asada M, Angeles JMM et al (2012) Cloning and characterization of Plasmodium vivax thioredoxin peroxidase-1. Parasitol Res 111:525–529. https://doi.org/10.1007/s00436-012-2864-3
Dayer R, Fischer BB, Eggen RIL, Lemaire SD (2008) The peroxiredoxin and glutathione peroxidase families in Chlamydomonas reinhardtii. Genetics 179:41–57. https://doi.org/10.1534/genetics.107.086041
Selkirk ME, Smith VP, Thomas GR et al (1998) Resistance of filarial nematode parasites to oxidative stress. Int J Parasitol 28:1315–1332. https://doi.org/10.1016/s0020-7519(98)00107-6
Prime (2020) Schrödinger Release 2020–4. Schrödinger, LLC, New York
Abraham MJ, Murtola T, Schulz R et al (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Oostenbrink C, Villa A, Mark AE, Van GWF (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem 25:1656–1676. https://doi.org/10.1002/jcc.20090
Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J (1981) Interaction models for water in relation to protein hydration. In: Pullman B (ed) Intermolecular forces. Springer, Dordrecht, pp 331–342. https://doi.org/10.1007/978-94-015-7658-1_21
Vrahatis MN, Androulakis GS, Lambrinos JN, Magoulas GD (2000) A class of gradient unconstrained minimization algorithms with adaptive stepsize. J Comput Appl Math 114:367–386. https://doi.org/10.1016/s0377-0427(99)00276-9
Berendsen HJCC, Postma JPMM, Van Gunsteren WF et al (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. https://doi.org/10.1063/1.448118
Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. https://doi.org/10.1063/1.328693
Hess B (2008) P-LINCS: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput 4:116–122. https://doi.org/10.1021/ct700200b
Miyamoto S, Kollman PA (1992) Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem 13:952–962. https://doi.org/10.1002/jcc.540130805
Darden T, York D, Pedersen L (1998) Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. https://doi.org/10.1063/1.464397
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38. https://doi.org/10.1016/0263-7855(96)00018-5
LigPrep (2020) Schrödinger Release 2020-4. Schrödinger, LLC, New York
Harder E, Damm W, Maple J et al (2015) OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput 12:281–296. https://doi.org/10.1021/acs.jctc.5b00864
Schrödinger Release 2020-4 (2020) SiteMap. Schrödinger, LLC, New York
Friesner RA, Banks JL, Murphy RB et al (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47:1739–1749. https://doi.org/10.1021/jm0306430
Frisch MJ, Trucks GW, Schlegel HB, et al (2016) G16_D09. Gaussian 16, Revision D.09, Gaussian, Inc., Wallin
Zhan C-G, Nichols JA, Dixon DA et al (2003) Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: molecular properties from density functional theory orbital energies. J Phys Chem A 107:4184–4195. https://doi.org/10.1021/jp0225774
Schüttelkopf AW, van Aalten DMF (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr 60:1355–1363. https://doi.org/10.1107/s0907444904011679
David CC, Jacobs DJ (2014) Principal component analysis: a method for determining the essential dynamics of proteins. Methods Mol Biol 1084:193–226. https://doi.org/10.1007/978-1-62703-658-0_11
Hayward S, de Groot BL (2008) Normal modes and essential dynamics. Methods Mol Biol 443:89–106. https://doi.org/10.1007/978-1-59745-177-2_5
Wolf A, Kirschner KN (2013) Principal component and clustering analysis on molecular dynamics data of the ribosomal L11·23S subdomain. J Mol Model 19:539–549. https://doi.org/10.1007/s00894-012-1563-4
Maddah M, Karami L (2021) An atomistic investigation on the interaction of distamycin A and its derivative with the telomeric G-Quadruplex as anticancer agents by molecular dynamics simulation. Arch Biochem Biophys 701:108797. https://doi.org/10.1016/J.ABB.2021.108797
Grant BJ, Rodrigues APC, ElSawy KM et al (2006) Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22:2695–2696. https://doi.org/10.1093/bioinformatics/btl461
Senanayake KS, Söderberg J, Põlajev A et al (2020) The genome of Setaria digitata: a cattle nematode closely related to human filarial parasites. Genome Biol Evol 12:3971–3976. https://doi.org/10.1093/gbe/evaa017
Perumal ANI, Gunawardene YINS, Dassanayake RS (2016) Setaria digitata in advancing our knowledge of human lymphatic filariasis. J Helminthol 90:129–138. https://doi.org/10.1017/s0022149x15000309
Mathew N, Karunan T, Srinivasan L, Muthuswamy K (2010) Synthesis and screening of substituted 1,4-naphthoquinones (NPQs) as antifilarial agents. Drug Dev Res 71:188–196. https://doi.org/10.1002/ddr.20357
Nisha M, Kalyanasundaram M, Paily KP et al (2006) In vitro screening of medicinal plant extracts for macrofilaricidal activity. Parasitol Res 100:575–579. https://doi.org/10.1007/s00436-006-0294-9
Senathilake KS, Karunanayake EH, Samarakoon SR et al (2017) Oleanolic acid from antifilarial triterpene saponins of Dipterocarpus zeylanicus induces oxidative stress and apoptosis in filarial parasite Setaria digitata in vitro. Exp Parasitol 177:13–21. https://doi.org/10.1016/j.exppara.2017.03.007
Mathew N, Misra-Bhattacharya S, Perumal V, Muthuswamy K (2008) Antifilarial lead molecules isolated from Trachyspermum ammi. Molecules 13:2156–2168. https://doi.org/10.3390/molecules13092156
Sureshan M, Prabhu D, Rajamanikandan S, Saraboji K (2023) Discovery of potent inhibitors targeting Glutathione S-transferase of Wuchereria bancrofti: a step toward the development of effective anti-filariasis drugs. Mol Divers 1:1–21. https://doi.org/10.1007/s11030-023-10617-7
Sureshan M, Prabhu D, Kadhirvel S (2023) Computational identification and experimental validation of anti-filarial lead molecules targeting metal binding/substrate channel residues of Cu/Zn SOD1 from Wuchereria bancrofti. J Biomol Struct Dyn 41:1–14. https://doi.org/10.1080/07391102.2022.2136245
Choi J, Choi S, Choi J et al (2003) Crystal structure of Escherichia coli thiol peroxidase in the oxidized state: insights into intramolecular disulfide formation and substrate binding in atypical 2-Cys peroxiredoxins. J Biol Chem 278:49478–49486. https://doi.org/10.1074/jbc.m309015200
Nguyen JB, Pool CD, Wong CYB et al (2013) Peroxiredoxin-1 from the human hookworm Ancylostoma ceylanicum forms a stable oxidized decamer and is covalently inhibited by conoidin A. Chem Biol 20:991–1001. https://doi.org/10.1016/j.chembiol.2013.06.011
Goyal B, Goyal D (2020) Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb Sci 22:297–305. https://doi.org/10.1021/acscombsci.0c00058
Gunderwala AY, Nimbvikar AA, Cope NJ et al (2019) Development of allosteric BRAF peptide inhibitors targeting the dimer interface of BRAF. ACS Chem Biol 14:1471–1480. https://doi.org/10.1021/acschembio.9b00191
Cardinale D, Guaitoli G, Tondi D et al (2011) Protein-protein interface-binding peptides inhibit the cancer therapy target human thymidylate synthase. Proc Natl Acad Sci U S A 108:E542-549. https://doi.org/10.1073/pnas.1104829108
Kiss R, Sandor M, Szalai FA (2012) http://Mcule.com: a public web service for drug discovery. J Cheminform 4:P17. https://doi.org/10.1186/1758-2946-4-S1-P17
Sindhu T, Venkatesan T, Prabhu D et al (2018) Insecticide-resistance mechanism of Plutella xylostella (L.) associated with amino acid substitutions in acetylcholinesterase-1: a molecular docking and molecular dynamics investigation. Comput Biol Chem 77:240–250. https://doi.org/10.1016/j.compbiolchem.2018.09.004
Acknowledgements
Authors are grateful to the management of SASTRA Deemed University for providing all necessary computational facilities. KS thankfully acknowledges DST-SERB for providing financial support in the form of research projects (No: EMR/2017/002841 and No: CVD/2020/000604) to conduct the study. MS is thankful for the funding as Senior Research Fellow grant from ICMR-SRF (No. Fellowship/96/2022-ECD-II, IRIS ID No. 2021-11346/F96). The authors thankfully acknowledge the high-performance computational facility in the School of Computing at SASTRA Deemed University for providing computational resources to complete the simulation studies. The authors like to thank Dr. U Venkatasubramanian, Associate Professor and Mr. J Adithyan, Research Scholar, School of Chemical and Biotechnology, SASTRA Deemed University for their help and suggestion in vitro experiments. Authors also thank to Dr. R. Velusamy, M. V. Sc., Ph.D., Assistant Professor and Head, Department of Veterinary Parasitology, Veterinary College and Research Institute Orathanadu, Thanjavur, Tamil Nadu, India for authenticating the filarial worm S. digitata.
Author information
Authors and Affiliations
Contributions
MS and SR: data curation, investigation, visualization, validation, writing—original draft. KS: conceptualization, writing—review and editing, resources, supervision, funding acquisition. The final version of the manuscript submitted was approved by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sureshan, M., Rajamanikandan, S. & Saraboji, K. Comprehensive approach to in silico identification and in vitro validation of anti-filarial hit molecules targeting the dimer interface of thioredoxin peroxidase 1 in Wuchereria bancrofti: a progress in anti-filariasis drug development. Mol Divers (2024). https://doi.org/10.1007/s11030-024-10922-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11030-024-10922-9