
Abstract
In this work, we investigate the nature of the O–O and O–N interactions in protonated 1,2-dioxirane-3-one derivatives and protonated 1,2-oxaziridine-3-one derivatives, respectively. The quantum theory of atoms in molecules and the natural bond orbital (NBO) method in conjunction with the localized molecular orbital energy decomposition analysis (LMOEDA) have been used. LMOEDA and NBO analyses reveal that the O–O and O–N interactions exhibit characteristics of dative covalent bonds. In addition, the L(r) = −∇2 ρ(r) function reveals that the O–O and O–N interactions can be categorized as strong hole–lump interactions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Molecular interactions play a key role in a wide range of biological, chemical, and physical fields including the development of new materials, new strategies for drug design, new supramolecular structures, and crystal engineering [1–6]. At present many molecular interactions as the lithium bond (LiB) [7, 8], the beryllium bond (BeB) [9, 10], the boron bond (BB) [11], the pnicogen bond (PB) [12, 13], the chalcogen bond (ChB) [14, 15], the halogen bond (XB) [16, 17], and the aerogen bond (AB) [18] are being extensively analyzed. Grabowski recently suggested that the term “Lewis acid–Lewis base interaction” seems to be more proper than the term “noncovalent interaction” for those interactions where the complex formation is connected with significant electron charge redistribution [19]. For all interactions, the electrostatic force is very important since the positively charged Lewis acid center interacts with the negatively charged Lewis base center. This is in line with the σ-hole concept which was applied to the halogen bond and to other noncovalent interactions [20, 21].
On the other hand, there is evidence from the Cambridge Structural Database, that the O···N interactions may play a significant role in the control of conformations of biphenyls carrying several nitro groups in ortho positions. In addition, the O···N interactions may involve a very small degree of covalent interaction [22]. Daszkiewicz [23] also stressed the importance of the O···N interaction between nitro groups in crystals. In his study of 104 structural fragments deposited in the Crystal Structure Database, he found that the O···N interactions, in most cases, are shorter than the sum of the van der Waals radii (2.84 Å). Ab initio, calculations of the interaction energy indicate attractive feature of this interaction, comparable to C–H···O interactions.
Curiously, O···O interactions have been detected in crystal of sodium nitroprusside. Nelyubina et al. [24] showed that nitroprusside anion is involved in relatively strong self-interactions (12.5 kJ mol−1) through the nitroso group. The strength of this interaction exceeds by far the corresponding interaction (5.9 kJ mol−1) in crystalline urea nitrate [25]. Pinkerton et al. [26] have studied through the quantum theory of atoms in molecules (QTAIM) a series of weaker and moderate inter- and intramolecular interactions O···O in the crystals of phthalic acid, 2,6-dinitrophenol [27] and 2-nitrobenzoic acid [28]. In the crystals of 2,6-dinitrophenol, they also found O···N interactions [27].
Recently, we have studied through the QTAIM, the nature of the bonds of the small-ring lactone and lactam 1,2-dioxirane-3-one and 1,2-oxaziridine-3-one (see Scheme 1 for the atom numbering) [29]. In that work, it was shown by QTAIM that there is not a bond critical point and bond path between the atoms O1 and O2, and O1 and N2, respectively. Also, the distance between O1 and O2 in 1,2-dioxirane-3-one is significantly longer than the O–O bond in hydrogen peroxide, 1.631 and 1.460 Å, respectively. Similarly, the interatomic distance between O1 and N2 in 1,2-oxaziridine-3-one is significantly longer than the N–O bond in hydroxylamine, 1.635 and 1.448 Å, respectively. In addition, the QTAIM topological parameters calculated at the bond critical point [electron charge density, ρ(r b ), Laplacian of electron charge density, ∇2 ρ(r b ) and total electronic energy density, H(r b )] showed that the C3–O2 and C3–N2 bonds in these molecules are slightly stronger than the C–O single bond in formic acid and C–N single bond in N-methylacetamide, respectively. However, when the heteroatoms and N2 in (1) and (2) species, respectively, are ring protonated, the C3–O2 and C3–N2 bonds are broken and the O1–O2 and O1–N2 bonds are formed, despite the strength of the C3–O2 and C3–N2 bonds in the neutral molecules [30]. That is, non-ring-protonated derivatives of the (1) and (2) species are in fact complexes of carbon dioxide and the cations OH+ and NH2 +, respectively.

Atom numbering scheme of the studied chemical reactions
Identification of the strongest bonds in chemistry as “bond dissociation enthalpies, BDH (or bond dissociation energies, BDE) values, although often used in chemistry to discuss the strength of the chemical bond, are not reliable bond strength descriptors” [31].
It is clear that the formation of the O1–O2 and O1–N2 bonds is somewhat unusual. With the aim to understand the nature of these bonding interactions, in this work, we performed a deeper electronic study of the species [OCO–OY]+ and [OCO–N(H)Y]+, with Y = –H, –F, and –CH3. The nature of the interatomic interactions O1–O2 and O1–N2 has been compared with the properties of conventional O–O and O–N bonds in hydrogen peroxide and hydroxylamine, respectively.
Computational details
The geometries of all species were fully optimized using the Møller–Plesset second-order perturbation theory [32] (MP2) with the 6-311++G(2d,2p) basis set. The minimum energy nature of the optimized structures was confirmed by the absence of imaginary frequencies. We have calculated the interaction energies (E Int), using the supermolecular approach, as the difference between the total energy of the species ([OCO–OY]+ or [OCO–N(H)Y]+) and the sum of total energies of the two isolated constituents (CO2 and OY+ or N(H)Y+). These energies and their components were obtained at the same level of theory using the localized molecular orbital energy decomposition analysis [33] (LMOEDA) partition method, according to the equation below:
where E ES is the electrostatic component, E EX is the attractive exchange component resulting from the Pauli exclusion principle, E REP is the interelectronic repulsion term and E PL and E DISP correspond to polarization and dispersion terms, respectively. These calculations have been carried out with the GAMESS program (version 2013-R1) [34]. In this work, the results have been interpreted jointly with a real physical property of the system: the electron charge density. The calculations of local topological properties of the electron charge density and its Laplacian function and the integrated atomic properties on the atomic basin, as well as the display of the molecular graphs were performed with the AIMAll software [35] with the wave function obtained at MP2/6-311++G(2d,2p) level of theory.
The natural bond orbital (NBO) method [36] has been used to analyze the population and energies of the chemical bonds O1–O2 and O1–N2. NBO analysis was performed at B3LYP/6-311++G(2d,2p) level of theory, with the NBO 3.1 program [37], as implemented in Gaussian 03 [38].
Results and discussion
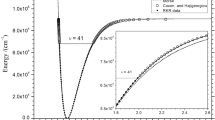
Figure 1 reports the minimum energy paths (MEPs) resulting from the protonation of an oxygen/nitrogen atom on 1,2-dioxirane-3-one and 1,2-oxaziridine-3-one, respectively. In both cases, it is observed that the reaction proceeds without any energy barrier (there is no transition state structure) into a very deep well for the [OCO–OH]+ and [OCO–NH2]+ species.

MEPs for the protonation of an oxygen/nitrogen atom of 1,2-dioxirane-3-one and 1,2-oxaziridine-3-one. ΔE denotes the electronic energy relative to the species [OCO–OH]+ and [OCO–NH2]+ (Color figure online)
Experimental and theoretical studies reveal that both, the hydroxyl cation (OH+) and the nitrenium ion (NH2 +), are ground state triplets [39]. According to Slipchenko et al. [39], the singlet–triplet gap (defined as the energy difference between the lowest energy singlet state and the lowest energy triplet state) is about −210 and −125 kJ mol−1, respectively. However, our calculations indicate that the species [OCO–OH]+ and [OCO–NH2]+ are ground state singlets. Singlet–triplet gap in [OCO–OH]+ is 82.3 kJ mol−1, while the triplet state for [OCO–NH2]+ is not an energy minimum on the potential energy surface. All attempts to find a stable structure lead to dissociation products (CO2 and NH2 + in singlet and triplet state, respectively).
The species OF+ and N(H)F+ have different electronic stabilities. The ground state of OF+ is triplet (ΔE S–T = −179.5 kJ mol−1), and N(H)F+ is ground state singlet (ΔE S–T = 15.8 kJ mol−1). The latter is in agreement with the reported values by Gobbi et al. [40] [ΔE S–T (average) = 26.1 kJ mol−1]. The species OCH3 + and N(H)CH3 + are ground state triplets. The species OCH3 + and N(H)CH3 + in their singlet state are not energy minima on the potential energy surface, both spontaneously rearrange to HOCH2 + and NH2CH2 +, respectively without any energy barrier (there is no transition state structure).
Although most of the cations OY+ and N(H)Y+ (with Y = –H, –F, –CH3) are ground state triplets [except N(H)F+], most of the species [OCO–OY]+ and [OCO–N(H)Y]+ are ground state singlets (except OCO–OCH3]+). In order to compare all species in the same electronic state, henceforth we only focus on species [OCO–OY]+ and [OCO–N(H)Y]+ (with Y = –H, –F, –CH3) in their singlet state.
In specie [OCO–OH]+, the O1–O2 interatomic distance (1.476 Å) is slightly longer than the O–O single covalent bond in hydrogen peroxide (1.460 Å). Similarly, the O1–N2 interatomic distance in specie [OCO–NH2]+ is longer than a conventional O–N single covalent bond as in hydroxylamine, 1.529 and 1.448 Å, respectively. It can be said that the interatomic distances are similar to that of a single covalent bond O–O and O–N, respectively.
The energy decomposition analysis (EDA) is a powerful tool for a quantitative interpretation of chemical bonds. In Table 1, it is reported the interaction energy components derived from the LMOEDA method. It is observed that the most important stabilization terms correspond to the exchange and polarization followed by the electrostatic and to a lesser extent by the dispersion term. This may indicate that the orbital interaction plays an important role in the stability of these complexes.
According to Su et al. [33], ionic bonds are characterized by high values of E ES and low values of the remaining components, for example, for the archetypal NaCl: E ES = −602.0 kJ mol−1 (81.3 % of the stabilizing components), E EX = −75.3 kJ mol−1 (10.16 % of the stabilizing components), E PL = −54.1 kJ mol−1 (7.30 % of the stabilizing components) and E DISP = −9.2 kJ mol−1 (5.23 % of the stabilizing components) [33]. Similar observations were made by Grabowski et al. [41]. On the other hand, covalent bonds are characterized by large exchange and polarization energies due to the effective reduction of the electron–electron Coulomb repulsion between pairs of bonded atoms and the significant orbital deformations, respectively. In Table 1, it can be observed high values of E EX (between 30 and 40 % of the stabilizing components) and E PL (between 33 and 46 % of the stabilizing components). These values suggest the formation of a covalent bond. In addition, Fig. 2 shows a good correlation between the polarization term and the interaction energies. Therefore, the electronic exchange and the orbital deformation play a key role in O–O and O–N bond formation of the studied species.

Correlation between interaction energies and the polarization component from the protonated species
The interaction energy, E int, provides a measure of the strength of the interaction between CO2 and the [OY]+ and [N(H)Y]+ species. The E Int values are typically of the same order of magnitude of that of single and dative covalent bonds [33, 42], are significantly higher than those found for conventional molecular interactions as HBs [43] and XBs [44], and are comparable in strength with the values reported by Grabowski [43] for the strongest hydrogen bond charge assisted (i.e., 166.8 kJ mol−1 for [F···H···F]− system calculated at the MP2/6–311++G(d,p) level of theory). These energies reveal that the O1–O2 interactions are stronger (except in the [OCO–OCH3]+ species) than the O–O single covalent bond in hydrogen peroxide. In turn, the O1–N2 interactions are weaker than the O–N single covalent bond in hydroxylamine.
Moreover, in Table 1, it can be observed that the substitution of a H atom by a F atom and by the CH3 group in the [OCO–OH]+ and [OCO–NH2]+ species has a big influence on the interaction energy. For example, the interaction energy in [OCO–N(H)F]+ is 46 % lower than that of [OCO–NH2]+, and the interaction energy of [OCO–N(H)CH3]+ is 51 % lower than that of [OCO–NH2]+. Something similar is observed in the [OCO–OY]+ species. Therefore, the substitutions of H by an electron-withdrawing atom (F) or by an electron-donating group (CH3) produce an important decrease of the stability of the O1–O2 and O1–N2 interactions.
In Table 2, we report selected geometric parameters and some topological properties of the electron charge density of all the studied species. It is well known that the QTAIM is a powerful tool to inquire about the nature of the chemical bonds [45–47]. In all species, it is observed that the O1–O2 and O1–N2 distances (~1.5 Å) are much smaller than the sum of van der Waals radii for the interacting atoms (i.e., 2.84 Å for oxygen–oxygen and 2.88 Å for oxygen–nitrogen) and slightly longer than conventional O–O and O–N single covalent bonds as in hydrogen peroxide (1.460 Å) and hydroxylamine (1.443 Å), respectively. This indicates clearly the presence of stronger bonding interactions between these atoms. Even more, the topological analysis of the electron charge density, ρ(r), shows the presence of a bond critical point (BCP) and a corresponding bond path in the O1–O2 and O1–N2 interactions, indicative of bonding interactions. The electron density values range from 0.2120 to 0.2467 a.u. for the O1–O2 interactions and from 0.1629 to 0.2054 a.u. for the O1–N2 interactions, thus being lower than the electron density values for the O–O interaction in hydrogen peroxide and O–N interaction in hydroxylamine, respectively. However, these values are significantly higher than those found for conventional molecular interactions as HBs [43], LiBs [48], BeBs [49], PBs [50], ChBs [51], and XBs [44].
It is important to note that in the O–O interaction in hydrogen peroxide and in the O–N interaction in hydroxylamine, ρ(r b ) is high and ∇2 ρ(r b ) < 0, that is, present typical properties of shared-shell interactions (covalent bond), while in O1–O2 and O1–N2 interactions, ρ(r b ) is relatively high, but ∇2 ρ(r b ) > 0. Therefore, these interactions are within the closed-shell regime and have characteristics similar to classic Lewis adducts OC–BH3 and H3N–BH3, where ρ(r b ) > 1 and ∇2 ρ(r b ) > 0.
Recently, in the framework of the HBs as well as the XBs, it was reported that the decrease of the total electronic energy density at the BCP, H(r b ), with increasing ρ(r b ), could be considered as an indicator of the interaction strengthening or interaction stabilization, in the same way as the decrease in the total interaction energy is an indicator of the complex stabilization [52, 53] In Tables 1 and 2, it can be observed that for the same pair of interacting atoms, H(r b ) decreases with increasing ρ(r b ) and decreasing E int. That is, these results also support the idea that H(r b ) is a good descriptor of the strength of the interatomic interactions.
In addition, in the context of the AIM theory, we have calculated the electrostatic interaction energy between the electron cloud of the O1 atom of the CO2 molecule and the nucleus of O2/N2 atom and vice versa (see Table 2). It is observed that |V e–n(O,X)| > |V e–n(X,O)|, that is, the electrostatic interaction between the electron cloud of the O1 atom and the nucleus of the O2/N2 atom contributes significantly to the formation of the O1–O2 and O1–N2 bonds.
Moreover, the NBO method provides a theoretical framework to interpret the formation of a chemical bond. It is a useful tool for the investigation of the relative stability and the nature of the covalent bonds. Table 3 reports the population and energies of the O1–O2 and O1–N2 chemical bonds. It is observed that in all species, the population of the O1–O2 and O1–N2 bonds is slightly less than in conventional O–O and O–N single covalent bonds as in hydrogen peroxide and hydroxylamine, respectively. However, the orbital energies of conventionals O–O and O–N single covalent bonds show less stability than the O1–O2 and O1–N2 bonds.
The analysis of population and orbital energies in the species [OCO–N(H)Y]+ and [OCO–OY]+ reveals that the degree of covalency decreases with Y in the order –H > –F > –CH3.
According to Bader et al. [54], “the value of the delocalization index, DI(A,B), always gives the number of electrons that are delocalized or exchanged between the basins of A and B, independent of any model.” This parameter, like the NBO analysis, allows us to assess the degree of covalency of the O1–O2 and O1–N2 bonds. Figure 3 shows a very good linear relationship between the population of the O1–O2 and O1–N2 bonds and DI(O1,O2/N2). Therefore, it appears that the degree of covalency of the O1–O2 and O1–N2 bonds varies with the O2/N2 atom, increasing in the same order as the interaction energies.

Correlation between population of O1–O2/O1–N2 bond and the delocalization index, DI(O1,O2/N2)
On the other hand, in Table 2, it can be seen that in all species upon the complexation, the C3–O4 and C3–O1 interatomic distances are shorter and longer, respectively, than the C–O covalent double bond in isolated, uncomplexed CO2 molecule (1.153 Å). Changes in the topological properties at the C3–O4 and C3–O1 BCPs (see Table 2) are also observed. That is, ρ(r b ) and H(r b ) in the C3–O4 bond are higher in magnitude than the values found in isolated CO2 [ρ(r b ) = 0.4619 a.u. and H(r b ) = −0.8626 a.u.]. In the C3–O1 bonds, it is observed the opposite. In other words, the C3–O4 bond increases its double bond character, while the C3–O1 bond decreases its double bond character with respect to CO2 isolated.
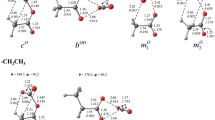
Moreover, the equilibrium bond angle, C3–O1–X2, ranges from 104.92° (in [OCO–OCH3]+) to 109.20° (in [OCO–OCH3]+). In addition, the L(r) function shows, on the valence shell concentration charge (VSCC) of the O1 atom, two nonbonded maxima [CP (3, −3) in L(r) function], whereas in O4, the L(r) function shows one nonbonded maxima [CP (3, −3) in L(r) function] in its VSCC. According to Bader [55] and Gillespie [56], these nonbonded maxima are associated with lone pairs of the O1 and O4 atoms. In other words, it appears that the O1 atom has characteristics of a tetrahedral atom and the O4 atom looks like the oxygen atom of carbon monoxide (see Fig. 4). A similar analysis shows that the O2 and N2 atoms in [OCO–OH]+ and [OCO–NH2]+ species, respectively, have a tetrahedral electronic structure.

Molecular graph and nonbonded maxima (yellow dots) of the L(r) function [CPs (3, −3) in L(r)] for the [OCO–NH2]+ specie (Color figure online)
The L(r) = −∇2 ρ(r) function has been used to characterized various interatomic interactions involving the hole–lump concept [9, 12, 13, 57, 58]. According to hole–lump theory, the areas with charge concentration [L(r) > 0] interact with the charge depleted areas [L(r) < 0] [45, 46]. Figure 5 shows the contour maps of the function L(r) for CO2 and the ionic species [OH]+, [OCO–OH]+, [OCO–NH2]+ and [NH2]+. It is observed that the molecules are oriented so that a local charge concentration (lump) in the valence shell concentration charge (VSCC) of the O1 atom is almost aligned with a local charge depletion (hole) in the VSCC of the O2 (in [OCO–OH]+ species) and N2 (in [OCO–NH2]+ species) atoms, respectively. In other words, the geometry of these species is strongly influenced by the positions of lump and hole in the constituents. Therefore, the L(r) function reveals a hole–lump interaction between O1 and O2 and between O1 and N2 atoms, respectively.

Contour map of the L(r) = −∇2 ρ(r) function. Blue lines denote L(r) > 0 and red lines, L(r) < 0. The black lines indicate the bond paths and the green dots indicate the BCP of the ρ(r) topology. Note the anisotropic distribution of the charge concentration in the VSCC of the O1 atom of the CO2 molecule (Color figure online)
Moreover, it is observed that the interacting atoms modify their VSCC. A polarization of the VSCC of O1 atom of CO2 in direction of the bond paths O1–O2 and O1–N2 is observed. Moreover, it is also seen an accumulation of electron charge density at the hole of the O2 and N2 atoms in species [OCO–OH]+ and [OCO–NH2]+ with respect to the free cations [OH]+ and [NH2]+, respectively. This is because of the charge transfer from the lone pair of O1 of CO2 to the [OH]+ and [NH2]+ species. It appears that there is a tendency to form continuous accumulation region of charge density between the interacting nuclei. These observations allow us to establish that the nuclei of atoms O2 and N2 cause a polarization of the electronic cloud on the VSCC of the O1 atom of CO2.
In Fig. 5, it is reported the atomic population in all the atoms of the species [OH]+, [OCO–OH]+, OCO, [OCO–NH2]+, and [NH2]+. A considerable rearrangement of the electronic charge density with respect to the free constituents is produced. Something similar happens in the rest of the studied species. The existence of a “hole” on the surface of the O2 and N2 atoms valence shell in the [OH]+ and [NH2]+ species is indicative of a local deficiency in electrons. It appears that it is through this “hole” that electronic charge transfer occurs during the formation of the [OCO–OY]+ and [OCO–N(H)Y]+ species. The total electron charge density transferred, in the framework of the AIM theory, CTAIM, was calculated as the CO2 electron charge density loss during the formation of the complex. These values are reported in the last column of the Table 2. It is observed that CTAIM in [OCO–OY]+ is higher than in [OCO–N(H)Y]+ for the same Y, like the interaction energies.
Conclusions
In this work, a deep theoretical study, within the QTAIM framework and NBO method in conjunction with LMOEDA, has been carried to determinate the nature of the interactions O–O and O–N in [OCO–OY]+ and [OCO–N(H)Y]+ species, respectively.
Based on LMOEDA analysis, we have found that the exchange and polarization components have the most significant contributions to the total interaction energy. Therefore, the electronic exchange and the orbital deformation play a key role in O–O and O–N bond formation in the studied species.
Topological analysis of the L(r) function reveals that O1 of the CO2 molecule and O2 and N2 of the [OCO–OY]+ and [OCO–N(H)Y]+ species have characteristics of tetrahedral atoms, while O4 atom of CO2 looks like the oxygen atom in carbon monoxide. Additionally, the L(r) function and calculating |V e–n(O,X)| reveal that the stability of the O–O and O–N bonds is established between the lone pair of the oxygen atom of the CO2 molecule and the charge density depletion region of the O2/N2 atom. That is, these interactions can be categorized as strong hole–lump interactions; a region charge concentration (lump) in the VSCC O1 (CO2) atom combines with that of a region of charge depletion (hole) in the VSCC of O2/N2 atom.
In addition, the geometry of these species is strongly influenced by the positions of lump and hole in the constituents CO2 and [OY]+ and [N(H)Y]+.
References
Moreira DRM, Leite ACL, Dos Santos RR, Soares MBP (2009) Approaches for the development of new anti-trypanosoma cruzi agents. Curr Drug Targets 10:212–231
Auffinger P, Hays FA, Westhof E, Ho PS (2004) Halogen bonds in biological molecules. Proc Natl Acad Sci USA 101:16789–16794
Bertani R, Sgarbossa P, Venzo A, Lelj F, Amati M, Resnati G, Pilati T, Metrangolo P, Terraneo G (2010) Halogen bonding in metal–organic–supramolecular networks. Coord Chem Rev 254:677–695
Bissantz C, Kuhn B, Stahl MA (2010) Medicinal Chemist’s guide to molecular interactions. J Med Chem 53:5061–5084
Bruce DW, Metrangolo P, Meyer F, Pilati T, Präsang C, Resnati G, Terraneo G, Wainwright SG, Whitwood AC (2010) Structure–function relationships in liquid–crystalline halogen-bonded complexes. Chem Eur J 16:9511–9524
Ding X, Tuikka M, Haukka M (2012) Halogen bonding in crystal engineering. In: Benedict JB (ed) Recent advances in crystallography. InTech, New York, pp 143–168
Kollman PA, Liebman JF, Allen LC (1970) The lithium bond. J Am Chem Soc 92:1142–1150
Del Bene JE, Alkorta I, Elguero J (2009) Characterizing complexes with F–Li+–F lithium bonds: structures, binding energies, and spin–spin coupling constants. J Phys Chem A 113:8359–8365
Eskandari K (2012) Characteristics of beryllium bonds: a QTAIM Study. J Mol Model 18:3481–3487
Villanueva EF, Mó O, Yáñez M (2014) On the existence and characteristics of π-beryllium bonds. Phys Chem Chem Phys 16:17531–17536
Esrafili MD (2012) Characteristics and nature of the intermolecular interactions in boron-bonded complexes with carbene as electron donor: an ab initio, SAPT and QTAIM study. J Mol Model 18:2003–2011
Alkorta I, Elguero J, Del Bene JE (2013) Pnicogen-bonded cyclic trimers (PH2X)3 with X = F, Cl, OH, NC, CN, CH3, H, and BH2. J Phys Chem A 117:4981–4987
Eskandari K, Mahmoodabadi N (2013) Pnicogen bonds: a theoretical study based on the Laplacian of electron density. J Phys Chem A 117:13018–13024
Esrafili MD, Mohammadian-Sabet F (2015) An ab initio study on chalcogen–chalcogen bond interactions in cyclic (SHX)3 complexes (X = F, Cl, CN, NC, CCH, OH, OCH3, NH2). Chem Phys Lett 628:71–75
Wang W, Ji B, Zhang Y (2009) Chalcogen bond: a sister noncovalent bond to halogen bond. J Phys Chem A 113:8132–8135
Legon AC (2010) The halogen bond: an interim perspective. Phys Chem Chem Phys 12:7736–7747
Politzer P, Murray JS (2013) Halogen bonding: an interim discussion. ChemPhysChem 14:278–294
Bauza A, Frontera A (2015) Aerogen bonding interaction a new supramolecular force? Angew Chem Int Ed 54:7340–7343
Grabowski SJ (2013) Non-covalent interactions—QTAIM and NBO analysis. J Mol Model 19:4713–4721
Murray JS, Lane P, Politzer P (2009) Expansion of the σ-hole concept. J Mol Model 15:723–729
Politzer P, Murray JS, Clark T (2013) Halogen bonding and other σ-hole interactions: a perspective. Phys Chem Chem Phys 15:11178–11189
O’Leary J, Wallis JD (2007) A competition between O···N and O···C through space interactions in the crystal structures of 3,3′-dinitro-2,2′-bipyridine N-oxides and N,N′-dioxides. CrystEngComm 9:941–950
Daszkiewicz M (2013) Importance of O···N interaction between nitro groups in crystals. CrystEngComm 15:10427–10430
Nelyubina YV, Lyssenko KA, Kotov VY, Antipin MY (2008) Anion–anion assembly in crystal of sodium nitroprusside. J Phys Chem A 112:8790–8796
Nelyubina YV, Lyssenko KA, Golovanov DG, Antipin MY (2007) NO3–NO3– and NO3–π interactions in the crystal of urea nitrate. CrystEngComm 9:991–996
Zhurov VV, Pinkerton AA (2014) Quantifying the inter- and intramolecular interactions in crystalline phthalic acid. Cryst Growth Des 14:5685–5691
Cenedese S, Zhurov VV, Pinkerton AA (2015) Charge density analysis of 2,6-dinitrophenol. Cryst Growth Des 15:875–883
Zhurov VZ, Pinkerton AA (2015) Inter- and intramolecular interactions in crystalline 2-nitrobenzoic acid—an experimental and theoretical QTAIM analysis. J Phys Chem A 119:13092–13100
Duarte DJR, Miranda MS, Esteves da Silva JCG, Liebman JF (2015) Theoretical characterization of the chemical bonds of some three-membered ring compounds through QTAIM theory. Struct Chem 27:663–670
Miranda MS, Duarte DJR, Esteves da Silva JCG, Liebman JF (2015) Protonated heterocyclic derivatives of cyclopropane and cyclopropanone: classical species, alternate sites, and ring fragmentation. Can J Chem 93:708–714
Kalescky R, Kraka E, Cremer D (2013) Identification of the strongest bonds in chemistry. J Phys Chem A 117:8981–8995
Møller C, Plesset MS (1934) Note on an approximation treatment for many-electron systems. Phys Rev 46:618–622
Su P, Li H (2009) Energy decomposition analysis of covalent bonds and intermolecular interactions. J Chem Phys 131:014102
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S et al (1993) General atomic and molecular electronic structure system. J Comput Chem 14:1347–1363
AIMAll (Version 11.12.19), Keith TA, TK Gristmill Software, Overland Park KS, USA, 2011 (aim.tkgristmill.com)
Reed AE, Curtiss LA, Weinhold F (1998) Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem Rev 88:899–926
NBO (Version 3.1) Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Weinhold F, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision E.01. Gaussian Inc., Wallingford, CT
Slipchenko LV, Krylov AI (2002) Singlet-triplet gaps in diradicals by the spin-flip approach: a benchmark study. J Chem Phys 117:4694–4708
Gobbi A, Frenking G (1993) The singlet-triplet gap of the halonitrenium ions NHX+, NX2 + and the halocarbenes CHX, CX2 (X = F, CI, Br, I). J Chem Soc Chem Commun 2:1162–1164
Grabowski SJ, Sokalski WA, Leszczynski J (2005) How short can the H···H Intermolecular contact be? New findings that reveal the covalent nature of extremely strong interactions. J Phys Chem A 109:4331–4341
Hopffgarten MV, Frenking G (2012) Energy decomposition analysis. Wiley Interdiscip Rev Comput Mol Sci 2:43–62
Grabowski SJ (2004) Hydrogen bonding strength—measures based on geometric and topological parameters. J Phys Org Chem 17:18–31
Duarte DJR, Angelina EL, Peruchena NM (2012) On the strength of the halogen bonds: mutual penetration, atomic quadrupole moment and Laplacian distribution of the charge density analyses. Comput Theor Chem 998:164–172
Bader RFW (1990) Atoms in molecules. A quantum theory. Clarendon, Oxford
Popelier P (ed) (2000) Atoms in molecules: an introduction. Prentice-Hall, Harlow
Matta CF, Boyd RJ (2007) The quantum theory of atoms in molecules: from solid state to DNA and Drug design. Wiley-VCH, Weinheim
Shahi A, Arunan E (2014) Hydrogen bonding, halogen bonding and lithium bonding: an atoms in molecules and natural bond orbital perspective towards conservation of total bond order, inter- and intra-molecular. Phys Chem Chem Phys 16:22935–22952
Yáñez M, Sanz P, Mó O, Alkorta I, Elguero J (2009) Beryllium bonds, do they exist? J Chem Theory Comput 5:2763–2771
Del Bene JE, Alkorta I, Sanchez-Sanz G, Elguero J (2011) Structures, energies, bonding, and NMR properties of Pnicogen complexes H2XP:NXH2 (X ≥ H, CH3, NH2, OH, F, Cl). J Phys Chem A 115:13724–13731
Esrafili MD, Mohammadian-Sabet F (2015) Does single-electron chalcogen bond exist? Some theoretical insights. J Mol Model 21:65
Angelina EL, Duarte DJR, Peruchena NM (2013) Is the decrease of the total electron energy density a covalence indicator in hydrogen and halogen bonds? J Mol Model 19:2097–2106
Duarte DJR, Angelina EL, Peruchena NM (2014) Physical meaning of the QTAIM topological parameters in hydrogen bonding. J Mol Model 20:2510
Bader RFW, Hernadez-Trujillo J (2010) Chemical bonding: from Lewis to atoms in molecules. J Comput Chem 31:2967–2970
Bader RFW, Johnson S, Tang TH, Popelier PLA (1996) The electron pair. J Phys Chem 100:15398–15415
Gillespie RJ (2008) Fifty years of the VSEPR model. Coord Chem Rev 252:1315–1327
Duarte DJR, Sosa GL, Peruchena NM (2013) Nature of halogen bonding. A study based on the topological analysis of the Laplacian of the electron charge density and an energy decomposition analysis. J Mol Model 19:2035–2041
Duarte DJR, Peruchena NM, Alkorta I (2015) Double hole–lump interaction between halogen atoms. J Phys Chem A 119:3746–3752
Acknowledgments
D.J.R. Duarte gratefully acknowledges the Secretaría General de Ciencia y Técnica de la Universidad Nacional del Nordeste (SEGCYT UNNE).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Duarte, D.J.R., Miranda, M.S., da Silva, J.C.G.E. et al. A theoretical study of the strong interactions between carbon dioxide and OH+ and NH2 + products resulting from protonation of 1,2-dioxirane-3-one and 1,2-oxaziridine-3-one, respectively. Struct Chem 27, 1743–1751 (2016). https://doi.org/10.1007/s11224-016-0794-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-016-0794-x