
Abstract
The mechanism of electrophilic aromatic substitution (SEAr) is still matter of debate and interest in the literature. In this work, the Friedel-Crafts alkylation and the acylation in the gas phase were investigated in the context of the unified mechanism for SEAr. In this unified proposal three kinds of intermediates can potentially be formed: oriented and unoriented π-complexes, intimate single electron transfer (SET) intermediates and σ-complexes. Quantum chemical calculations at M06-2X/6–311++G(d,p) level were carried out for the investigation of the reaction of benzene with acetyl and tert-butyl ions as model non-oxidant electrophiles for acylation and alkylation, respectively, in the gas phase. It was found that both the tert-butyl and the acetyl cations prefer to form oriented π-complexes. Both electrophiles do not react through a SET pathway with benzene. The π-complex between tert-butyl cation and benzene can evolve to a σ-complex, while in the case of the acetyl cation and benzene the σ-complex was not found as a minimum on the potential energy surface. Instead, it corresponds to a transient species or a very shallow minimum. The outcome of this is that the π-complex would only react with the aromatic ring evolving to the product with nucleophilic assistance by a species of the reaction medium, in either through a concerted mechanism or a specific interaction. This is also observed for aromatics with low ionization energies/nucleophilicities. However, very electron rich aromatic systems afford σ-complexes, and as their ionization energies increases (i.e., less nucleophilic), the more the resulting complex resembles a π-complex, more or less continuously. This suggests out that electrophilic aromatic substitution reactions cannot be rationalized within a single mechanistic framework. Instead, a continuum of mechanistic possibilities may be involved.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The Friedel-Crafts alkylation and acylation are important classes of the electrophilic aromatic substitution (SEAr) reaction. Their mechanism has been initially rationalized within the framework of the SEAr concerted mechanism [1] (Scheme 1A). Within this model, aromaticity is preserved during the process of addition of the electrophile. Some years later, a stepwise mechanism was proposed to explain the isotope effects found for the nitration, the bromination and for the protonation of aromatics obtained by Melander [2, 3]. Nowadays the generally accepted mechanism (Scheme 1B), passes through two different intermediates. The first one, the so-called π-complex 1 (Scheme 1B), is formed by the electrostatic interaction between the electrophile and the electronic belt of the aromatic ring. The electrophile then attacks the aromatic ring leading to a second intermediate, called a σ-complex, 2 (Scheme 1B), where the aromaticity of the ring is disrupted. Experimental evidence supporting the existence of both intermediates have been found since then (π-complex [4–7], σ-complex [8, 9]). On the other hand, previous studies of the mechanism of nitration of aromatics led to an alternative proposition of an unified mechanism for SEAr (Scheme 1C) involving an unoriented π-complex (1), an intimate single electron transfer (SET) intermediate (3) and the σ-complex [10–15]. Regioselectivity and reactivity could be easily explained by this mechanism [16], even in difficult cases such as the ipso substitution preferences in a number of cases, which could not be fully previously understood [17]. This unified mechanism involving a SET step is proposed to be the main mechanism for the SEAr whenever electron rich nucleophiles and oxidizing electrophiles react with each other.

Mechanistic proposals for SEAr
In this scope, we have been investigating the extension of the SET concept for other electrophilic aromatic substitutions. In order to investigate the role of a less oxidizing electrophile in the reaction mechanistic profile, we have carried out calculations for the SEAr involving acetyl and tert-butyl cations as electrophiles, and benzene, in the gas phase. This would correspond to a Friedel-Crafts acylation and a Friedel-Crafts alkylation respectively.
Results and discussion
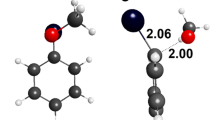
The reaction of tert-butyl cation with benzene was used as a model to study the mechanism of Friedel-Crafts alkylation. DFT calculations at M06-2X/6–311++G(d,p) level were carried out (see Computational Details section for details). In this case, two minima were found, namely an oriented π- (4) and a σ- (6) complexes (Fig. 1). The analysis of the π-complex geometry shows that the electrophilic carbon in the tert-butyl cation is about 3 Å to the closest carbon atom of the ring. This is within the typical range found for electrophiles interacting with aromatic systems determined by X-ray diffraction [18–20]. The transition state for the interconversion of such intermediates (5) is also shown in the same figure. Figure 2 shows the pictorial representation of the potential energy surface for the reaction, together with some other possible isomeric intermediates. The π-complex 4 was found to be 0.9 kcal/mol more stable in relation to the σ-complex 5. This is different from what is found in the case of the nitration of benzene, where the σ-complex is the more stable of the key intermediates. This result agrees well with earlier calculations [22, 23] and gas phase ion-molecule reactions, carried out by Cacace et al. [21] The theoretical and experimental for the reaction on the gas phase results are in good agreement. The theoretical activation enthalpy for the interconversion of the π-complex to the σ-complex is 2.0 kcal/mol.

Geometries of the π-complex between benzene and tert-butyl ion (4), the transition state connecting π- and σ-complexes (5) and the respective σ-complex (6), calculated at M06-2X/6–311++G**. CHelpG charges over the electrophile are also shown. Distances are given in Angstroms and angles in degrees

Pictorial representation of the calculated potential energy surface of the reaction of benzene with the tert-butyl cation (represented by E+) and its comparison with experimental results. M06-2X/6–311++G(d,p) enthalpies are shown in black and the experimental results from Cacace et al. [21] are shown in red dashed line
Analysis of the CHelpG charges shows that most of the positive charge (+0.787e) resides over the tert-butyl cation. This indicates that the SET process is not occurring at this system, what could be anticipated evaluating the ionization potentials of the tert-butyl radical (IP = 6.70 ± 0.03 eV [24]) and benzene (9.24372 ± 0.00005 eV [25]), which predicts that single electron transfer from the benzene to the tert-butyl cation would be endothermic by 58.7 kcal/mol (2.54 eV). The proton shift into the aromatic ring from the σ-complex can occur (Scheme 2), despite of the high barriers, these being the rate determining step in the gas phase [21]. The results correlate well with the experimental ones, as shown in Fig. 2.

Proton migration after the attack of the tert-butyl cation to benzene
Thus, these results indicate that the electrophilic aromatic substitution may have different mechanisms depending on the electrophile, since nitration, which involves an oxidizing electrophile (NO2 +) seems to prefer the SET mechanism for benzene [10, 11], further affording a σ-complex in a very exothermic step, while the tert-Bu+ seems to prefer to form a π-complex, eventually forming the σ-complex in an endothermic step. Thus, this suggests that non-oxydizing electrophiles tend to afford σ-complexes that are less stable than the parent π-complex, in the case of the reaction with benzene.
Another kind of electrophile that may be non-oxidazing is the acyl cation. These carbocationic intermediates are the key intermediates involved on the Friedel-Crafts acylation [13, 14]. In order to investigate this aspect the reaction of the acetyl cation to benzene ring was used as a model reaction. Figure 3 shows the geometries of the complexes found. It is noteworthy that only the π-complexes 7 (oriented) and 8 (unoriented) are found from the interaction of the acetyl cation with the benzene ring. The most stable species is 7a, with the methyl group pointing toward the aromatic ring, with the π-complex 7b being only 1.2 kcal/mol less stable than 7a. On the other hand, unoriented the π-complex 8 lies 9.4 kcal/mol above 7a. All the attempts of locating a σ-complex itself resulted only into the π-complexes. Neither intrinsec reaction coordinate (IRC) or calculations involving solvent (IEFPCM) resulted in success. We could, though, locate a transition state that would lead to the σ-complex, which is 8.6 kcal/mol above π-complex 7. Animation of the imaginary frequency showed the correct reaction coordinate. Other structures for proton migration within the aromatic ring were computed. Their relative energies are shown in Fig. 4. Detailed geometries of such structures are given in he supporting information. One can see from these results that the proton migration is more energetic in the acetylation of benzene than the relative conterparts in the tert-butyl alkylation, with barriers about 16–17 kal/mol, relatively close to to the energy of the separated reagents (17 kcal/mol). Noteworthy, previous experimental attempts of our group of reacting the acetyl cation with of benzene in the gas phase in a pentaquadrupole mass spectrometer only resulted in proton transfer, leading to the protonated benzene as only product observed [26]. No other adducts were observed in these experiments. This can now be rationalized, once the σ-complexes are either elusive or kinetically unachieaveble in the gas phase. The IRC calculations of the transition states of the electrophilic addition and the proton transfer between ipso and ortho position, which in theory are connected through the σ-complex, allowed us to estimate that this elusive intermediate would be about 8.8 kcal/mol higher in energy in relation to the π-complex 7a.

The optimized geometries of the complexes between CH3CO+ and benzene obtained at M06-2X/6–311++G(d,p) level. The oriented π-complexes (7a and 7b), an unoriented π-complex (8), and the transition state leading to an elusive σ-complex (9) have their relative enthalpies shown below the respective structures. Distances are given in Angstroms and angles in degrees

Pictorial representation of the potential energy surface of the reaction of benzene with the acetyl cation (represented by Ac+) calculated at M06-2X/6–311++(d,p) level
These results have puzzled us, since that would mean that the electrophile could only insert into the benzene ring with assistance of an additional species from the reaction media acting as a base (Scheme 3), which would result in kinetic models different of what one would expect. The strongest Lewis base (B) present at the beginning of the reaction is probably the precursor of the acyl cation, typically the parent acyl chloride. It is noteworthy that this mechanism rescue some aspects of Ingold’s original proposition [1] involving a concerted mechanism for SEAr, in order to preserve the aromaticity of the benzenoid ring (Scheme 1A).

Concerted mechanism for electrophilic aromatic acylation
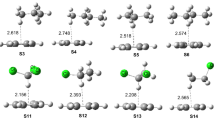
In order to investigate this, we have carried out DFT calculations including the presence of a molecule that could potentially act as a base. In the Friedel-Crafts reaction conditions, the aromatic (ArH), AlCl3 and the acetyl chloride (RCOCl), are usually mixed, and the solvent often is the aromatic compound itself or a weakly nucleophilic solvent, such as CH2Cl2 or CS2. Under these conditions the most basic species present in the reaction media would be a carbonyl compound, either RCOCl or the reaction product. ArC(=O)R. Since in the beginning of the reaction there is no product or it is in low concentration, we have decided to consider the acetyl chloride, as a potential Lewis base. Initial interaction of the acetyl chloride with the acetyl cation and benzene afforded several π-complexes, shown in Fig. 5. Several initial orientations were considered, affording structures 10 to 14 after geometry optimization. All structures correspond to minima on the potential energy surface.

Optimized geometries of π-complexes interacting with acetyl chloride. Relative energies are given in kcal/mol, and refer to ΔH298 (ΔG298 in brackets)
Excepting structure 13, all structures of the π-complexes are very similar energetically. Upon the assistance of a nucleophilic species, i.e. the acetyl chloride, one could finally obtain the corresponding hydrogen bonded σ-complex (16) as a true minimum at the potential energy surface. The σ-complex (16), is 10.6 kcal/mol higher in energy in relation to the π-complex 11, and 9.2 kcal/mol less table than 12, this being an endothermic process. This is once again the opposite of what is observed for nitration, where this step is quite exothermic. The σ-complex (16) is eventually converted to protonated final product 18, through the transition state 17. The final product corresponds to the protonated acetophenone hydrogen bonded to the acetyl chloride, and this step is exothermic by 10.6 kcal/mol in relation to the π-complex and 37.5 kcal in relation to the σ-complex (Fig. 6).

Optimized geometries of the TS for electrophilic addition, σ-complex, transition state for deprotonation and reaction product. Relative energies are given in kcal/mol, and refer to ΔH298 and ΔG298 in brackets
These results for this particular system suggest shifts in the general picture of the mechanism for electrophilic aromatic substitution. According to these results, second order on the acyl chloride and primary isotope effects or general base catalysis should be observed, conversely to what has been widely proposed in the textbooks [27]. Actually, experimental primary isotope effects for the reaction of a CH3CH2CO+. Sb2F11 − salt with benzene and related systems have been reported [28–31], as well as complex kinetics, where second order on the acyl chloride-AlCl3 complex is observed [32]. This fully agrees with the predictions from our calculations. We would like to stress that SET does not take place in this type of SEAr involving the carbon atom as electrophile, as could also be anticipated by the differences between ionization potentials of acetyl radical (7.0 eV [33]) and benzene (9.24372 ± 0.00005 eV [25]), which predicts that this process would be endothermic by 51.7 kcal/mol (experimental). This agrees very well with the value of 51.3 kcal/mol computed at M06-2X/6–311++G(d,p) level.
Computations at the same level of theory, i.e., M06-2X/6–311++G(d,p) level of key intermediates (π- and σ-complexes), considering the acetyl cation interacting with other substituted aromatic rings in the gas phase were carried out. There is an apparent correlation between the experimental ionization energy (IE) of the aromatic compound and the computed distance between the electrophilic carbon of the acetyl cation (Table 1), as well as the acetyl bond angle (Fig. 7). The charge-transfer capacity of the aromatic ring plays a role in the capacity of forming the σ-complex. Actually, a continuous between the σ- and π-complexes seems to exist, depending on the nucleophilicity of the aromatic ring. Thus, this may indicate that the mechanism of the Friedel-Crafts reaction can be both the base assisted one, for low nucleophilicity aromatics, such as benzene, affording substantial isotope effects and complex kinetics, indicating that the deprotonation is rate determining, whereas highly nucleophilic aromatics can follow the traditional stepwise mechanism. In the case the aromatic ring is electron rich, which is reflected in its relatively low potential ionization energy, there is much charge-transfer to the acetyl cation, which then gets closer to the aromatic ring, eventually forming a σ-complex as intermediate, without aid from an external base. This also leads to larger degree of bending of the acetyl cation. This would afford the classical picture, in which the electrophile addition to the ring, without assistance of a base, would evolve to a σ-complex, which is quickly deprotonated in a subsequent step.


Correlation between experimental ionization energies of several substituted aromatics with selected geometrical parameters
Conclusions
Thus, the SEAr may be rationalized as a continuum mechanistic, where stronger electrophiles, such as H+ , primary and methyl cation, react with the aromatic ring without going through π-complex [35–38], while weaker electrophiles would not be strong enough to disrupt the benzene aromaticity. In this case, the substitution on the ring requires the assistance of a base in either a concerted or base-assisted mechanism, as originally proposed by Ingold [1] (Scheme 1A). Additionally to these two extremes, there are the cases where oxidizing electrophiles are involved, which would react through a SET mechanism (Scheme 1C). Thus a very rich mechanistic universe is possible. Further studies are necessary to establish its boundaries aiming to organize this whole universe as a single mechanistic model for this amazing class of reactions.
Computational details
Geometry optimizations were performed by the Gaussian 09 package [39] using M06-2X hybrid functional [40] with the 6–311++G** basis set. The optimized geometries were characterized as minima on the potential energy surface by the absence of imaginary vibrational frequencies, whereas the transition states were characterized by the presence of one imaginary frequency. In order to evaluate whether the transition states found correspond to the reaction studied, intrinsic reaction coordinates calculations (IRC) were carried out. Energies correspond to enthalpies at 298 K and 1 atm.
References
Hughes ED, Ingold CK, Reed RI (1946) Nature 158:448
Melander L (1949) Acta Chem Scand 3:95
Melander L (1949) Nature 163:599
Benesi HA, Hildebrand JH (1949) J Am Chem Soc 71:2703
Christen M, Koch W, Simon W, Zollinger H (1962) Helv Chim Acta 45:2077
Hassel O, Stromme KO (1958) Acta Chem Scand 12:1146
Holman RW, Gross ML (1989) J Am Chem Soc 111:3560
V. A. Koptyug (1984) Contemporary problems in carbonium ion chemistry III, Vol. 122, Springer Berlin / Heildelberg
Shteingarts VD (1981) Russ Chem Rev 50:735
Esteves PM, Carneiro JWDM, Cardoso SP, Barbosa AGH, Laali KK, Rasul G, Prakash GKS, Olah GA (2003) J Am Chem Soc 125:4836
Chen Z, Mo Y (2013) J Chem Theor Comp 9:4428
T. Brink, M. Liljenberg (2016) in Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, J. Mortier (Ed.), Wiley, Chapter 4, pg 83
Kim EK, Bockman TM, Kochi JK (1993) J Am Chem Soc 115:3091
Davlieva MG, Lindeman SV, Neretin IS, Kochi JK (2005) J Org Chem 70:4013–4021
Kochi JK (1992) Acc Chem Res 25:39
Queiroz JFD, Carneiro JWDM, Sabino AA, Sparrapan R, Eberlin MN, Esteves PM (2006) J. Org. Chem. 71:6192
Moodie RB, Schofield K (1976) Acc Chem Res 9:287
Rosokha SV, Kochi JK (2002) J Org Chem 67:1727
Vasilyev AV, Lindeman SV, Kochi JK (2002) New J Chem 26:582
A. V. Vasilyev, S. V. Lindeman, J. K. Kochi (2001) Chem. Commun 909
Cacace F, Crestoni ME, Fornarin S (1992) J Am Chem Soc 114:6776
Heidrich D (2002) Angew Chem 114:3343
Heidrich D (2002) Angew Chem Int Ed 41:3208
Houle FA, Beauchamp JL (1979) J Am Chem Soc 101:4067
Chewter LA, Sander M, Muller-Dethlets K, Schlag EW (1987) J Chem Phys 86:4737
P. M. Esteves, M. N. Eberlin, unpublished results
Smith MB, March J (2007) March’s advanced organic chemistry: reactions, mechanism and structure, sixth edn. John Wiley & Sons, Inc., New Jersey
Olah GA, Lukas J, Lukas E (1969) J Am Chem Soc 91:5319
Grovenstein E, Aprahamian NS (1962) J Am Chem Soc 84:212
Miller LL, Watkins BF (1976) J Am Chem Soc 98:1515
Effenberger F, Maier AH (2001) J Am Chem Soc 123:3429
Hoornaert G, Slootmaekers PJ (1968) Bull Soc Chim Belges 77:295
Lias SG, Bartmess JE, Liebman JF (1988) J Phys Chem Ref Data 17(Suppl. 1):1
Webbook chemistry database (webbook.nist.gov) and references therein.
Kuck D (1990) Mass Spectrom Rev 9:583
Miklis PC, Ditchifield R, Spencer TA (1998) J Am Chem Soc 120:10482
Olah GA, Schlosberg RH, Porter RD, Mo YK, Kelley DP, Gheorghe D, Mateescu J (1972) Am Chem Soc 94:2034
Sieber S, Schleyer PVR, Gauss J (1993) J Am Chem Soc 115:6987
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, 2009.
Zhao Y, Truhlar DG (2008) Theor Chem Accounts 120:215
Acknowledgements
This work is dedicated to the Prof. George A. Olah in the occasion of his 90th Anniversary. Work was financially supported by CNPq, FAPERJ, CAPES.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Absolute energies and selected geometries are available in the supporting information.
ESM 1
(DOCX 1495 kb)
Rights and permissions
About this article

Cite this article
Oliveira, F.G., Rodrigues, F.L., de Oliveira, A.V.B. et al. Thoughts about the electrophilic aromatic substitution mechanism: the Friedel-crafts alkylation and acylation of benzene with acetyl and t-butyl cations in the gas phase. Struct Chem 28, 545–553 (2017). https://doi.org/10.1007/s11224-017-0915-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-017-0915-1