
Abstract
A series of interatomic interactions interpretable as halogen bonds involving I…I, I…O, and I…C(π), as well as the noncovalent interactions I…H and O…O, were observed in the crystal structures of trans-1,2-diiodoolefins dimers according to ab initio calculations and the quantum theory of “atoms in molecules” (QTAIM) method. The interplay between each type of halogen bond and other noncovalent interactions was studied systematically in terms of bond length, electrostatic potential, and interaction energy, which are calculated via ab initio methods at the B3LYP-D3/6-311++G(d,p) and B3LYP-D3/def2-TZVP levels of theory. Characteristics and nature of the halogen bonds and other noncovalent interactions, including the topological properties of the electron density, the charge transfer, and their strengthening or weakening, were analyzed by means of both QTAIM and “natural bond order” (NBO). These computational methods provide additional insight into observed intermolecular interactions and are utilized to explain the differences seen in the crystal structures.

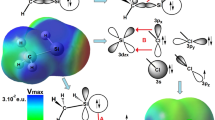
The contour map presents the regions of electronic concentration and depletion along each bond in one dimer. The blue points denote the BCPs. The blue lines denote positive Laplacian of electron density, which indicate the ionic interactions, van der Waals or intermolecular interactions, and the red lines denote negative Laplacian of electron density which indicate the covalent bonds.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Halogen bonds have recently been extensively studied as they play an essential role in medicinal chemistry [1, 2], molecular recognition [3], material science [4, 5], and crystal engineering [6,7,8]. Halogen bonds are noncovalent interactions between an electrophilic region of a halogen and a nucleophile [9, 10]. These interactions actually cover a range from very weak to fairly strong depending on the nature of the halogen, its environment, and the nature of the negative site. Halogen bonds can schematically be depicted R–X⋯Y, where X is a halogen (typically I, Br, Cl, and rarely F), and Y is defined as the halogen bond donor [11, 12]. X acts as an electron acceptor for the interaction, whereas Y is typically an electron-rich O, N, S, or Y–donor functional group (e.g., π–electron systems or aromatic surfaces). A halogen atom may be covalently bound to one or several atoms and can additionally form one or several halogen bonds simultaneously [9, 13, 14].
Halogen bonds have similarities with hydrogen bonds and involve the same mechanisms [15]. In 2008, Metrangolo et al. summarized the similarities and differences between halogen bonding and hydrogen bonding complexes [6]. Kirk et al. investigated the competition between hydrogen bonding and halogen bonding for the (Y = Cl, Br, I, At)/halogenabenzene/NH3 systems and concluded that hydrogen bonding has an advantage when the halogen is Cl, while halogen bonds tend to be formed when the halogen atom Y = I [16]. Halogen bonds have been observed in crystal structures containing halogen atoms [17]. Due to their geometric properties, halogen bonds are considered efficient tools in designing the structures of crystals [6, 18]. The concept of halogen bonds in crystal engineering attracts increasing attention as they can be pivotal in the stability of crystals [19, 20].
Although halogen bonding was first observed two centuries ago [11], the fundamentals of its nature and its potential applications in crystal engineering have remained unexplored until recently. In most cases reported in the literature, halogen bonds were studied in cocrystals between two different compounds, one of which being the halogen bond donor and the other is the acceptor [17, 21]. On the basis of computational studies, this type of interaction was shown to predominantly originate from charge transfer and electrostatics [22,23,24]. Later, the interaction was also found to possess polarization and dispersion contributions. In 2014, Deepa et al. [25] carried out a theoretical study of a series of organic crystal structures containing various halogen bonds and found the strongest halogen bonds involved iodine as both halogen bond donor and acceptor. In the following year, Koskinen et al. [26] carried out a detailed study of unexpectedly strong I+…S halogen bonds in [I(2-imidazolidinethione)2]+ with the results supporting the coordinative nature of the halogen bond.
Furthermore, topological properties, vibrational frequencies, interaction energies, and charge transfer in halogen bond–containing systems have been studied using both Bader’s quantum theory of atoms in molecules (QTAIM) [27] and Weinhold’s “natural bond orbital NBO” methods. A series of important studies are through QTAIM theory [28,29,30,31]. Clark et al. [32] calculated the electrostatic potential of the series of molecules CF3X (X = F, Cl, Br, and I) and found that the three unshared pairs of electrons produced a belt of negative electrostatic potential around the central region of the X atom (except for F), leaving the outermost region positive, which designated the “σ-hole.” They also discovered that the strength of the σ-hole depends on the nature of the halogen atom. The more polarizable and the lower the electronegativity of the halogen atom, the more positive the σ-hole. Thus, the interaction strength of halogen bonds increases in the order of F < Cl < Br < I. It is the σ-hole that allows the halogen atom to form a halogen bond with a Lewis base [33] and that makes the A…X-R angle tend towards a linear configuration [15]. In 2010, Zeng et al. [34] comparatively analyzed the properties of halogen bonds and hydrogen bonds by QTAIM calculations and concluded that the two interactions were coincident in topological properties. In the following year, they analyzed halogen bonds between sulfides and dihalogen molecules and found that electrostatic interactions played an important role in these halogen bonds [35]. Grabowski [36] calculated the QTAIM characteristics of halogen bonds, dihalogen bonds, and halogen-hydride bonds. In 2018, Bauzá et al. [37] analyzed the interplay between π-hole and lone pair⋯π/X-H⋯π interactions through QTAIM calculations. In 2019, Benito et al. [38] first reported the cocrystallization of an adenine derivative which acts as a halogen bond acceptor; the calculations were carried out via DFT calculations and the QTAIM method. In 2020, Wzgarda-Raj et al. [39] investigated several observed types of halogen bonding interactions in a series of cocrystals in detail based on QTAIM. In addition to the traditional halogen bonds, Domagała et al. [40] built a model of a double (+/−)-charge-assisted halogen bridge for a set of quinuclidine-like cation derivatives and anions; these charged fragments were observed to form strong halogen bonding complexes, with interaction energies high as 100 kcal/mol.
In this work, calculations carried out on new crystal structures of trans-1,2-diiodoolefins are reported, including nine monomers and nine dimers. These crystal structures were previously synthesized by Hettstedt et al. [41] in 2015 (see Fig. 1). The purpose of this study is to investigate the characteristics and properties of halogen bonds (i.e., I…I, I…O, and I…C(π), where C(π) can be aromatic, aliphatic, or acetylenic π-systems) and other noncovalent interactions such as I…H and O…O observed in these crystal structures.

1,2-Diiodoolefines that have been studied in this work
Computational methods
Data for halogen bonds observed in crystal structures of trans-1,2-diiodoolefins reported by Hettstedt et al. [41] have been used as references for quantum chemical calculations to analyze noncovalent interactions. The structures of monomers and dimers were obtained from the Cambridge Crystal Structure Database (CCSD). Geometry optimization, molecular electrostatic potential, and interaction energy calculations were carried out using the Gaussian09 program package [42,43,44,45]. The DFT-D3 method, which is recommended in studying noncovalent interactions [46,47,48,49], was applied to the monomer and dimer optimizations. Both Kolář et al. [50] and Bauzá et al. [51] verified that the B3LYP-D3 method combined with the “def2” basis set series can be used to successfully examine halogen bonds and the properties of σ-holes. Therefore, the B3LYP-D3/6-311++G(d,p) and B3LYP-D3/def2-TZVP levels of theory were used to optimize the structures of monomers. Frequency calculations were run to be sure that the geometry was a potential energy minimum (no negative frequencies were obtained). The keyword “counterpoise” was used for the calculations of corrected interaction energies (∆E(AB)) including the inherent basis set superposition error (BSSE) [52] according to Eq. (1).
Here, E(AB) is the total energy of dimer AB and E(A) and E(B) are the energies of monomers A and B, respectively.
The electrostatic potential on the molecular surfaces of all monomers was analyzed in order to gain insight into the nature and directionality of the halogen bond interactions being considered herein. The electrostatic potentials were considered to be an outer contour of the electron density, and were cut off at the 0.001 au (electrons/bohr−3) surface, as proposed by Bader et al. [53]. The most positive value of the potentials (the local maximum) is referred to as VS, max. Natural bond orbital (NBO) calculations were performed using the NBO 3.1 program [54] as implemented in Gaussian09. The QTAIM theory was applied to find critical points and these were analyzed in terms of electron density and the Laplacian. The topological properties at the bond critical points (BCPs) of halogen bonds were computed with the program-AIMALL 2000 [55].
Results and discussion
Monomers
Geometries
In the work reported in ref. [41], trans isomers were obtained for all systems but one: acetal 7, for which a mixture of cis (7a) and trans (7b) derivatives were formed in the synthetic process. Therefore, there are in total nine monomers reported on in this study. All of their geometries were optimized at the B3LYP-D3/6-311++G(d,p) and B3LYP-D3/def2-TZVP levels of theory. Figure 2 shows the nine trans-1,2-diiodoolefin structures optimized at the B3LYP-D3/6-311++G(d,p) level of theory. The C=C bond lengths (the common part in the nine trans-1,2-diiodoolefin structures) in both the optimized and crystal structures are listed in Table 1. It can be seen that the bond lengths of the C=C in the nine monomers calculated at the B3LYP-D3/6-311++G(d,p) and B3LYP-D3/def2-TZVP levels of theory are all in good agreement with the values seen in the crystal structures. Also, there is only a marginal difference between the C=C bond lengths optimized at the two levels of theory. Therefore, considering the computational cost, the B3LYP-D3/6-311++G(d,p) level of theory was applied to carry out all molecular electrostatic potential (VS, max) calculations.

Overview over the molecular structures of the compounds. The purple atoms stand for I, the red atoms are O, the gray atoms are C, and the white ones are H
Electrostatic potentials
Figure 3 shows contour maps of the electrostatic potential for monomers 1–8. The σ-hole of the two iodine atoms in all nine monomers is positive (see the blue region in Fig. 3). The VS, max values for the two iodine atoms are listed in Table 2. The maximum VS, max value of the nine monomers is 27.30 kJ mol−1 for monomer 8. Monomers 5 and 6 had the second and third largest VS, max values of 22.87 kJ mol−1 and 22.32 kJ mol−1 respectively. The VS, max values for monomers 7(a) and 7(b) are the smallest among all monomers, which are 14.96 kJ mol−1 and 15.18 kJ mol−1, respectively. Based on a comparison of all the studied monomers, the iodine atom’s chemical environment most strongly affects the electrostatic potential of iodine’s σ-hole.

Electrostatic potentials mapped on the surface of monomer molecules electron density (0.001 e au−3). The electrostatic potential varies from negative (red) to positive (blue)
Dimer
Geometries
Six dimers of diiodoalkene were provided in the experimental supplementary data of ref. [41], including dimers 2, 3, 5, 6, 7b, and 8. Each of these dimer structures was partially geometry-optimized at the B3LYP-D3/6-311++G(d,p) and B3LYP-D3/def2-TZVP levels of theory. Because the dimers were too large to be fully geometry-optimized, partial atoms or groups were fixed to ensure optimization is successful, with the fixed atoms or groups chosen to be far away from the locations of the noncovalent bonds (e.g., I…I, I…O, I…C(π), I…H and O…O) formed. The details of fixed atoms or groups for each dimer are listed in the Supporting Information. The geometries optimized at the B3LYP-D3/6-311++G(d,p) level of theory are shown in Fig. 4. The noncovalent bond lengths of each dimer, including the values in crystal structures and the values calculated at the B3LYP-D3/6-311++G(d,p) and B3LYP-D3/def2-TZVP levels of theory, are listed in Table 3. The deviations between the crystal structure values and the values calculated at the two levels of theory are quite small. The average deviation between the noncovalent bond lengths optimized at the B3LYP-D3/6-311++G(d,p) level of theory and the values in crystal structures are 0.123 Å, and the corresponding deviation is 0.101 Å between the values optimized at the B3LYP-D3/def2-TZVP levels of theory and the crystal structure values. Again, the noncovalent bond lengths optimized at these two levels of theory are similar. Because the electron charge distribution of the halogen atom is anisotropic, the halogen can act both as the Lewis acid and as the Lewis base [56,57,58,59]. This is why dihalogen bonds are possible in the 6 dimers.

Organic crystal structures of the dimers concerned with intermolecular interactions given by dotted line
Interaction energies
There are five different types of intermolecular noncovalent interactions in the six diiodoalkene dimers (see Fig. 4): I…I, I…O, I…C(π), I…H, and O…O. Interaction energy is an important measure of the strength of an intermolecular interaction. The interaction energy in the dimers can be regarded as the energy difference between the dimer and the monomers as captured via Eq. (1). Table 4 summarizes the interaction energies of the halogen bonds in the six dimers. All results were corrected for BSSE by using counterpoise methods. Naturally, the effect of BSSE correction is prominent for halogen bonds. Calculations are carried out for crystal and optimized dimers, respectively, to explore whether if it is necessary to optimize the crystal structure geometries.
Of all the dimers shown in Fig. 4, dimer 2 possesses the smallest number of intermolecular noncovalent bonds: two halogen bonds of I…I and I…H. Its interaction energies calculated at the B3LYP-D3/6-311++G(d,p) level of theory are − 9.58 kJ mol−1 and − 11.67 kJ mol−1 respectively for crystal and optimized geometries; and the respective values are − 11.7 kJ mol−1 and − 12.57 kJ mol−1 at the B3LYP-D3/def2-TZVP level of theory. Dimer 3 contains three halogen bonds of I…I, I…C(π), I2…C27(π), and I17…C12(π). Dimer 3 has the second highest interaction energy (Table 4) due to the two strong I…C(π) noncovalent bonds. Similarly, the I…C(π) noncovalent bond was also found in dimers 5 and 6; I23…C11(π) in dimer 5 and I1…C33(π) in dimer 6. Moreover, the halogen bond of type I…O was found in dimers 5 (I2…O25) and 6 (I27…O3). The respective interaction energies computed at the two levels of theory are − 29.44 kJ mol−1 and − 32.02 kJ mol−1 for crystal dimer 5 and − 34.19 kJ mol−1 and − 31.79 kJ mol−1 for the geometry-optimized dimer 5. Equally, the respective interaction energies computed at the two levels of theory are − 24.59 kJ mol−1 and − 26.45 kJ mol−1 for crystal dimer 6 and − 29.86 kJ mol−1 and − 27.00 kJ mol−1 for the optimized dimer. The interaction energy in dimer 7b is also very high because there are five halogen bonds in dimer 7b: one I…I, one I…C(π), and three I…H, the details are shown in Fig. 4 and Table 4. The highest interaction energy occurs in dimer 8, and the corresponding interaction energies computed at the two levels of theory are − 36.75 kJ mol−1 and − 39.09 kJ mol−1, respectively, for crystal dimer 8, and are − 44.55 kJ mol−1 and − 39.95 kJ mol−1 for optimized dimer 8. This dimer has three halogen bonds of I…O, I…C(π), and I…H and one O…O noncovalent bond. To summarize, in Table 4, both the interaction energies (E_Int) and the BSSE energies (E_BSSE) for crystal dimers calculated at the two levels of theory are very close to the values for the optimized dimers. Therefore, the properties of halogen bonds can be calculated directly using the crystal structures without geometry optimization.
Natural bond orbital analysis
To better understand the intermolecular interactions, natural bond orbital (NBO) analysis was carried out to characterize the weak interactions. Formation of complexes involving noncovalent bonds is associated with an orbital interaction between the bonding orbital in the electron donor and the antibonding orbital in the electron acceptor. Table 5 lists the second-order perturbation energy (E(2)) and the charge transfer (∆q) obtained by NBO analysis. Both E(2) and ∆q represent the transfer from one molecule (donor) to the other molecule (acceptor) in the six dimers. Owing to the time-consuming nature of the B3LYP-D3/def2-TZVP level of theory, all NBO calculations were carried out at the B3LYP-D3/6-311++G(d,p) level of theory. The second-order perturbation energy represents the degree of charge transfer from the bonding orbital to the antibonding orbital, which is the degree of electron delocalization. Ultimately, the second-order perturbation energy allows us to quantitatively evaluate the charge transfer due to the formation of the halogen bond.
The results listed in Table 5 show that there is a positive relationship between the second-order perturbation energy E(2) and the charge transfer ∆q in the studied systems. Due to the centrosymmetry of dimer 3, the charge transfer from one monomer to another in both the crystal and optimized dimers is zero. Figure 5 presents the strong linear relationship between ∆q and E(2) with the exception of 7b. Dimer 7b forms more I…H halogen bonds compared to other dimers. The linear relationship between ∆q and E(2) indicates that charge transfer is an important factor in the noncovalent bonds seen in crystal systems.

The charge transfer (au) vs. second-order perturbation energy (kJ mol−1)
Topological properties
The topological and energetic properties at the BCPs of the interactions between the two molecules in the six dimers were analyzed by comparing the following parameters; the electron density ρ(b), the Laplacian of electron density ∇2ρ(b), the kinetic electron energy density Gb, the potential electron energy density Vb, the sum of Gb and Vb (Hb), and the ratio of Gb to Vb (− Gb/Vb). The results are collected in Table 6. A positive value of ∇2ρ(b) implies an interaction between closed shell complexes; ionic interaction, van der Waals force, or hydrogen bonding, while a negative value of ∇2ρ(b) indicates a shared interaction as in a covalent bond [60]. Rozas’s study [61] concluded that ∇2ρ(b) and Hb may be useful in characterization of the strength of interactions. This means that for weak A…B interactions, where ∇2ρ(b) > 0 and Hb > 0, the interactions are mainly electrostatic; for medium strength interactions, where∇2ρ(b) > 0 and Hb < 0, the interactions are partly covalent in nature, while strong interactions show ∇2ρ(b) < 0 and Hb < 0; these are characteristically covalent. Measures of the covalency in noncovalent interactions include the kinetic electron energy density Gc (positive), the potential electron energy density Vc (negative), and the ratio − Gc/Vc. Values of − Gc/Vc > 1 generally indicate a noncovalent interaction, whereas when − Gc/Vc is < 1, the interaction is covalent in nature. For the dimers investigated here (in Table 6), all ∇2ρ(b) values are positive, the Hc values are positive, and the − Gc/Vc values are greater than 1. This means that these interactions belong to weak interactions of an electrostatic nature.
The electron density, ρ(b), at the bond critical point is used to describe the strength of a bond, where the larger the value of ρ(b), the stronger the bond. In dimer 2 (see Fig. 5), the intermolecular halogen bond of I1…I18 is the strongest with ρ(b) = 0.008 au. In dimer 3, the halogen bonds of I2…C27(π) and I17…C12(π)) were strongest; their ρ(b) was 0.007 au. In dimer 5, the I2…O25 bond was the strongest with a ρ(b) value of 0.01 au. In dimer 6, the I1…C33(π) interaction was the strongest and its ρ(b) was 0.008 au. In dimer 7b, the I35…C16(π) bond was the strongest and its ρ(b) was 0.009 au. In dimer 8, the I12…O33 was the strongest and its ρ(b) was 0.015 au. The results concluded from the topological measures of interaction properties are coincident with the experimental results reported in Table 3.
Figure 6 presents the regions of electronic concentration and depletion along each bond in the six dimers; both the contour maps (left) and relief maps (right) of ∇2ρ(b) are shown. The blue points denote BCPs. The blue lines denote positive Laplacian of electron density, which indicate interactions of an ionic character (e.g., van der Waals or intermolecular interactions), and the red lines denote negative Laplacian of electron density which indicate covalent bonds. The corresponding values of the Laplacian of electron density are listed in Table 6. Relief maps provide an intuitive view of the Laplacian of electron density, the curves above the plane show the positive Laplacian of electron density, and the curves below the plane show the negative Laplacian of electron density.

Molecular contour maps (left) and relief maps (right) of Laplacian of the electron density for the six dimers
Figure 7 shows the bond lengths of halogen bonds (e.g., I…I, I…O, I…C(π) in Table 3) and their relationship with electron densities ρ(b). For the halogen bonds, the lower the electron density, the longer the bond length. Therefore, the electron density might serve as a rough measure to estimate the strength of halogen bonding interactions.

Relationship between the halogen bonds lengths (Å) and the electron densities at the BCPs (au)
Conclusions
In this study, ab initio and QTAIM studies were performed to explore the nature of halogen bonds and some other noncovalent bonds in a series of crystal structure geometries of trans-1,2-diiodoolefins. The ab initio calculations were carried out at the B3LYP-D3/6-311++G(d,p) and B3LYP-D3/def2-TZVP levels of theory for both crystal and optimized monomers and dimers. Firstly, the calculation results show deviations between the two levels of theory to be quite small. Secondly, the computational values for optimized structures are close to the values for crystal structures.
The reported results provide important information concerning the physical chemistry of these materials. In particular, the crystal geometrical architecture and intermolecular bonding properties were shown to be reproducible with the calculations. The interaction energy and electron density appear to be appropriate tools to judge the stabilities of the crystal structures. Quantification of the noncovalent bonding energy between the molecules in dimers was evaluated both on the crystal and optimized structures, and the interaction energies were within 11.67 kJ mol−1 and 44.55 kJ mol−1 with B3LYP-D3/6-311++G(d,p). The intermolecular interactions responsible for the formation of the dimers are weak to moderate in strength, these interactions were clearly of enough local significance to guide the solid state crystallization process. Moreover, for the halogen bonds of type I…I, I…O, and I…C(π), there is a strong linear relationship between the electron densities ρ(b) and the bond lengths. This confirms the relationships between electron density and the stability of halogen bonds.
References
Shing HP (2017) Halogen bonding in medicinal chemistry: from observation to prediction. Future Med Chem 9:637–640
Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM (2013) Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Mol Model 56:1363–1388
Jungbauer SH, Bulfield D, Kniep F, Lehmann CW, Herdtweck E, Huber SM (2014) Toward molecular recognition: three-point halogen bonding in the solid state and in solution. J Am Chem Soc 136:16740–16743
Parlane FGL, Mustoe C, Kellett CW, Simon SJ, Swords WB, Meyer GJ, Kennepohl P, Berlinguette CP (2017) Spectroscopic detection of halogen bonding resolves dye regeneration in the dye-sensitized solar cell. Nat Commun 8:1761–1768
Priimagi A, Cavallo G, Metrangolo P, Resati G (2013) The halogen bond in the design of functional supramolecular materials: recent advances. Acc Chem Res 46:2686–2695
Metrangolo P, Resnati G, Pilati T (2008) Halogen bonding in crystal engineering. Struct Bond 126:105–136
Raatikainen K, Rissanen K (2012) Breathing molecular crystals: halogen- and hydrogen-bonded porous molecular crystals with solvent induced adaptation of the nanosized channels. Chem Sci 3:1235–1239
Mukherjee A, Tothadi S, Gautam R (2014) Desiraju, Halogen bonds in crystal engineering: like hydrogen bonds yet different. Acc Chem Res 47:2514–2524
Desiraju GR, Ho PS, Kloo L, Legon AC, Marquardt R, Metrangolo P, Politzer P, Resnati G, Rissanen K (2013) Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl Chem 85:1711–1713
Peter P, Jane SM (2020) Electrostatics and polarization in σ- and π-hole noncovalent interactions: an overview. Chemphyschem 21:579–588
Cavallo G, Metrangolo P, Milani R, Pilati T, Priimagi A, Resnati G, Terraneo G (2016) The halogen bond. Chem Rev 116:2478–2601
Erdelyi M (2012) Halogen bonding in solution. Chem Soc Rev 41:3547–3557
Parisini E, Metrangolo P, Pilati T, Resnati G, Terraneo G (2011) Halogen bonding in halocarbon-protein complexes: a structural survey. Chem Soc Rev 40:2267–2278
Metrangolo, Resnati G (2012) Halogen bonding: where we are and where we are going. Cryst Growth Des 12:5835–5838
Carlsson AC, Gräfenstein J, Budnjo A, Laurila JL, Bergquist J, Karim A, Kleinmaier R, Brath U, Erdélyi M (2012) Symmetric halogen bonding is preferred in solution. J Am Chem Soc 134:5706–5715
Shuman L, Tianlv X, van Mourik T, Früchtl H, Kirk SR, Jenkins S (2019) Halogen and hydrogen bonding in halogenabenzene/NH3 complexes compared using next-generation QTAIM. Molecules 24:2875–2886
Walsh RB, Padgett CW, Metrangolo P (2001) Crsytal engineering through halogen bonding: complexes of nitrogen heterocycles with organic iodides. Cryst Growth Des 1:165–175
Siram RBK, Karothu DP, Row TNG, Patil S (2013) Unique type II halogen···halogen interactions in pentafluorophenyl-appended 2, 2′-bithiazoles. Cryst Growth Des 13:1045–1049
Grabowski SJ (2013) Hydrogen and halogen bonds are ruled by the same mechanisms. Phys Chem Chem Phys 15:7249–7259
Metrangolo P, Resnati G (2008) Halogen versus hydrogen. Science 321:918–919
Cinčić D, Friščić T, Jones W (2011) Experimental and database studies of three-centered halogen bonds with bifurcated acceptors present in molecular crystals, cocrystals and salts. Cyst Eng Comm 13:3224–3231
Politzer P, Murray JS (2013) Halogen bonding: an interim discussion. Chem Phys Chem 14:278–294
Brinck T, Borrfors AN (2019) Electrostatics and polarization determine the strength of the halogen bond: a red card for charge transfer. J Mol Model 25:125–133
Clark T, Murray JS, Politzer P (2018) A perspective on quantum mechanics and chemical concepts in describing noncovalent interactions. Phys Chem Chem Phys 20:30076–30082
Deepa P, Pandiyan BV, Kolandaivel P, Hobza P (2014) Halogen bonds in crystal TTF derivatives: an ab initio quantum mechanical study. Phys Chem Chem Phys 16:2038–2047
Koskinen L, Hirva P, Kalenius E, Jääskeläinen S, Rissanen K, Haukka M (2015) Halogen bonds with coordinative nature: halogen bonding in a S–I+–S iodonium complex. Cryst Eng Comm 17:1231–1236
Bader RFW (1991) A quantum theory of molecular structure and its applications. Chem Rev 91:893–928
Foroutan-Nejad C, Shahbazian S, Marek R (2014) Toward a consistent interpretation of the QTAIM: tortuous link between chemical bonds, interactions, and bond/line paths. Chem Eur J 20:10140–10152
Grimme S, Muck-Lichtenfeld C, Erker G, Kehr G, Wang H, Beckers H, Willner H (2009) When do interacting atoms form a chemical bond? Spectroscopic measurements and theoretical analyses of dideuteriophenanthrene. Angew Chem Int Ed 48:2592–2595
Spackman MA (2015) How reliable are intermolecular interaction energies estimated from topological analysis of experimental electron densities? Cryst Growth Des 15:5624–5628
Politzer P, Murray JS (2019) A looking at bonds and bonding. Struct Chem 30:1153–1157
Clark T, Hennemann M, Murray JS, Politzer P (2007) Halogen bonding: the σ-hole. J Mol Model 13:291–296
Politzer P, Lane P, Concha MC, Ma Y, Murray JS (2007) An overview of halogen bonding. J Mol Model 13:305–311
Zeng Y, Zhang X, Li X, Zheng S, Meng L (2010) Ab initio and AIM studies on typical π-type and pseudo-π-type halogen bonds: comparison with hydrogen bonds. Int J Quantum Chem 111:3725–3740
Zhang X, Zeng Y, Li X, Meng L, Zheng S (2011) A computational study on the nature of the halogen bond between sulfides and dihalogen molecules. Struct Chem 22:567–576
Grabowski SJ (2012) QTAIM characteristics of halogen bond and related interactions. J Phys Chem A 116:1838–1845
Bauzá A, Seth SK, Frontera A (2018) Molecular electrostatic potential and “atoms-in-molecules” analyses of the interplay between π-hole and lone pair···π/X–H···π/metal···π interactions. J Comput Chem 39:458–463
Roselló Y, Benito M, Molins E, Barceló-Oliver M, Frontera A (2019) Adenine as a halogen bond acceptor: a combined experimental and DFT study. Crystals 9:224–233
Wzgarda-Raj K, Rybarczyk-Pirek AJ, Wojtulewski S, Palusiak M (2020) C—Br⋯ S halogen bonds in novel thiourea N-oxide cocrystals: analysis of energetic and QTAIM parameters. Acta Crystallogr C 76:170–176
Domagała M, Lutynska A, Palusiak M (2018) Extremely strong halogen bond. The case of a double-charge-assisted halogen bridge. J Phys Chem A 122:5484–5492
Hettstedt C, Mayer P, Karaghiosoff K (2015) Halogen bonding in the crystal structures of 1,2-diiodo alkenes. New J Chem 39:8522–8533
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7:3297–3305
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132:154104–154123
Grimme S, Ehilich S, Goerigk L (2011) Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 32:1456–1465
Frisch MJ, Truchs GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Schmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JJA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2015) Gaussian 09, Revision E.01. Gaussian, Inc., Wallingford, CT
Zhao H, Chang J, Du L (2016) Effect of hydrogen bonding on the spectroscopic properties of molecular complexes with aromatic rings as acceptors. Comput Theor Chem 1084:126–132
Zhang Q, Du L (2016) Hydrogen bonding in the carboxylic acid–aldehyde complexes. Comput Theor Chem 1078:123–128
Tang S, Zhao H, Du L (2016) Hydrogen bonding in alcohol–ethylene oxide and alcohol–ethylene sulfide complexes. RSC Adv 6:91233–91242
Bauzá A, Quinonero D, Deya PM, Frontera A (2013) Halogen bonding versus chalcogen and pnicogen bonding: a combined Cambridge structural database and theoretical study. Cryst Eng Comm 15:3137–3144
Kolář M, Hostaš J, Hobza P (2014) The strength and directionality of a halogen bond are co-determined by the magnitude and size of the σ-hole. Phys Chem Chem Phys 16:9987–9996
Bauzá A, Ramis R, Frontera A (2014) Computational study of anion recognition based on tetrel and hydrogen bonding interaction by calix[4]pyrrole derivatives. Comput Theor Chem 1038:67–70
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19:553–566
Bader RFW, Carroll MT, Cheeseman JR, Cheng C (1987) Properties of atoms in molecules: atomic volumes. J Am Chem Soc 109:7968–7979
Glendening ED, Reed AE, Carpenter JE, Weinhold F (1990) NBO 3.1, QCPE Bull 1990, 10, 58
Keith TA (2011) AIMAll, 15.09.27, TK Gristmill Software
Zordan F, Brammer L, Sherwood P (2005) Supramolecular chemistry of halogens: complementary features of inorganic (M− X) and organic (C− X ‘) halogens applied to M− X...X ‘− C halogen bond formation. J Am Chem Soc 127:5979–5989
Domagała M, Matczak P, Palusiak M (2012) Halogen bond, hydrogen bond and N⋯ C interaction–on interrelation among these three noncovalent interactions. Comput Theor Chem 998:26–33
Domagała M, Palusiak M (2014) The influence of substituent effect on noncovalent interactions in ternary complexes stabilized by hydrogen-bonding and halogen-bonding. Comput Theor Chem 1027:173–178
Domagała M, Lutyńska A, Palusiak M (2017) Halogen bond versus hydrogen bond: the many-body interactions approach. Int J Quantum Chem 117:e25348–e25356
Popelier PLA (2000) Atoms in molecules: an introduction. Prentice Hall, Pearson Education Limited, New York
Rozas I, Alkorta I, Elguero J (2000) Behaviour of ylides containing N, O and C atoms as hydrogen bond acceptors. J Am Chem Soc 122:11154–11161
Funding
This work was supported by the Fundamental Research Funds for the Central Universities (Grant No. lzujbky-2019-cd05), the National Natural Science Foundation of China (No. 81973786), and the Science Foundation of Guangxi (AA17204096, AD16380076).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 17 kb)
Rights and permissions
About this article

Cite this article
Yuan, Y., Mills, M.J.L., Li, F. et al. Halogen bonds and other noncovalent interactions in the crystal structures of trans-1,2-diiodo alkenes: an ab initio and QTAIM study. J Mol Model 26, 331 (2020). https://doi.org/10.1007/s00894-020-04591-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04591-2