
Abstract
Hydrogen peroxide (H2O2) is a mild, environmentally friendly, and versatile oxidizer. The current main production process, the anthraquinone method, has high energy consumption and pollution. However, the photocatalytic production of H2O2 is considered to be a green and sustainable process. In this paper, the research progress of photocatalytic production of H2O2 was reviewed. Firstly, the basic principle of photocatalytic production of H2O2 and the three production pathways, namely, oxygen reduction, water oxidation, and dual-channel were introduced. Then, advanced photocatalysts for H2O2 production were introduced with emphasis. From the reactants and products, several new reaction systems and improvement strategies were introduced, including the construction of a spontaneous system without a sacrifice agent, a gas–liquid–solid three-phase system, and inhibition of H2O2 decomposition. The in-situ applications of photocatalytic H2O2 production in the environmental field, including in-situ disinfection and in-situ degradation of pollutants are discussed. Finally, the research progress and future research directions in this field are summarized and prospected.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hydrogen peroxide (H2O2) is one of the most important basic chemicals [1]. It is widely used in chemical production [2], healthcare [3], and environmental treatment [4]. The global demand for hydrogen peroxide is expected to grow to about 5.7 million tons per year by 2027 [5]. As a green and efficient oxidizer, H2O2 can oxidize various substrates under mild conditions without any toxic by-products, and the only by-product is H2O. Hydrogen is a sustainable energy carrier that can solve the world’s energy problems [6]. However, H2O2 is a promising new type of liquid fuel, and the energy density of 70 wt% H2O2 (3.1 MJ L−1) is close to that of compressed H2 (2.8 MJ L−1) at 35 MPa. H2O2 is water-soluble, which makes it easy to store and transport, and is considered an ideal substitute for hydrogen [7]. In addition, H2O2 is an environmentally friendly disinfectant that can be used to inactivate disease-causing microorganisms without causing secondary pollution [8]. In modern society where epidemics are rampant, the human demand for H2O2 is increasing, and finding an efficient, clean, and sustainable H2O2 production is an important research topic.
Anthraquinone oxidation (AQ) is currently the most dominant H2O2 production process, accounting for about 95% of the total [9]. Although H2O2 is a green oxidant, the AQ process is not environmentally friendly. The whole process consists of multiple steps such as hydrogenation, hydro-oxidation, extraction, and purification, which consume a large amount of energy, organic reagents, and pressurized hydrogen, and generate a lot of wastewaters, waste gas, and solid waste [10]. Alcohol oxidation [11] and Direct synthesis [12] are considered alternative methods, but the alcohol oxidation method requires an oxygen-rich and high-pressure environment and will produce organic by-products such as aldehydes or ketones. Direct synthesis has safety hazards, requires precise control of H2/O2, fills a large amount of inert gas to prevent the explosion, and requires the use of precious metal catalysts to increase the cost of the reaction, which limits its industrial application. Therefore, there is an urgent need for an efficient, safe, and clean method to produce H2O2.
Photocatalytic production of H2O2 is a green and safe production process. It uses abundant water and oxygen in nature as raw materials, sunlight as an energy source, and semiconductors as catalysts, with almost no pollutant emission and without the use of hazardous H2. Currently, the photocatalysts used for H2O2 production include metal oxides (TiO2, ZnO, WO3, etc.), carbon-nitrides (g-C3N4), metal sulfides (In2S3, CdS, etc.) [13], bismuth-based oxides (BiVO4, etc.), metal–organic frameworks (MOFs), and covalent organic frameworks (COFs), and hydrogen-bonded organic frameworks (HOFs), etc. Although the photocatalytic production of H2O2 has a bright future, the current yield is still low and cannot meet the industrial application [14]. In addition to the low activity of the photocatalyst itself, the photocatalytic H2O2 production also faces the problems of low oxygen reduction activity and selectivity, the need for a high concentration of dissolved oxygen and expensive sacrificial agents, and the decomposition of the generated H2O2, which have constrained the development of this technology.
This paper summarizes the recent research progress of photocatalytic production of H2O2, focusing on the basic principles and pathways of photocatalytic H2O2 production, advanced photocatalysts and their modification strategies, and advanced production systems that can improve the overall efficiency and in-situ application of photocatalytic H2O2 production in environmental field from the viewpoints of reactants and products (Fig. 1). Finally, an outlook on the future research direction is presented, which inspire the design of high-performance photocatalysts and their in-situ environmental applications by considering the photocatalytic H2O2 production systems comprehensively.

Advanced systems and elements of photocatalytic H2O2 production system
Principle of photocatalytic H2O2 production
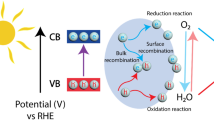
Photocatalytic H2O2 production follows the basic principle of photocatalysis, which is mainly divided into three steps (Fig. 2) [15]. (i) the photocatalyst excites electrons (e−) from the valence band (VB) to the conduction band (CB) by absorbing ultraviolet or visible light, simultaneously creating holes (h+) in the VB, thus forming electron–hole pairs (e−−h+); (ii) electrons and holes are separated and migrated to the surface of the photocatalyst, and some of them are recombined in the process; (iii) photogenerated carriers which are separated and migrated to the surface will react with chemical substances on the surface of the catalyst to selectively generate H2O2, and at the same time, participate in the oxidation and reduction reaction, while the carriers that do not participate in the reaction will recombine.

Copyright 2019 Wiley
a Photoexcitation and charge decay pathway in photocatalyst. b A schematic representation of the photocatalytic system for H2O2 production.
The photocatalytic H2O2 production occurs through three pathways: oxygen reduction (ORR), water oxidation (WOR), and dual-channel [16], as analyzed in detail below.
Oxygen reduction reaction (ORR) for H2O2 production
ORR is generally recognized as the main pathway for H2O2 production and is divided into a one-step two-electron oxygen reduction reaction (2e−ORR, O2 → H2O2) and a two-step one-electron oxygen reduction reaction (e−ORR, O2 → ·O2− → H2O2). ORR occurs at the CB of the material and holes (h+) can be consumed at the VB through the action of the electron donor (ED), thus reducing the recombination of electrons and holes and enhancing the photocatalytic activity (Fig. 3a). Specifically, h+ on the VB oxidizes water or other ED to O2 and H+ (Eq. 1) or ED+, while e− on the CB reacts with adsorbed O2 to generate H2O2. Equation (2) shows the 2e−ORR process, where O2 combines the two H+ and the two e− to directly generate H2O2 (Eq. 2). Alternatively, H2O2 can also be obtained by the e−ORR process, where O2 receives an electron to become ·O2− (Eq. 3), followed by accepting another electron and two protons to generate H2O2 (Eq. 4). Where the reduction of O2 to ·O2− is the rate-limiting step, and this reaction can only occur when the CB edge position is more negative than − 0.33 V. The reaction potential of 2e−ORR (+ 0.68 VNHE) is greater than that of e−ORR (− 0.33 VNHE), which suggests that 2e−ORR has a good thermodynamic property. However, from the reaction kinetics point of view, e−ORR requires only one electron per step, so it is easier than 2e−ORR.

Three pathways of photocatalytic H2O2 production: a oxygen reduction pathway; b water oxidation pathway; c Dual-channel approach
The e−ORR produces intermediates such as 1O2 (Eq. 5) that are unfavorable to the H2O2 yield, and the generated ·O2− may also interact with organics in solution and reduce the H2O2 yield. In addition, the 4e−ORR (+ 1.23 VNHE, Eq. 6) is thermodynamically more favorable due to its more positive electrode potential, which would reduce the selectivity of the 2e−ORR (+ 0.68 VNHE), and the 4e−ORR reaction to generate H2O detracts the H2O2 yield.
Water oxidation reaction (WOR) for H2O2 production
Similar to ORR, the water oxidation reaction (WOR) occurs at the VB of the material, which is divided into one-step two-electron water oxidation reaction (2e−WOR, H2O → H2O2, Eq. 7) and two-step single-electron water oxidation reaction (e−WOR, H2O → ·OH → H2O2). At the same time, electrons (e−) are consumed through electron acceptors (EA) at the CB, thereby reducing the recombination of electrons and holes and enhancing the photocatalytic activity (Fig. 3b). e−WOR synthesizes H2O2 in two consecutive steps using ·OH as an intermediate (Eqs. 8 and 9). The lower reaction potential of 2e−WOR means that it is more thermodynamically favorable, but e−WOR is more kinetically favorable. WOR eliminates the need for organic sacrificial agents to capture h+, which reduces production costs. However, 4e−WOR generates O2 (+ 1.23 VNHE, Eq. 10) which is thermodynamically more favorable than 2e−WOR (+ 1.76 VNHE), so 2e−WOR is often less likely to occur.
Dual channel path for H2O2 production
The synergistic dual-channel production of H2O2 by 2e−ORR and 2e−WOR is the most optimal pathway to increase the utilization efficiency of h+ and e− (Fig. 3c), with an atomic utilization of up to 100% (Eq. 11). The production of H2O2 by O2 and H2O in the absence of a sacrificial agent is thermodynamically an “uphill” reaction, whereby the solar energy is stored in H2O2. However, a fully dual-channel pathway is difficult to achieve because the 4e−WOR is thermodynamically more favorable, resulting in the 2e−WOR being less likely to occur, and the reaction produces an excess of O2.
Advanced photocatalysts for the photocatalytic H2O2 production
In general, the catalytic activity of a material depends on its structural composition, surface area, number of active sites, physical, chemical and thermal stability, good controllability and strong electron transfer ability. The following describes several photocatalysts that can be used for photocatalytic synthesis of H2O2, as well as modification methods through various principles [17].
TiO2-based photocatalysts
TiO2 has become the most widely studied photocatalyst due to its low cost, non-toxicity, and good chemical stability [18]. Since the TiO2 CB bottom (− 0.19 V vs. NHE, pH = 0) is more negative than the 2e− ORR potential (+ 0.68 V), which can generate H2O2 via ORR, it has received much attention in the field of photocatalytic H2O2 production. However, TiO2 has poor light absorption due to its large band gap (~ 3 eV) and only responds to UV light (< 400 nm), which accounts for 4–5% of the solar spectrum [19, 20]. The product H2O2 is easily adsorbed on the surface of TiO2 and decomposed, and reacts with Ti–OH group to form the Ti–OOH complex, which is electronically decomposed into Ti–OH and OH− (Ti–OOH + H+ + e− → Ti–OH + OH−) [21]. In addition, the slow oxidation of water leads to the easy accumulation of h+ at VB resulting in a high electron–hole recombination rate. Therefore, the photocatalytic activity of TiO2 is low, and the modification of TiO2 by deposition of metal nanoparticles, modification of carbon nanomaterials, and construction of heterojunctions can improve the yield of H2O2. When sunlight is the only irradiation source, the photocatalytic activity of TiO2 is not sufficient for industrial applications. In order to improve the photocatalytic performance of TiO2, many efforts have been made to develop new TiO2 materials [22].
Metal nanoparticles (NPs) with O2 affinity and high electron density can act as electron traps to trap photogenerated electrons [23], thus promoting the efficient separation of photogenerated charges and inhibiting the reduction of Ti–OOH by photogenerated electrons. Teranishi et al. deposited Au NPs on anatase TiO2 with H2O2 yields exceeding 10 mM. This was attributed to the electron transfer at the interface of Au and TiO2 to improve the charge separation efficiency, and Au NPs have an affinity for O2 as reduction sites for ORR [24]. The work function of metal nanoparticles is related to their size, and the interfacial barrier between metal nanoparticles and TiO2 can be adjusted by changing the size to promote photogenerated charge generation. Kim et al. deposited Au on porous TiO2 membranes by physical vapor deposition, and the millimolar level of H2O2 could be rapidly produced in a few minutes. Au NPs of different sizes formed a potential gradient with TiO2 to reduce the interfacial potential between the small-sized Au and TiO2 interface barrier (Fig. 4a), and the porous film facilitates the scattering of UV light and promotes the production of more photogenerated electrons and holes (Fig. 4b) [25]. However, it has been reported that H2O2 is strongly adsorbed and reduced by the captured electrons on Au NP [26], and the modulation of Schottky barriers and electron density through the formation of alloys is beneficial to increase the final yield of H2O2. Tsukamoto et al. utilized Au–Ag/TiO2 to efficiently promote the formation of H2O2 and inhibit its decomposition. Compared with Au/TiO2, the appropriately lowered Schottky barrier height of Au–Ag/TiO2 promotes effective charge separation and facilitates electron transfer from TiO2 to Au–Ag (Fig. 4c). In addition, the alloy leads to an increase in the electronegativity of Au, and the transfer of electrons from Ag to Au increases the electron density of Au and thus promotes the 2e−ORR. Meanwhile, H2O2 is more easily adsorbed by Ag in the alloy, whereas the positively charged Ag is inactive for the reduction of H2O2 (Fig. 4d) [27]. Promoting charge transfer by introducing electron-rich organic ligands is beneficial to increase the electron density of metal nanoparticles and improve the yield and selectivity of H2O2. Chu et al. modulated the electron density of Pd by ligating Pd NP deposited on TiO2 with surface amine groups, and the yield of H2O2 was 10 times higher than that of Pd/TiO2. The TiO2/Pd Schottky barriers are lower than those of TiO2/H2O thus forming an asymmetric electric field, and the band edge shift drives electron transfer to Pd, the introduction of amine group coordination further reduces the Schottky barrier and enhances the charge separation. The amine group and Pd attract protons to migrate to the surface of TiO2 to promote the formation of peroxides (Fig. 4e), which improves the selectivity of the ORR to generate H2O2 [28].

Copyright 2019 American Chemical Society. c Schematic energy-band diagrams for Au/TiO2, Ag/TiO2, and Au–Ag/TiO2 heterojunction. d Mechanism for photocatalytic production of H2O2 on Au–Ag/TiO2 catalyst [27]. Copyright 2012 American Chemical Society. e Preparation process and surface oxygen reduction mechanism of Pd/APTMS/TiO2 [28]. Copyright 2019 American Chemical Society
a Size effect of the Au NPs-loaded TiO2 on H2O2 generation. b UV light scattering in porous TiO2 films [25].
Photocatalytic intermediate water decomposition (PIWS) (2H2O → H2 + H2O2) is considered a promising method as it can obtain both H2 and H2O2 at the same time, and H2 gas and H2O2 liquid can be separated automatically. Piao’s team used in-situ photodeposition of Pt/TiO2 photodegraded water to obtain a high yield of H2O2 and H2 in pure water, with the yield of H2O2 (5096 µmol g−1 h−1) was 1.9 times that of the commercial anatase. The anatase TiO2 VB position (2.97 eV) provides sufficient oxidation potential for WOR production of H2O2 [29]. The team then loaded Pt onto porous platelet titanite TiO2 nanoflutes, and the H2O2 yield was further increased to 8.2 mmol h−1 g−1. The Pt NP was uniformly distributed on the TiO2 surface to promote charge separation. The abundant hydroxyl groups on the surface promote the adsorption of water, and the adsorbed water provides protons for the TiO2 oxygen sites to form *OH, and the two *OH combine to form H2O2 [30]. Sun et al. developed a new titanium dioxide/single layer Ti3C2Tx/MXene/Au composite material for photocatalytic synthesis of H2O2. The optimal addition ratios of MXene and Au are 0.4% and 15%, respectively, which help to separate and transfer photogenerated electron–hole pairs and inhibit carrier recombination, thereby improving the production efficiency of H2O2.
Compared with expensive metal-loaded modifications, the modification of TiO2 with carbon-containing materials such as graphene and Ti3C2 can reduce the synthesis cost while increasing light absorption and improving the charge transfer. Zero-dimensional quantum dots (QDs) are often used as photosensitizers to extend the light absorption range and as co-catalysts to promote photogenerated charge separation. Zheng et al. found that sulfur-nitrogen co-doped graphene quantum dots (SN-GQD) modification significantly red-shifted the light absorption edge (Fig. 5a), positively charged S and C in the quantum dots enhanced the adsorption of O2 and the formation of internal peroxides, and the negatively charged N and C capture protons to promote the proton-coupled electron transfer (PCET) process (Fig. 5b) [31]. Two-dimensional reduced graphene oxide (rGO) has a lower Fermi energy level than the CB edge of TiO2, so it is often used as an electronic mediator for facilitating the electron transfer of TiO2. Moon et al. found that rGO/TiO2 has a higher H2O2 generating activity than that of TiO2, and the in-situ generation of CoPi on rGO/TiO2 further improves the performance, which is attributed to the promotion of WOR by CoPi (Fig. 5c) [32]. Zeng et al. developed the Z-scheme heterojunction TiO2/rGO/WO3. rGO, as a mediator for interfacial electron transfer, promoted the migration of CB e− from WO3 to TiO2, leading to electron enrichment of TiO2 to promote ORR [33]. Due to the fast excitation or vibrational excursion of hot electrons in graphene (0.4–3 ps), it is difficult to trigger the ORR efficiently. Hu et al. found that constructing a Schottky junction at the interface of graphene and TiO2 promotes the rapid injection of excited hot electrons from graphene into TiO2, which significantly improves the charge carrier lifetime (0.5 ps → 4.0 ns) (Fig. 5d). The light response range of TiO2/Gr has been extended from the original visible light region to 800 nm, and H2O2 can be efficiently produced by visible light or even near-infrared light (Fig. 5e) [34]. By adjusting the Schottky barrier of graphene/rutile TiO2, the yields of H2O2 under visible and near-infrared light were further improved [35]. Ti3C2 is a typical MXene, which can be used as a co-catalyst to promote carrier separation and migration in combination with TiO2. Chen et al. synthesized the H2O2 yield of Ti3C2/TiO2 under UV light is 21 times higher than that of P25, which is uniformly distributed on the surface of Ti3C2 nanosheets, and the strong interfacial interactions induce the transfer of electrons from P25 to the surface of Ti3C2, which prevents the formation of Ti–OH and thus inhibits the decomposition of H2O2. In addition, Ti3C2 significantly increases the light absorption intensity (Fig. 5f), reduces the complexation rate of photogenerated carriers, and accelerates the separation and transfer of photogenerated charges [36].

Copyright 2018 Elsevier. c Linear sweep voltammetry with the electrodes of TiO2, TiO2/CoPi, rGO/TiO2, and rGO/TiO2/CoPi [32]. Copyright 2014 Royal Society of Chemistry. d Transient PL spectra of TiO2/Gr samples at 460 nm excitation wavelength. e Uv–vis absorption of TiO2/Gr and TiO2 [34]. Copyright 2022 Wiley. f UV–vis DRS spectra of P25, Ti3C2 MXene, and their composites [36]. Copyright 2021 Elsevier
a UV–vis DRS of TiO2 and the three GQDs modified TiO2, inset: the energy of absorbed light for TiO2 and SN-GQD/TiO2; b Mechanism for H2O2 formation on the photoactivated SN-GQD/TiO2 [31].
A composite of TiO2 with another semiconductor to form a heterojunction is a common modification strategy, which can effectively promote charge separation and transfer. Feng et al. found that Au-modified Bi2O3-TiO2 Type-II heterojunction can effectively promote charge separation by retaining the electrons and holes on the CB of Bi2O3 and on the VB of TiO2 under the irradiation of UV–visible light [37]. Z-scheme heterojunction not only achieves spatial separation of oxidation and reduction sites but also maintains strong oxidation and reduction capabilities. Behera et al. found that heterojunctions constructed of TiO2 (n-type semiconductor) and B-doped g-C3N4 (BCN, p-type semiconductor) follow the Z-scheme electron transfer mechanism, with high conductivity and low electron–hole complexation rate, and the highest generation rate of H2O2 under visible light is 110 μmol h−1. The excited state e− in TiO2 migrates into the VB of BCN, the CB e− of BCN reduces O2 to generate H2O2, and h+ in the VB of TiO2 oxidizes phenol to CO2 and H2O (Fig. 6a, b). Simultaneous occurrence of the Z-scheme and p-n heterojunction charge transfer mechanisms decreases electron–hole complexation rate and enhances charge separation and migration [38]. When the two semiconductors have an interlaced band configuration, the resulting heterojunction can be divided into Type-II heterojunction and Z-scheme heterojunction. However, there are many types of Z-scheme heterojunction and the concept is fuzzy, which is easy to cause confusion. S-scheme heterojunction is a new concept which is based on Type-II heterojunction and Z-scheme heterojunction to describe the photocatalytic mechanism more clearly and vividly. Additionally, the relative positions of the original Fermi levels of the two semiconductors before contact differ between Z-scheme heterojunction and S-scheme heterojunction. For Z-scheme heterojunction, the Fermi levels of the two semiconductor materials are close, indicating that their work functions are similar. In contrast, for S-scheme heterojunction, there is a larger difference in the Fermi level positions of the two semiconductor materials, meaning there is a greater difference in their work functions. This difference in energy levels results in a variation in their band structures when forming heterojunctions. In the S-scheme heterojunction, the band structure of the two semiconductor materials presents an arrangement that is close to an “S”. The S-scheme heterojunction is an emerging heterostructure consisting of oxidizing photocatalysts (OP) and reducing photocatalysts (RP) with staggered energy band structures, which can retain photogenerated carriers with superior redox capabilities. In2S3 has a narrow bandgap of 2.0–2.3 eV and has been considered as a potential material to form heterojunctions with TiO2. Yang et al. synthesized S-scheme heterojunction TiO2/In2S3 with TiO2 nanofibers (NFs) as the core and ultrathin In2S3 nanosheets (NSs) as the shell. WOR and ORR occurred on the TiO2 and In2S3 sides, respectively, and the H2O2 yields were more than 5 times those of pristine TiO2. When TiO2 is in close contact with In2S3, e− transfers from In2S3 to TiO2 at lower Fermi levels, forming an interface built-in electric field (BIEF) from In2S3 to TiO2. The energy band edge of TiO2 bends downward due to the accumulation of electrons, whereas that of In2S3 bends upward due to the loss of electrons. Upon photoexcitation e− is again reverse transferred from TiO2 back to In2S3, and TiO2 CB e− spontaneously complexes with In2S3 VB h+ (Fig. 6c). As a result, electrons and holes with superior redox capacity are retained in the CB of In2S3 and VB of TiO2, respectively [39]. Polydopamine (PDA) has a π-conjugated structure and abundant quinone groups that can accept and give electrons and facilitate electron transfer, so PDA is widely used to construct S-scheme heterojunctions. Wang et al. designed an antiproteinite TiO2/PDA (TP0.5) that follows the S-scheme charge transfer mechanism. The anti-opal structure provided a large surface area and enhanced light scattering. Due to the higher Fermi energy level of PDA, free electrons are transferred from PDA to TiO2, a built-in electric field (BIEF) is formed and bent at the interface, and the electrons in the BIEF-driven CB of TiO2 are rapidly transferred (~ 5 ps) to the highest occupied molecular orbital (HOMO) of PDA, which is subsequently photoexcited to the lowest unoccupied molecular orbital (LUMO) of PDA, while the holes stay in the VB of TiO2 (Fig. 6d). The PDA has electron storage and charge/discharge properties, accumulating electrons under light exposure, and the accumulated electrons flow back from the PDA into the TiO2 after stopping light exposure. In addition, PDA forms a coating on the surface of TiO2, which effectively inhibits the decomposition of H2O2 [40]. Jiang et al. prepared an S-scheme heterojunction photocatalyst by coupling TiO2 with 3D-ordered macroporous sulfur-doped graphite carbon nitride (3DOM SCN/T) using the electrostatic self-assembly method. The H2O2 yield of the optimized photocatalyst is 2128 μmol h−1g−1 without adding a hole scavenger. This remarkable performance is due to the synergy of the 3DOM framework and the S-scheme heterojunction. The former enhances the light-trapping ability and provides abundant active sites for surface reactions, while the latter promotes the spatial separation of photogenic carriers and improves the redox ability [41]. Elika et al. coupled TiO2 nanotubes (NT) with a wide band gap to a 3D flower-like ZnIn2S4 with a narrow band gap to form an S-scheme heterojunction. Using ZnIn2S4 as a reduction photocatalyst and TiO2 NT as an oxidation photocatalyst, efficient carrier separation and enhancement of photocatalytic activity were realized [42] (Table 1).

Copyright 2021 Royal Society of Chemistry. c S-scheme photocatalytic mechanism in TiO2/In2S3 heterojunction [39]. Copyright 2023 Springer. d S-scheme charge transfer mechanism of TiO2 and PDA [40] Copyright 2022 American Chemical society
Charge-transfer mechanism of BCN/TiO2 photocatalyst under visible light a p-n heterojunction mechanism and b Z-scheme charge-transfer pathway of double charge [38].
g-C3N4-based photocatalysts
Graphitized carbon nitride (g-C3N4) is the most widely studied nonmetallic semiconductor, where C and N are hybridized with sp2 to form π-conjugated bonds, and most of them have a stacked two-dimensional structure with C6N7 as the structural unit. g-C3N4 is considered a promising photocatalyst because of its visible-light responsiveness (bandgap of about 2.7 eV), special band structure, and stable chemical structure. The 2e−ORR selectivity of g-C3N4 is high because the 1,4-endoperoxide to which O2 is reduced on g-C3N4 can inhibit the formation of the e−ORR intermediate *OOH and promote the two-electron reduction of O2 [43]. However, the layered stacking structure, insufficient ability to capture visible light, and high electron–hole complexation rate constrains their activity for the production of H2O2. Morphology control, defect control, elemental doping, and construction of heterostructures can be used to modify g-C3N4, and the use of multiple modifications can lead to photocatalysts with better performance.
Increasing the specific surface area by shape modulation can provide more potential reaction sites and diffusion channels, which can help accelerate the mass transfer. Shiraishi et al. synthesized mesoporous g-C3N4 by thermal polymerization using SiO2 as a hard template and exhibited higher H2O2 generating activity than the nonporous g-C3N4. However, H2O2 yield decreased when the surface area of mesoporous g-C3N4 was larger than 228 m2 g−1. The authors suggested that the primary amine defects on the surface of the mesopore were the active site of the 4e−ORR and accelerated H2O2 decomposition [44]. Wang et al. synthesized g-C3N4 aerogel by sol–gel method, and the H2O2 precipitation rate under visible light irradiation was much higher than that of pure g-C3N4. The porous aerogel structure can rapidly transport reactants and products through the pores, and it has a more effective photogenerated carrier separation efficiency under visible light excitation, which is conducive to the formation of 1,4-endoperoxides and thus selectively promotes the 2e−ORR [45]. Ultrathin two-dimensional structures (< 10 nm) such as ultrathin nanosheets often have fully exposed active sites and more effective charge separation efficiency [24]. Ultrathin carbon nitride nanoplates (m-CNNP) prepared by Liu et al. using glucose-assisted mechanical layering had thicknesses and average lateral dimensions of 2.98 and 100.4 nm, with a specific surface area (89.5 m2/g) that was 9 times higher than that of the original graphitized carbon nitride (GCN), which provided more potential reactive sites and altered the pathway for the production of H2O2. H2O2 is produced mainly by 2e−ORR under visible light irradiation, and the yield is increased by four times (Fig. 7) [46]. The H2O2 generating activity of phosphate-doped ultra-thin hierarchical mesoporous carbon nitride nanosheets synthesized by our research group is 7 times that of non-porous carbon nitride. The special morphology of the coexistence of mesopores and macropores not only provides abundant reactant sites and diffusion channels but also shortens the charge transfer distance to facilitate carrier transfer to the interface. After the introduction of phosphorus, the charge is redistributed, and P replaces part of the C atoms, resulting in carbon defects that trap electrons and thus inhibit photogenerated carrier complexation, improving the photoelectric performance. In addition, P doping induces a positive shift of the CB, and the generation pathway of H2O2 is transformed from e−ORR to 2e−ORR [47]. Xu et al. constructed a hollow core–shell OCN@In2S3 composite photocatalyst by growing In2S3 ultra-thin nanosheets on the surface of O-doped hollow g-C3N4 nanospheres by a two-step hydrothermal method. The hollow structure provides a high specific surface area and enhanced light absorption. O doping increases the number of active sites, and heterojunctions promote the rapid separation and transfer of photogenerated carriers. The synthesized OCN@In2S3 maintained a high selectivity of double electron ORR (n = 1.67) and inhibited the recombination of photogenerated carriers during the catalytic reaction. Under visible light irradiation, the H2O2 yield of OCN@In2S3 reaches 632.5 μmol h−1 g−1, which is 5.7 times that of g-C3N4 and 12.3 times that of In2S3, which is also higher than that of most g-C3N4-based photocatalysts [48].

Copyright 2021 Elsevier
The size statistics of a thickness and b lateral size over m-CNNP. c ESR spectra of DMPO-·O2− in isopropanol and d the effect of reactive species during the H2O2 production by GCN and m-CNNP [46].
Proper introduction of defects is not only conducive to the inhibition of photogenerated carrier complexation, forming active centers to promote redox reactions but also forming intermediate gap states to change the electronic energy band structure and expand the visible light absorption range. The introduction of CB and nitrogen vacancies are two common defect engineering strategies. By forming carbon vacancy through thermal annealing in an argon gas stream, Li et al. improved the photogenerated carrier separation efficiency and expanded the visible light region, and the H2O2 yield was much higher than that of g-C3N4 [49]. Xie et al. rapidly prepared oxygen-doped g-C3N4 (O-CNC) with carbon vacancies by microwave heating in 7 min, and the photocatalytic H2O2 yield under simulated sunlight irradiation was more than four times that of the g-C3N4 [50]. Our group introduced both an inverse opal (IO) structure and carbon vacancies (Cv) into g-C3N4. The IO structure increased the specific surface area and promoted the absorption and utilization efficiency of visible light, while the carbon vacancies effectively facilitated the separation and migration of photogenerated carriers, thus improving the production efficiency of H2O2. At high temperatures, carbon atoms gained energy from Ar and escaped from the triple azide ring to form carbon vacancies, which was confirmed by the electron spin resonance (ESR) spectra and decrease of C/N after the introduction of carbon vacancies (Fig. 8a, b) [51]. An appropriate amount of nitrogen vacancies can not only shorten the band gap by forming a medium gap state but also enrich electrons as a trapping site and facilitate O2 adsorption, which improves the catalyst’s light absorption and enhances the surface charge separation and migration. Zheng et al. calcined graphitic carbon nanospheres (NVCNS) containing nitrogen vacancies in H2 plasma, and the yields of H2O2 were 2.5 times higher than those of graphitic carbon nanospheres (CNS) [52]. Wang et al. obtained a nitrogen-rich vacancy g-C3N4 (CNK0.2) by thermal decomposition after KOH treatment, with a shortened band gap and a 30-fold higher yield of H2O2 than that of g-C3N4 under visible light [53]. Nitrogen defects introduced alone are capable of enriching electrons, while double defect sites have the potential to further promote charge density distribution localization. Zhang et al. introduced cyano (–C≡N) and N vacancies sequentially into g-C3N4 (Nv-C≡N–CN) (Fig. 8c), and experimental and theoretical calculations show that N vacancies effectively adsorb and activate O2, while the –C≡N group enhances the adsorption of H+. The synergistic interaction of –C≡N and nitrogen vacancies not only enhances visible light absorption and carrier separation but also promotes the formation and hydrogenation of *OOH, ultimately facilitating the production of H2O2 [54]. Cong et al. used a feasible method to synthesize oxygen-modified graphite-carbon nitrogen compounds (g-C3N4-ND4-OM3) with N defects. Due to the presence of N vacancy and oxygen-containing functional group, the absorption band generated by the electron transition is enhanced, thus expanding the visible light response range of the catalyst. Nitrogen defects can trap electrons and effectively inhibit the recombination of photogenerated electrons and holes. The introduction of oxygen-containing functional groups can improve the hydrophilicity of g-C3N4 and facilitate the adsorption of dissolved oxygen. The introduction of N vacancies and oxygen-containing functional groups also changed the electrostatic potential distribution of the structural units of G-C3N4-based photocatalysts and improved the electron-donating ability of g-C3N4. As a result, the precipitation rate of H2O2 catalyzed by g-C3N4-ND4-OM3 was as high as 146.96 μmol g−1 L−1 under visible light irradiation. Photocatalyzed hydrogen peroxide is produced by direct 2e−ORR [55].

Alkali metal (Na, K) doping synergized with defect engineering (vacancies, functional groups) can further promote the production of H2O2. Wu et al. introduced alkali metal dopants (KOH or NaOH) and nitrogen vacancies on g-C3N4 by ion-thermal method. The bandgap was shortened from 2.85 to 2.63 eV, and the nitrogen vacancies rapidly trapped photogenerated electrons to promote charge separation. The synthesis rate of H2O2 was 89.5 times higher than that of the g-C3N4 [56]. The Na-doped cyanide-deficient porous carbon nitride photocatalyst developed by Chen et al. showed about 220 times higher yield of H2O2 than that of g-C3N4 under visible light. The introduction of cyano defects (–C≡N) can narrow the band gap, and –C≡N acts as an oxygen adsorption site via local charge polarization to promote the adsorption and protonation of O2 under the assistance of Na+ [57]. It can be seen that the defect-engineered modified g-C3N4 has excellent photocatalytic activity, and the synergistic effect with alkali metal element doping can further promote the efficient production of H2O2. What is often overlooked is that the general g-C3N4 material contains a large number of suspension bonds and defects, which are the recombination centers of photogenerated carriers, greatly hindering their catalytic activity. Yang et al. proposed a new approach to solving this problem by reasonably customizing ordered g-C3N4 nanorods (CNR) through molten salt-assisted anti-defect engineering. The resulting highly crystalline CNR exhibits high efficiency in the artificial photosynthesis of H2O2. The experimental results show that increasing the crystallinity of g-C3N4 and decreasing the defect concentration can effectively promote charge separation and transport. The results of this study reveal the effectiveness of molten salt-assisted anti-defect engineering in improving catalyst activity [58].
Doping of non-metallic and/or metallic elements can modulate the energy band structure of g-C3N4, enhance the light absorption ability, and change the redox potential to promote ORR and/or WOR. Wang et al. synthesized oxygen-doped g-C3N4 (OCN) with positively shifted CB and VB, which enhanced water oxidation ability and achieved dual-channel production of H2O2 [59]. Feng et al. developed boron-doped g-C3N4 using KBH4-assisted thermal polymerization, the leaf vein-like special morphology exposes a large number of reaction sites, and boron doping expands the photoresponsive range and improves the charge transfer ability, and enhances the 2e−ORR selectivity [60]. In another study, Feng et al. prepared sulfur-doped g-C3N4 by sulfide-assisted annealing and found that sulfur doping promotes light absorption and locally enriches electrons, which is beneficial to charge separation and transfer [61]. The introduction of P element into carbon nitride can help to reduce the band gap and inhibit the recombination of photogenerated charges. Xue et al. prepared P-doped carbon nitride by calcining urea and (NH4)2HPO4, and the yield of H2O2 was 6.6 times higher than that of CN. P doping can effectively inhibit the electron–hole complexation, and density functional theory (DFT) calculations and oxygen temperature-programmed desorption (O2-TPD) characterization demonstrated that P doping facilitates the adsorption of O2 and the generation of intermediates [62]. Kim et al. used a bottom-up approach to dope O and P elements into g-C3N4, which not only increased the visible light absorption but also prevented the decomposition of H2O2, and the C–O and P–O bonds acted as active sites for charge trapping and adsorption of *OOH [63]. Zhang et al. doped halogens (Br or Cl) into g-C3N4 by hydrothermal treatment with NH4Br or NH4Cl, respectively, and the doped halogens broadened the visible absorption edge and effectively promoted charge transfer. After Br doping and Cl doping, the H2O2 yield is increased by 2 times and 1.6 times, respectively [64]. Che et al. found that iodide ions could terminate the polymerization process of polymeric carbon nitride (PCN), and prepared ultrathin fragments with small size and large specific surface area, and the yield of H2O2 (3265.4 μM h−1) was 25.4 times higher than that of PCN. The abundant cyano and hydroxyl groups on the surface of fragmented PCN photocatalyst by the terminating polymerization (TP-PCN) significantly increased the adsorption capacity of O2, and DFT calculations demonstrated that the electrons in β spin orbitals could directly jump to the π* orbitals of O2 to promote O2 activation and reduce the energy barrier generated by H2O2 (Fig. 9) [65]. The lack of Lewis acid sites in carbon nitride hinders the adsorption and oxidation of alcohols, so it is usually necessary to add a high concentration of alcohols (≥ 5%) for the photocatalytic production of H2O2. Zhang et al. significantly enhanced the yield of H2O2 after the introduction of K intercalation into carbon nitride by the salt-melting method. Only 0.5% isopropyl alcohol (IPA) was added to produce 1800 μM H2O2 within 2.5 h, and the H2O2 yield was positively correlated with the K content. This was attributed to the fact that K doping modulated the active sites of the CN framework, and the Lewis acid sites generated on the catalyst surface formed bonds with the O atoms in IPA to promote the adsorption of IPA, and the protons released from IPA tended to bind to Lewis base sites to promote the activation of O2 [66]. Hu et al. synthesized Cu-doped g-C3N4 hollow microspheres by a template-assisted method and found that the band gap was narrowed and the photogenerated carrier separation was enhanced by Cu doping. As the active site, Cu(I)–N adsorbs and activates O2, promoting electron transfer to O2 to generate ORR [67]. Du et al. developed O-doped carbon nitride loaded with Ni nanoparticles to produce H2O2 efficiently under visible light. The tubular hollow structure provides a large number of active sites for the reaction and promotes the absorption of visible light, and the O doping and Ni loading improve the charge separation efficiency and exhibit a high selectivity for 2e−ORR [68].

Copyright 2021 Wiley
a TEM image of TP-PCN. b, c and d represent O2-TPD, Eads of O2 on different sites and the free-energy diagrams for PCN and TP-PCN [65].
In general, metal particle doping showed better performance, and the researchers attributed this performance boost to the additional reaction center induced by precious metal particles. However, due to the high cost of precious metal particles, the metal-free approach is promising. Metal-free modification has inherently lower productivity compared to metal. To further increase the yield, a basic understanding of the reaction pathway is required. Hu et al. have developed a range of cost-effective metal-free H2O2 photocatalysts consisting of graphitized carbon nitride (melem) and common imine groups, including 4,4′-diphenyl phthalic anhydride (ODPA), 1,4,5,8-naphthalene tetracarboxylic dihydride (NPDA) and 4,4′-diphenyl phthalic anhydride (BPDA). Using ODPA-modified g-C3N4 as a catalyst, which also showed a higher H2O2 yield than other non-metal photocatalysts [69]. Transient absorption, off-site fourier transform infrared spectroscopy (FT-IR), and in-situ electron paramagnetic resonance (EPR) show that ultrafine charges transfer from the melem core to water at ~ 3ps to form unique N–OH intermediates. This high wellbore transfer rate ensures high production rates. The electron absorption capacity of the anhydride group controls the electron transfer rate and ensures effective charge separation. Our strategy represents a new way to achieve renewable energy applications with low material costs, simple synthesis strategies, good environmental impact, and high H2O2 yields.
Co-catalysts can modulate the electronic structure, enhance light absorption, and promote photogenerated carrier separation. The addition of appropriate co-catalysts to g-C3N4 is a simple and effective modification strategy. Carbon nanomaterials such as carbon nanotubes and carbon nanofibers have the unique property of sp2 hybridized carbon bonds, which are often used as photosensitizers and electron transfer agents to broaden the absorption edge of incident light and enhance the photogenerated electron mobility. The carbon nanomaterials fixed on g-C3N4 can receive and transport g-C3N4 CB e− and improve ORR activity. Carbon nanotubes (CNTs) have a π-conjugated structure that is capable of receiving, transmitting, and storing electrons. The H2O2 synthesis rate of CNTs (48.7 μmol h−1) was significantly higher than that of g-C3N4 (2.5 μmol h−1) when CNTs were introduced into g-C3N4 nanoplates to form g-C3N4. CNTs promote electron generation by enhancing the reducing capacity and contributing to the transfer of CB e− [70]. The surface of cellulose nanofibers is rich in functional groups such as –OH and –COOH, which can be tightly linked to g-C3N4 edge amino groups. The amino-rich g-C3N4 was first synthesized by two-component thermal polymerization and then assembled by pyrolysis of functional groups on the surface of cellulose nanofibers connected with g-C3N4 edge aminos to produce carbon/g-C3N4, and the synthesis rate of H2O2 under visible light (1100 μM h−1) was 4.2 times higher than that of the single-component g-C3N4. Carbon nanofibers not only improve the porosity of the material but also strongly affect the photophysical properties of the material, that is, the narrowing of the band gap, the enhancement of visible light absorption, the increase of photogenerated carrier, and the acceleration of electron transfer rate [71]. Transition metal phosphides (TMPs) are considered to be good alternatives to precious metals due to their abundant reserves, low cost, metal-like, and excellent electronic properties. The electron transfer properties of bimetallic phosphides are superior to those of monometallic phosphides. The NiCoP/g-C3N4 photocatalytic production of H2O2 prepared by continuous in-situ growth and bimetallic phosphide modification method performed much better than CoP/g-C3N4 and Ni2P/g-C3N4. As a co-catalyst, NiCoP nanoparticles are uniformly dispersed on the surface of g-C3N4 nanosheets with good electronic conductivity, which not only enhances visible light absorption but also effectively transfers charge and improves charge separation efficiency [72]. Transition metal carbides or carbon nitrides (MXenes, such as Ti3C2, Ti2C, Nb2C, and Mo2C) can be used as electron traps to capture photogenerated electrons, significantly improving carrier separation and migration, in addition to their surface functional groups to promote binding to g-C3N4. The large specific surface area and a large amount of exposed transition metal ions provide a rich variety of active catalytic sites for the reaction. The photocatalytic H2O2 yield of Ti3C2/g-C3N4 (TC/pCN) under visible light irradiation is about 2.1 times that of g-C3N4. The formation of the Schottky junction between Ti3C2 nanosheets and host porous g-C3N4 nanosheets facilitates the space charge separation and provides the active sites for the production of H2O2, which enhances the photocatalytic activity [73]. The 0D Ti3C2 quantum dots (QDs) have better dispersion and richer active edge sites than 2D Ti3C2 sheets. The Ti3C2 quantum dots modified defective antiproteinite g-C3N4 (TC/CN) with a high concentration of carbon vacancies, prepared by our group by electrostatic self-assembly method, showed an optimal H2O2 yield of 560.7 μmol L−1 h−1 under visible light, which is 9.3 times higher than that of CN. Schottky junctions were formed between the intimate interfaces of Ti3C2 QDs and defective g-C3N4, which realized the spatial separation of electrons and holes. The recombination of carriers in the defect sites is effectively avoided (Fig. 10) [74]. Metal single-atom catalysts are emerging photocatalysts that act as co-catalysts to trap photogenerated carriers and promote charge separation. Chu et al. used Co single atoms and anthraquinone molecules as oxidation and reduction centers, respectively, to achieve spatial segregation of photogenerated carriers. The water molecules were strongly adsorbed on Co single atoms leading to the enhancement of WOR, while anthraquinone molecules attached to the edge amino group of g-C3N4 improved the ORR selectivity [75]. Zhang et al. report a carbon nitride highly loaded Ni single-atom photocatalyst with a porous ultra-thin structure that achieves an apparent quantum yield of 10.9% and a solar-chemical conversion efficiency of 0.82% at 420 nm. By using in-situ synchrotron X-ray absorption spectroscopy and Raman spectroscopy, the author directly observed the changes in Ni sites after O2 adsorption. Comprehensive theoretical calculation found that Ni load reduced the energy barrier of *OOH formation and inhibited the dissociation of O=O bonds, which was conducive to the formation of key intermediate *OOH, and further promoted the generation activity and selectivity of H2O2 [76].

Copyright 2021 American Chemical Society.
Photogenerated electron and hole behavior in carbon vacancies of a inverse opal g-C3N4 with carbon vacancies (IOCNCV) and b TC/CN-20 and c mechanism of H2O2 generation from TC/CN-20 [74].
The design and synthesis of heteroatom sites aim to improve photocatalytic selectivity by introducing heteroatoms with different electronic structures and regulating the electronic states on the catalyst surface. Tan et al. designed a macroporous inverse opal-type carbonitride (CNIO-GaSA) with a Ga–N5 atomic site. The photocatalyst showed high reactivity, H2O2 yield of 331.7 μmol L−1 h−1, and solar chemical conversion efficiency of 0.4%. It is much higher than the efficiency of natural photosynthesis in plants (~ 0.1 percent). Combined experimental evidence and density functional theory simulations reveal that mixed states at the Ga–N5 site, including hybrids from Ga 4p and N 2p, can not only enhance charge carrier separation and transfer but also promote water adsorption, activation and *OH intermediates formation, which is the rate-determining step of 2e−ORR [77] (Table 2).
Other novel photocatalysts
BiVO4
BiVO4 is considered to be an ideal narrow bandgap (2.4 eV) photocatalyst for the production of H2O2 with a suitable energy band structure for ORR and WOR. In addition, BiVO4 can resist the oxidation of ·OH (H2O2 + hν → 2·OH or H2O2 + e− + H+ → ·OH + H2O) accompanying the production of H2O2, so it has long-term stability. However, due to severe photogenerated charge complexation, the yield of H2O2 from BiVO4 in the presence of a sacrificial agent was less than 12 µM h−1 and less than 5 µM in pure water. In 2016, Hirakawa et al. used BiVO4 loaded with gold nanoparticles to produce H2O2 from O2 and pure water under visible light, significantly increasing the yield to 40.2 μM (10 h). Since the CB bottom of BiVO4 (0.02 V vs. NHE, pH = 0) was between the e−ORR potential (− 0.13 V) and the 2e−ORR potential (+ 0.68 V), the authors concluded that the special CB position could selectively promote the 2e−ORR to generate H2O2, and the electron spin resonance (ESR) and rotating disk electrode (RDE) tests confirmed that the main production pathway of Au/BiVO4 was 2e−ORR [78]. Precise loading of co-catalysts on the crystal faces by photodeposition promotes the spatial separation of electrons and holes, which is conducive to the enhancement of photocatalytic activity. Liu et al. developed a molybdenum-doped faceted BiVO4 (Mo:BiVO4), which was accurately loaded with CoOx and Pd as co-catalysts for the 4e−WOR and the 2e−ORR on the {110} and the {010} facets, respectively, by photodeposition, which adjusted the two crystalline faces’ interfacial energies and enhanced charge separation (Fig. 11). In the sacrificial-free system, the H2O2 synthesis rate of CoOx/Mo:BiVO4/Pd up to 1425 µM h−1 without sacrificial agent [79]. On this basis, the research group also developed CoOx/BiVO4/(Ag/Pd) to construct Ag/Pd binary cocatalyst with core/shell structure on the {010} surface of BiVO4. Ag nuclei significantly reduce the Schottky barrier on the {110} reduction surface, enhancing the overall asymmetry of energetics and charge separation [80]. Dr.Dai et al. prepared BiVO4 photocatalyst doped with rare earth element yttrium (Y) by hydrothermal method. Y doping can improve the adsorption of oxygen on the surface of BiVO4 photocatalyst, and this doping process induces the in-situ formation of monoclinic/tetragonal BiVO4 heterojunction, which further improves the photogenerated carrier separation efficiency. The optimized Y-doped BiVO4 photocatalyst has 4 times higher production activity than BiVO4 under simulated sunlight [81].

Copyright 2022 Nature.
a Schematic deposition processes of CoOx and Pd on Mo:BiVO4 and the corresponding SEM images of b Mo:BiVO4, c CoOx/Mo:BiVO4, and d CoOx/Mo:BiVO4/Pd. e, f Energy-dispersive X-ray spectroscopy (EDS) elemental mapping and line profile along with the white arrow of CoOx/Mo:BiVO4/Pd [79].
Organometallic frameworks (MOFs)
Metal–organic frameworks (MOFs) are a class of porous materials composed of metal oxide clusters and organic ligands with large surface area, controlled open channels and pores, and good chemical stability. Some amine-functionalized MOFs have been reported to be capable of absorbing visible light. MIL-125-NH2 consisting of Ti8O8(OH)4 clusters and 2-aminoterephthalic acid is one of the common photocatalysts. In 2018, Yamashita’s group used MOFs for the first time for the photocatalytic production of H2O2. Whether in triethanolamine (TEOA), benzyl alcohol, or aqueous solution, the deposition of NiO nanoparticles on MIL-125-NH2 can significantly increase the synthesis rate of H2O2 [82]. However, the process of separating benzaldehyde and the product H2O2 is energy intensive. To avoid the additional energy consumption caused by the separation of the products, the group also prepared MIL-125-R7 with hydrophobicity by grafting alkyl chains onto MIL-125-NH2, which not only significantly increased the synthesis rate of H2O2, but also promoted the spontaneous separation of benzaldehyde and H2O2 in benzyl alcohol/water (BA/H2O) phase [83]. However, the grafted alkyl chains would block some pores, resulting in a decrease in the surface area of the material. To preserve the original pores of the MOF, the group alkylated MIL-125-NH2 with octadecyl phosphonic acid (OPA) to develop OPA/MIL-125-NH2 with hydrophobicity, which further enhanced the yield of H2O2 in the two-phase system (Fig. 12a). Alkylation using OPA modified only Ti8O8(OH)4 while retaining most of the pores. Due to the blockage of the pores by the alkyl chains, the BET surface area of MIL-125-R7 (560.7 m2 g−1) was only about 37% of that of the original MIL-125-NH2 (1498 m2 g−1), whereas OPA/MIL-125-NH2 was able to maintain about 83% of the surface area (1242 m2 g−1) [84].

Copyright 2019 Royal Society of Chemistry. b Proposed mechanism for photocatalytic H2O2 production by hydrophobic OPA/Fe-Zr-MOF in a BA/H2O two-phase system [86]. Copyright 2021 American Chemical Society
a Structures of MIL-125-R7 and OPA/MIL-125-NH2. The two-phase system is composed of a BA/H2O phase containing OPA/MIL-125-NH2 in the BA phase [84].
Zirconium-based MOFs are structurally stable, and the charge separation can be promoted by doping Ti ions as an electronic medium to partially replace Zr ions in zirconium MOF. Chen et al. developed hydrophobic titanium-zirconium-based MOFs (OPA/Zr100-xTix-MOFs), in which the H2O2 yield of metal Zr clusters after alkylation by OPA is 4.5 times that of Zr100-MOF in BA/H2O two-phase system. Ti species effectively promote charge transfer and inhibit photogenerated charge complexation. The hydrophobicity promotes the spatial separation of MOF in BA and H2O2 in water, which is conducive to the reduction of H2O2 decomposition, and the generation of benzaldehyde in the organic phase, so there is no need to separate the sacrificial agent and the product H2O2 again [85]. The team then developed a hydrophobic solution for benzaldehyde in the organic phase. Subsequently, the team developed a hydrophobic iron-doped zirconium-based MOF (OPA/Fe–Zr-MOF), and the yield of H2O2 was further increased to 13.1 mM h−1. Doped Fe3+ promoted visible light absorption and participated in the ORR as an electron donor, whereas the hydrophobic material and the product H2O2 were located in BA and aqueous phases, respectively, thus inhibiting the subsequent Fenton-like reaction of Fe3+ (Fig. 12b) [86]. Li et al. developed a boron-containing zirconium MOF with UiO-66 topology (UiO-66-B) by replacing terephthalic acid (PTA) with 4-carboxyphenylboronic acid (PBA) in zirconium-based MOF (UiO-66). The adsorption capacity of O2 was significantly improved by the introduction of boron elements and promoted the proton-coupled double electron transfer process. In addition, the carrier separation and charge transfer rates were accelerated, and the synthesis rate of H2O2 (314 μM h−1) was 3.2 times higher than that of the original UiO-66 [87].
Covalent organic frameworks (COFs)
COFs are emerging porous covalent crystalline polymers connected by covalent bonds with visible light response. The highly ordered porous crystal structure effectively prevents electron and hole recombination, has high charge separation and migration efficiency, and can be regulated at the molecular level. In addition, COFs have the advantages of low density, high specific surface area, thermal stability, adjustable skeleton and pores, and abundant active sites, which are considered promising photocatalysts [88]. There is much research on photocatalytic hydrogen production and CO2 reduction of COFs, but the research on photocatalytic H2O2 production has just started. In 2020, Krishnaraj et al. were inspired by the Wurster system, and for the first time, COFs were used in the photocatalytic production of H2O2, and two (diaryl amino) phenyl covalent organic frameworks with strong reducing properties, TAPD-(Me)2 and TAPD-(OMe)2, whose high crystallinity (Kagome lattice) and large specific surface area (1165 m2 g−1) allow for efficient charge transfer and diffusion, and the catalysts are stable and reusable [89].
COF photocatalysts prepared by structural design and optimization showed high stability and catalytic activity. Hydroxyl groups are active sites for O2 adsorption, contributing to the conjugation effects of electron donation as well as ORR in the photocatalytic synthesis of H2O2. Zhou et al. reported the preparation of “segmented” COFs (TxHy-COFs) photocatalysts and their application in the synthesis of H2O2. The results show that by changing the feed ratio of TAPT and THTA monomers, the functional design of hydroxyl (-OH) modification on the surface of fragment TxHy-COFs is realized, which greatly improves the surface hydrophilicity and effectively improves the separation and transport of photoexcited electrons and holes. Under visible light irradiation without a sacrifice agent, fragmented T1H1.8-COF showed an increased H2O2 yield of 2567 μmol L−1 h−1, which was 4.2 times higher than that of the original T1H1-COF. In addition, metal ions (Fe3+, etc.) were introduced into the COF framework by impregnation method to achieve in-situ activation of H2O2, and benzene was directly hydroxylated to phenol in the water system with a yield of 9.4% and a selectivity of 99% [90].
Covalent triazine frameworks (CTFs) are a class of nitrogen-rich COFs consisting of alternating ultra-stable triazine bonds and phenyl units with high conjugation degree, good tenability, and chemical stability. However, the strong π–π interactions between the layers can seriously affect the photoexcitation and separation of carriers. Effective charge separation can be achieved by introducing functional groups on CTFs to form oxidation and reduction centers, respectively, at the molecular level. Yu et al. used SiO2 nanospheres as templates and introduced benzothiazole (BT), which has a strong electron-withdrawing effect, into covalent triazine framework nanoshells via aldolamine condensation reaction, which significantly promoted charge separation. When the content of BT is 5 mol%, the optimal yield of H2O2 (1630 μmol h−1 g−1) is about 3 times that of undoped BT covalent triazine frame nanoshells. In the absence of BT, the HOMO is mainly located on the benzene ring, while the LUMO is uniformly off-domain on the conjugated skeleton and overlaps with the HOMO. However, after the introduction of BT, LUMO is concentrated around BT due to its strong electron-withdrawing ability. This results in the transfer of electrons from within the phenyl molecule to the neighboring BT unit via the π-conjugated backbone under photoexcitation [91]. The triazine unit has electron-rich properties, and precise design at the molecular level of CTFs and grafting of polar groups can help to achieve photogenerated charge separation. Wu et al. grafted polar thiourea functional groups onto covalent triazine frameworks (CTFs) to fabricate Bpt-CTFs, which not only significantly facilitated charge separation and translocation, but also significantly facilitated proton transfer. The synthesis rate of H2O2 is as high as 3268.1 μmol h−1 g−1, which is about 10 times higher than that of the unfunctionalized CTF. Photogenerated electrons and holes were concentrated in the triazine unit and thiourea sites, respectively. The high concentration of hole-oxidized water in the thiourea site provides O2 and protons for the ORR. The thiourea site has a high proton conductivity (Fig. 13a), which facilitates proton transfer and promotes the proton-coupled electron-transfer process, which accelerates the 2e−ORR process on the perovskite in the triazine unit. Thus, the overall reaction kinetics was improved (Fig. 13b) [92]. To improve the stability and charge separation of COFs to enhance the photocatalytic production of H2O2, Wang et al. synthesized fluorine-substituted imine-containing triazine covalent organic frameworks (TF50-COF) with a synthesis rate of up to 1739 μmol g−1 h−1 and an apparent quantum efficiency of 5.1% under > 400 nm light and 10% ethanol solution. F substitution produces a large number of Lewis acid sites, regulates the electron distribution of neighboring carbon atoms, and the strong electron-withdrawing ability promotes the in-plane charge transfer. An appropriate amount of F substitution regulates the π-electron interactions by increasing the stacking energy, improving the crystallinity and porosity of the material, promoting the chemisorption of O2, and enhancing the visible light absorption and photostability [93].

Copyright 2022 Wiley. c Free energy diagram of two-electron water oxidization pathway toward H2O2 production on diacetylene linker in CTF-BDDBN [94]. Copyright 2019 Wiley. d Photocatalytic mechanism of H2O2 synthesis in the presence of COF-TfpBpy [96]. Copyright 2022 Wiley
a Temperature-dependent proton conductivity of Bpt-CTF, Bpu-CTF and Dc-CTF. b Mechanism for photocatalytic H2O2 production on the surface of Bpt-CTF [92].
The dual-channel production of H2O2 is the most optimal pathway with 100% atom utilization. Since it is thermodynamically more favorable for 4e−WOR to produce O2 (+ 1.23 eV vs. NHE) than 2e−WOR (+ 1.78 eV vs. NHE), 2e−WOR is often not easy to occur, which greatly limits the yield of H2O2. However, in recent years, it has been found that several COFs have already realized dual-channel production of H2O2, which will provide ideas for a more sustainable and efficient production of H2O2. Chen et al. synthesized H2O2 via a dual-channel pathway by introducing either acetylene or diethylacetylene into covalent triazine frameworks (CTF-EDDBN and CTF-BDDBN). Acetylene and diethylacetylene not only promote charge separation in the conjugated structure, but also can significantly reduce the energy required for the generation of *OH, which promotes the generation of H2O2 from 2e−WOR (Fig. 13c). The occurrence of 2e−ORR was confirmed by comparing the H2O2 yields under different atmospheres, RDE tests and EPR tests. First-principles calculations demonstrated that acetylene and diethylacetylene promote O2 adsorption. In addition, the Gibbs free energy change (ΔG) of *OH was examined using the triazine ring, carbon atom on phenyl, acetylene, and diethylacetylene as active sites, respectively, and it was found that *OH adsorbed on acetylene or diethylacetylene was more readily formed. The occurrence of 2e−WOR was confirmed by water oxidation half-reaction experiments and rotating ring disk electrode (RRDE) tests [94]. Subsequently, the team built on this foundation by precisely designing a novel covalent heptazine framework (CHF-DPDA) with spatially separated redox centers using dual channels of pure water and oxygen to produce H2O2. This material has periodically arranged and spatially separated redox centers and the 2e−ORR and 2e−WOR reactions take place on the s-heptazine and on the ethyne or diethyl acetylene bonds, respectively. s-heptazine consists of three s-triazine rings with a high content of nitrogen atoms in the backbone (C/N ≈ 0.86) and a high electron affinity. Due to the significant difference in electronegativity between s-heptazine and biphenyl compounds, this promotes efficient electron transfer and concentration on the central s-heptazine, while holes remain on the acetylene or diacetylene bond. This spatial separation property significantly reduces exciton binding to 24 meV, promoting more free charge generation and participation in redox reactions [95]. Kou et al. found that nitrogen atoms on the bipyridine of bipyridine-based polymers facilitated the adsorption of H2O to undergo protonation and promote 2e−WOR, which then accelerated the 2e−ORR by Yeager-type oxygen adsorption, allowing for dual-channel production of H2O2. The optimal photosynthetic rate of H2O2 (1042 μmol h−1) was 496 times higher than that of the g-C3N4. COF-TfpBpy first undergoes a light-induced structural transformation in which two bipyridine N atoms adsorb two water molecules and hydrogen bonds are formed between the water molecules. Photoexcitation of the 2e−WOR generates H2O2 leaving two protonated pyridine substituents (PyH+). O2 is adsorbed on the PyH+ substituent to form an endoperoxide intermediate (N–H–O–O–H–N). The protonation of bipyridine facilitates the adsorption of oxygen by internal peroxide intermediates and accelerates the 2e−ORR process (Fig. 13d) [96].
To promote charge separation, in addition to modifying COFs by precise design at the molecular level, some scholars have also investigated the effect of constructing heterostructures on the production of H2O2. Zhang et al. prepared S-scheme heterojunctions of ZnO/COF (TpPa-Cl) by electrostatic self-assembly, and the synthesis rate of H2O2 under simulated solar irradiation (2443 μmol h−1 g−1) was 3 and 8.7 times higher than those of ZnO and TpPa-Cl, respectively. The imine-based COFs represented by TpPa-1 have high photocatalytic activity but still face the problem of rapid recombination of photogenerated carriers. By introducing inorganic semiconductors with suitable energy band structures to construct S-scheme heterojunctions, effective photogenerated charge separation can be promoted and high redox capacity can be guaranteed under the driving of the built-in electric field [97]. Wang et al. prepared a Type II heterojunction (TpMa/CN) consisting of CN and b-ketoenamine COF with 49 times higher yield of H2O2 than that of the CN. On the one hand, b-ketoenamine and triazine in TpMa act as an electron donor and an acceptor, respectively. The electron donor–acceptor (D–A) is favorable for causing intramolecular charge transfer, promoting exciton separation, and enhancing visible light absorption. On the other hand, an embedded electric field was formed at the interface of CN and COF, which accelerated charge transfer and separation [98]. Luo et al. introduced a sulfone unit (a strong electron-receiving nucleus) into the COF matrix to promote photocatalytic H2O2 generation from pure water and air. The sulfone unit accelerates the separation of photogenerated electron–hole pairs, enhances the protonation of COFs, promotes the “side pair” Yeager adsorption of O2, jointly inhibits the formation of traditional *OOH intermediates, and converts the two-step e−ORR route to an efficient one-step 2e−ORR route. Under LED visible light (λ = 400 nm), the yield of H2O2 in pure water is 1501.6 μmol g−1 h−1, which is about 3 times higher than that of COFs with similar structure but no sulfone site (487.6 μmol g−1 h−1). The H2O2 yield of FS-COFs under xenon lamp (λ = 420 nm) reached a record high of 3904.2 μmol g−1 h−1, exceeding that of most metal-free catalysts reported under similar conditions. In addition, FS-COFs can effectively photocatalyze H2O2 in a variety of natural waters (including tap water, Pearl River water, and seawater), and the generated H2O2 can be used for in-situ degradation of organic pollutants (such as Rhodamine B) through Fenton reaction [99].
Resorcinol-phenolic resins (RFs)
RFs are formed by polycondensation of phenol and formaldehyde and are commonly used as sensitizers to increase visible light absorption in the photocatalytic field. The large surface area and abundant pore structure have strong adsorption capacity for organic pollutants, and the large number of reactive hydroxymethyl and phenolic hydroxyl groups in the structure are easy to be reacted by other groups, which can be used to adjust the material properties from the molecular level. There are many studies on photocatalytic water decomposition and pollutant degradation. In 2019, Shiraishi et al. first used RF for photocatalytic H2O2 production by alkali-catalyzed high-temperature (~ 523 K) hydrothermal method (RF523). Composed of methylene-linked benzene-quinone π-conjugated electron donor–acceptor (D–A) units, the solar-to-chemical conversion (SCC) efficiency for the stable production of H2O2 under simulated sunlight exceeds 0.5%. The synthetic reagent of RF523 is cheap and has a narrow band gap (about 2.0 eV), which can absorb long-wave light with a wavelength of about 700 nm, achieve effective separation of photogenerated carriers, and has low decomposition activity of H2O2 [100]. The band gap energy (1.69 eV) of RF-(COOH)2–523 resin produced by the acid catalysis method was further reduced and the conductivity was higher than that of the base catalysis method. Under the condition of pH < 4, the H2O2 yield of RF acid resin prepared by high-temperature hydrothermal is 1.5 times that of the original RF resin, and the SCC is increased to 0.7%. The acid-catalyzed reaction is triggered by formaldehyde protonation, and the lower cross-linking leads to a more flexible structure, resulting in a strongly π-stacked benzoquinone D–A structure [101]. Since the individual D–A units in RF resins are connected by insulating methylene groups, charge transfer (CT) occurs via π-stacking, but the dissipative discontinuity bands severely inhibit charge transfer. Polythiophene is a π-conjugated conducting polymer with high charge transport properties. To further improve the charge transfer efficiency, the group prepared RF resin powder photocatalysts RF/P3HT doped with poly (3-hexylthiophene-2,5-diyl) by high-temperature hydrothermal method. P3HT was highly dispersed within the resin particles and acted as a π-conjugated conductive linker to promote electron transfer. The HOMO on the thiophene unit gave electrons to resin CB to generate the CT complexes, and the resin CB electron transfer enhances the separation of electron–hole pairs, further promoting efficient ORR and WOR (Fig. 14). Under simulated sunlight irradiation, the SCC efficiency of RF/P3HT resin was further increased to 1.0% [102]. The pioneering work of Shiraishi et al. opens up a pathway to explore the photocatalytic production of H2O2 by resorcine-formaldehyde (RF) resins. Since then, remarkable progress has been made in research based on RF523 resins, such as mesoporous RF balls, RF nanobowls, polythiophene/phenol/anthraquinone doped RF and graphene quantum dot-RF composites [103, 104].

Copyright 2021 American Chemical Society
Physical and electronic structure of the materials. a Cross-linking structure of the RF resins. b π-conjugated and π-stacked D–A architecture of the undoped RF resins. c Electronic band structure of the undoped RF resins. d Structure of P3HT. e The architecture of the P3TH-doped RF resins. f Electronic band structure of the P3HT-doped RF resins [102].
In addition, alternative resin materials with well-defined structures can be developed through molecular-level design and polymer structures can be designed to regulate catalytic reaction kinetics, so as to explore the potential and prospect of phenolic resins in H2O2 photosynthesis. Wang et al. reported an alternative phenolic resin beyond RF523 as a special photocatalyst for long-term photocatalytic H2O2 production. M-aminophenol and formaldehyde were selected as precursors. Aldol condensation, Mannich reaction, and cyclic condensation form m-aminophenol-formaldehyde (APFac) resin under environmental conditions, avoiding the cumbersome and dangerous preparation process of expensive precursors and photocatalysts. The good affinity of the benzoxazine structure for reactants and intermediates and good product selectivity enables APFac resin to have a high H2O2 production efficiency under visible light without sacrificing agents. In particular, the H2O2 yield of APF resin is negatively correlated with the hydrothermal temperature due to the destruction of the benzoxazine structure, which indicates that hydrothermal treatment is not a necessary method for phenolic resin activation [105].
Hydrogen-bonded organic framework (HOFs)
The concept of hydrogen-bonded organic frameworks (HOFs) was proposed in the early 1990s [106]. HOFs achieve self-assembly through hydrogen bonds, and hydrogen bonds are significantly more adaptable and flexible than covalent bonds in COFs in terms of bond energy [107], making HOFs easy to recycle [108], mild synthesis conditions [109], strong versatility, and clear structure [110, 111]. The high crystallinity of HOFs can narrow the band gap and improve the light collection efficiency, thus accelerating the rapid transport of carriers and improving the mobility and conductivity of carriers.
However, there have been few reports on HOFs of H2O2 generation by photocatalysis. Designing highly active HOFs combined with excellent photostability for photocatalyzed H2O2 generation applications remains a big challenge. Hu et al. designed highly crystalline perylene HOF (PTBA) and amorphous simulated sample (PTPA) for photocatalytic H2O2 precipitation. By connecting perylene with benzoic acid and forming intermolecular hydrogen bonds, PTBA has high crystallinity and simple topology, thus promoting the separation and transfer of photocarriers. Under visible light irradiation, the photocatalytic H2O2 yield of PTBA is as high as 2699 µmol g−1 h−1, which is more than 500 µmol g−1 h−1 higher than that of its analog PTPA. The study reveals the great potential of HOF for efficient and stable photocatalytic generation of H2O2 [112] (Table 3).
Advanced system for photocatalytic H2O2 production
Establishment of a spontaneous system without sacrificial agents
Sacrificial agents are ethanol or other organic alcohol electron donors that react with photogenerated holes, contribute to charge separation, and provide oxygen and protons for ORR. However, sacrificial agents are expensive and increase the cost of producing H2O2. In addition, the impurities generated after introducing the sacrificial agent will mix with H2O2, causing difficulties in separating and purifying H2O2.
The photocatalytic H2O2 production using pure water not only reduces the synthesis cost but the product H2O2 can also be directly applied in situ. Due to the unfavorable adsorption of O2 on the catalyst surface and slow charge transfer, the photocatalytic performance of H2O2 in pure water is usually low (apparent quantum yield (ΦAQY) < 8%, λ = 420 nm), and the synthesis rate is less than 0.1 mmol h−1 g−1. Improving the 2e−ORR activity and selectivity is an effective strategy for the efficient production of H2O2 in pure water. As a key reactant of ORR, O2 adsorption performance is essential for the efficient production of H2O2 in pure water. The Pauling-type (end-pair) adsorption of O2 by single-atom catalysts makes it a highly active and selective site for 2e−ORR. Teng et al. synthesized an Sb single-atom photocatalyst (Sb-SAPC) with d10 electronic configuration for efficient H2O2 production (ΦAQY = 17.6% (λ = 420 nm)) in pure water. The electron enrichment of single Sb sites under visible light favors the adsorption of O2, and the Pauling-type adsorption reduces O–O bond breaking, which significantly inhibits 4e−ORR, thus improving the selectivity of 2e−ORR and promoting the formation of Sb-μ-peroxide (Sb-OOH). In addition, the Sb site in the neighboring melem unit of the N-atom-enriched hole promotes 4e−WOR [113]. The polyfuran structure formed by hydrothermal carbonization of biomass facilitates oxygen adsorption and activation and has superior electron transfer properties to promote ORR for efficient production in pure water. Xu et al. used a dilute sulfuric acid-assisted hydrothermal method to convert cellulose into hydrothermal carbon with high photocatalytic properties (HTCC), with yields up to 1,160 μmol g−1 h−1 of H2O2 in visible light and pure water [114]. Kang’s group hydrothermalized cellulose with the assistance of sulfuric acid, and the carbon dots (CDs) formed during the carbonization process effectively promoted the photogenerated charge generation, separation, and transfer, and the yield of H2O2 was as high as 2093 μmol g−1 h−1 in visible light and pure water. Interestingly, the authors achieved efficient screening and optimization of the synthesis and catalytic conditions through the construction of machine-learning models and the TPV test [115]. Chu et al. prepared a cross-linked polymer photocatalyst (CDA) with β-cyclodextrin aldehyde and 4-amino-6-hydroxy-2-mercaptopyrimidine with an H2O2 yield of 557.2 μM g−1 h−1 in air and pure water, and the catalyst maintained performance over a wide pH range (pH = 1–11). This was attributed to the enhanced oxygen adsorption capacity after doping with cyclodextrin, and the production of large amounts of H+ during the WOR process, which made the catalyst have good pH adaptability [116].
Seawater accounts for 97% of the water resources on the earth’s surface, and utilizing seawater to produce H2O2 is more consistent with the Sustainable Development Goals than pure water. This not only helps alleviate water scarcity but also significantly reduces response costs. However, salts in seawater usually hinder the light absorption and electron transfer ability of catalysts, and even lead to catalyst deactivation. As a good electron donor–acceptor material, carbon point (CDs) has been widely used in the field of photocatalysis. Wu et al. prepared a metal-free photocatalyst (PM-CDs-30) for H2O2 production in seawater by phenol condensation using CDs, proanthocyanidins (organic dyes), and 4-methoxybenzaldehyde with high photocatalytic activity (1776 μmol g−1 h−1) under visible light. In natural seawater, it is about 4.8 times that of pure polymer (PM-CDs-0). The authors found that metal cations in seawater can enhance the ionization of functional groups on the surface of CDs, which enhances the electron-absorbing effect of CDs. As a result, CDs can attract and inhibit more electrons, thereby prolonging the separation of electrons and holes. Biopolymers such as cellulose or lignin can not only be directly used as photocatalysts after hydrothermal reaction but also as ideal carriers of multiphase catalysts [117]. Gopakumar et al. used alkaline hydrolyzed lignin-loaded BiOBr nanosheets (LBOB) to produce H2O2 directly from air and seawater under visible light and showed good activity (4085 μM (6 h)). The addition of lignin polymer lowered the reduction potential while maintaining the same visible light absorption capacity. When metal ions in seawater are ionized, organic functional groups on the lignin carrier can act as electron sinks to enrich electrons. The extraction of protons by Bronsted-Lorry bases makes LBOB more active in seawater than in pure water for the production of H2O2 [118].
In addition to the spontaneous photocatalytic H2O2 production in pure water and seawater systems, simultaneous CO2 reduction and H2O2 production with suitable photocatalysts is of research value in the current two-carbon background. Soltani et al. found a photocatalytic system in which calcium titanate (CaTiO3) photocatalyst modified with silver-manganese oxide (Ag–MnOx) double co-catalyst simultaneously promoted the reduction of CO2 to CO and the oxidation of H2O to H2O2. The system has high activity and selectivity. No external voltage and no compound consumption is required. Although Ag/CaTiO3 (CTO) photocatalyst can react with water to reduce CO2 to produce CO and O2, MnOx and silver nanoparticles (Ag NPs) as double cocatalysts not only increase the CO generation rate by more than 2 times, but also change the selectivity of oxidation reaction. Produces H2O2 with a high selectivity of 99%. Ag NPs promotes the reduction of CO2 to CO in the conduction band (CB) by photoexcited electrons, while Mn(III) oxide deposited on the surface of CaTiO3 promotes the oxidation of H2O to H2O2 in the valence band (VB) with the help of the HCO3− reaction medium. The product CO migrates from the aqueous solution to the gas phase, while H2O2 is stably stored in the HCO3− aqueous solution. This collaborative photocatalytic system utilizes stable, ubiquitous CO2 and H2O, reducing CO2 while also promoting the formation of H2O2, successfully converting light energy into chemical energy [119].
Increase the oxygen concentration at the interface
Photocatalytic H2O2 production is basically a solid–liquid two-phase system, and the yield of H2O2 is directly affected by the O2 content in the suspension. The higher concentration of O2 facilitates rapid binding with photogenerated electrons to promote the ORR process and inhibits the recombination of photogenerated carriers, so photocatalytic production is usually carried out in oxygenated saturated water. However, due to the low solubility of O2 in water (25°, 1 atm, 8 mg L−1) and slow diffusion rate in the liquid phase (2.13 × 10−5 cm−2 s−1), which not only severely limits the yield of H2O2, but also leads to additional energy consumption by continuous aeration. The gas–solid-liquid three-phase system constructed by depositing semiconductors on a porous hydrophobic substrate is conducive to increasing the O2 concentration and oxygen transfer rate at the reaction interface and enhancing the photocatalytic kinetics. The porous hydrophobic substrate provides a gas diffusion channel for O2, and the diffusion coefficient of O2 (2.03 × 10−1 cm−2 s−1) is four orders of magnitude higher than that of the liquid phase, which dramatically increases the interfacial O2 concentration. In 2019, Feng’s team constructed a gas–solid-liquid three-phase system by fixing Au-TiO2 nanoparticles on a super-hydrophobic carbon fiber porous membrane, and the steady-state concentration of H2O2 in UV light was 44 times higher than that of a conventional solid–liquid two-phase system [120]. In another study, the group found that the gas–liquid-solid three-phase system driven by visible light enhanced the plasma photocatalytic synthesis of H2O2, and the steady-state concentration of H2O2 in the three-phase system (2.9 mM) was three times higher than that in the two-phase system [121]. Chen et al. constructed a gas–liquid-solid three-phase reaction system with tubular confined space, and the synthesis rate of H2O2 (375 μM h−1) was much higher than that of the two-phase system (48 μM h−1). Firstly, the porous melamine foam (MF) was coated with graphene and polytetrafluoroethylene (PTFE) to form a hydrophobic porous skeleton, and then sodium doped and N-defective graphitic carbon nitride (Na-CvCN) was loaded onto the hydrophobic porous skeleton to form a tubular confined space with a thickness of about 100 μm. The product H2O2 can diffuse rapidly into the aqueous solution through the tubular confined space, avoiding decomposition in contact with the catalyst and thus increasing the steady-state concentration [122]. Li et al. made use of the hydrophobic and mesoporous properties of triphenyl-dimethoxy-terephthalic acid COFs and loaded them onto a porous substrate to form a gas–liquid-solid three-phase system (CFP/COF). The synthesis rate of H2O2 under visible light and pure water was up to 2882 μmol h−1 g−1, which was 15 times higher than that of the conventional liquid–solid two-phase system. The water contact angle of the CFP/COF was 119°, the hydrophobicity of the catalyst layer provides conditions for the formation of a three-phase interface, and the 78% porosity of the carbon fiber provides abundant channels for gas diffusion (Fig. 15a). Due to the strong conjugation of TPB to enrich electrons, O2 is activated and reduced in the TPB region. In addition, CFP/COF can produce H2O2 in a wide pH range (pH = 3–10) and shows high stability. The H2O2 yield of the three-phase system formed after the hydrophilic oxygen-doped carbon nitride is fixed on the fiber paper is only 2.3 times that of the two-phase system, which indicates that the hydrophobicity of the catalyst may be important for the formation of a stable three-phase system [123]. Sun et al. designed a super-hydrophobic photocatalyst (Pd/A/BiVO4) with a yield of 805.9 μmol h−1 g−1 of H2O2 in visible light and pure water. Pd/A/BiVO4 uses aminopropyl trimethoxysilane (APTMS) as a bridge ligand between BiVO4 and Pd, increases the charge density of Pd through partial charge transfer of the electron-giving group of APTMS-NH2, adsorbs O2 and forms the active site of Pd-Ox. In addition, the long chain dominated by C-H in APTMS leads to the hydrophobicity of Pd/A/BiVO4. The constructed gas–liquid-solid three-phase system can directly utilize O2 in the gas phase, and the separation of the catalyst and product system is beneficial to inhibit the decomposition of H2O2 (Fig. 15b, c) [124]. Photocatalytic oxygen reduction reaction (ORR) to generate H2O2 with lower energy input is a promising strategy. Chen et al. devised a simple method to adjust the hydrophobicity and oxygen accessibility of TiO2 photocatalysts by stearic acid (SA) modification at a controllable crystalline ratio and studied the adsorption structure of SA and TiO2 in detail. It was found that SA molecules formed a stable ester bond with the surface of TiO2, resulting in increased hydrophobicity with the increase of SA load. The 2e−ORR photocatalytic H2O2 yield on SA-modified TiO2 was up to 3160 μM h−1 g−1, which was 1.69 times higher than that of the TiO2. From the electrochemical impedance spectroscopy and molecular dynamics simulation results, the increase of O2 concentration in the interfacial microenvironment after SA modification is the reason for the enhanced 2e−ORR activity rather than the enhancement of hydrophobicity [125].

Reduction of decomposition of the product H2O2
The final yield of photocatalytic H2O2 production depends on the two components of H2O2 formation and decomposition, which follow zero-order and one-order kinetics, respectively. The overall kinetic equation is shown in Eq. (12). Where, Kf and Kd represent the rate constants for H2O2 formation (µmol L−1 min−1) and decomposition (min−1), respectively.
Photogeneration of e− and h+ promotes H2O2 production, but may further induce the decomposition of H2O2 into ·OH or ·O2− by e− or h+ (Eqs. 13 and 14). It was found that UV light induces the decomposition of H2O2, and H2O2 is strongly adsorbed on the surface of TiO2 under visible light (λ > 420 nm) to form a Ti–OOH surface complex. The e− excited by visible light in Ti–OOH can migrate to the CB of TiO2, and can also reduce the adsorbed H2O2 to ·OH and OH−.
By modifying the surface of the material to block the interaction between H2O2 and the photocatalyst surface, the decomposition of H2O2 can be effectively prevented. Due to the strong adsorption of TiO2 on H2O2, the inhibition of H2O2 decomposition on TiO2-based photocatalysts is the most widely studied. Surface complexation is a common strategy to inhibit H2O2 decomposition by covering the surface of TiO2 to prevent the formation of Ti–OOH. Complexation of TiO2 with cations (such as Cu2+, Zn2+) or anions (F−, PO43−) can inhibit H2O2 decomposition [126,127,128]. Zuo et al. found that SnO2 can passivate TiO2 surface [129]. Zheng et al. found that coating a Nafion layer on the surface of TiO2 prevented the formation of Ti–OOH, and the negatively charged shell layer concentrated O2 and protons on the surface of TiO2 [130]. Li et al. found that the addition of fluoride to Au/TiO2 could form Ti–F complexes to inhibit the decomposition of H2O2, and the yield of H2O2 on Au/F–TiO2 was four times higher than that of Au/TiO2. Lee et al. showed that the formation of Ti–OOH on Au/TiO2 was inhibited by the addition of fluoride [131]. Lee et al. thermally decarboxylated monolayers of benzoic acid or naphthoic acid organic molecules adsorbed on the surface of TiO2 to produce TiO2@C core/shell nanocomposites. The chemical bond between the TiO2 core and the carbon shell is not easy to break, and the carbon shell not only does not hinder the transfer of carriers but also inhibits the adsorption of H2O2 on the surface of the TiO2 core, which prevents the consumption of H2O2 [132]. Ma et al. reported a plasmonic titanium dioxide nanotubes-carbon dots (HTNT-CD) composite catalyst with a high yield of H2O2 of up to 3.42 mmol g−1 h−1 under visible light irradiation. HTNT-CD possesses both Lewis and Brønsted acidic sites, and the abundant plasmon formation on the catalyst surface facilitates the coupled electron transfer (PCET) to promote the generation of H2O2 by ORR. The acidic protons stabilized H2O2 under the reaction conditions and ultimately achieved a high yield [133].
Compared with metal oxides, g-C3N4 can theoretically inhibit the oxidative decomposition of H2O2 to a certain extent due to its lower VB potential (1.4 VNHE). However, it has been reported that the H2O2 generation activity of g-C3N4 will be greatly decreased under conditions rich in ·OH, so the inhibition of the decomposition of H2O2 into ·OH is very important to improve the final yield. Zhang et al. used oxidized red phosphorus (ORP) to modify g-C3N4, and efficiently synthesized H2O2 (125 μM h−1) under visible light and in pure water. Red phosphorus has a strong light absorption ability and promotes charge separation and H2O2 decomposition. The decomposition rate of RP/GCN was as high as 80% under light, with an obvious ·OH signal after 30 min of light, while the decomposition rate of OPR/GCN was only 10%. The direct contact between H2O2 and P atoms was prevented by oxidized red phosphorus, H2O2 was adsorbed on the O atoms of ORP through hydrogen bonding, and the photogenerated electrons were transferred from H2O2 to ORP, which significantly inhibited the decomposition of H2O2 (Fig. 16) [134]. Our group developed g-C3N4 nanotubes (ACNT-5) with in-situ grafted > OH groups on the surface, and the yield of H2O2 reached 240.36 μmol h−1 g−1 in the absence of a sacrificial agent. The photocatalysts with > OH groups grafted on the surface not only trapped photogenerated holes to promote charge separation and provide protons for ORR, but also significantly inhibited the decomposition of H2O2. ACNT-5 can still maintain 65% yield after 1 h of the experiment. The > OH group grafted on the surface can act as an electron trap to generate ·OH (2 > OH· → H2O2) on the surface, thus inhibiting the decomposition of H2O2 [135].

Copyright 2021 Elsevier
a H2O2 decomposition over ORP/GCN and RP/GCN in the dark and under visible-light irradiation. b Detection of radical ·OH during the photocatalytic H2O2 decomposition after 30-min irradiation. c Side view of electron density difference of H2O2 adsorption on ORP. Colors: P (blue), O (red), and H (white), yellow and light blue parts represent electron accumulation and depletion regions. d The xy-planar-averaged differential charge density (black line) along the z-axis and the plane-integrated differential electron density (red line) [134].
In addition to the modification of the material surface, the construction of hydrophobic photocatalysts can also effectively prevent the adsorption and decomposition of H2O2. Hong et al. developed a hydrophobic fluorinated polymer/TiO2 with a yield of 110.4 μM of H2O2 in pure water, and atomistic simulations confirmed that the adsorption energy of H2O2 on the surface of the material was low, and the hydrophobicity of the fluorinated polymer/TiO2 effectively inhibited the decomposition of H2O2 [136]. Lee et al. successfully prepared CdS/sulfur-doped carbon nanomaterials with a concentration of H2O2 up to 17.1 mM in KOH solution with 2-propanol, which is much higher than that of commercial CdS. The carbon layer on the photocatalyst significantly reduces the adsorption energy of H2O2, and the sulfur-doped carbon further reduces the adsorption energy. The authors concluded that the sulfur-doped carbon modification increased the water contact angle of the material, thus preventing H2O2 adsorption and decomposition [137].
Alternatively, the decomposition of H2O2 can be effectively inhibited by changing the reaction system, and the construction of a two-phase reaction system (water-benzyl alcohol) is one solution. Sun et al. synthesized four imine-conjugated COFs with pyrene-building units 1,3,6,8-tetraacylphenylpyrene (Py-CHO). The synergistic effect of tetrakis (4-aminophenyl) diamine (TAPD) and bipyridine (Bpy) units combined with pyrene units in COFs was studied. By schiff’s base condensation reaction was prepared by Py-Py-COF package (Py-CHO with 4,4′,4″,4‴-(pyrene-1,3,6,8 tetrayl) tetrakisaniline (Py-NH2)) and Py-Da-COF (Py-CHO with 1,4-diaminobenzene (Da-NH2)), The photocatalytic performance of four pyridinyl COFs for H2O2 production in the absence/presence of sacrificial agent was investigated. It was found that the distribution of pyrene units on the large surface area of COFs played an important role in the photocatalytic performance. COFs with electron-rich pyrene units favored H2O2 formation, but a large number of neighboring pyrene units led to severe H2O2 decomposition. Interestingly, the authors found that the two-phase reaction system (BA/H2O) inhibited the H2O2 decomposition, thus realizing the accumulation of H2O2 in the reaction system [138].
The decomposition rate of H2O2 is not only related to the surface properties of the catalyst but also to the reaction conditions (temperature, pH, sacrificial agent, etc.). Burek et al. found that high temperatures (> 40 °C) and alkaline conditions (pH > 8) significantly accelerated the decomposition of H2O2 [139]. Teranishi et al. also found that lower temperatures inhibited the decomposition of H2O2. Compared with aliphatic alcohols, alcohols with aromatic structures can significantly inhibit the decomposition of H2O2 produced by TiO2-based photocatalysts [140]. Zhang et al. found that the H2O2 decomposition constants (Kd) of benzyl alcohol (BA) and furfuryl alcohol (FFA) were significantly lower than those of isopropyl alcohol (IPA) at all concentrations. The authors concluded that BA and FFA could combine on the surface of TiO2 to form a more stable complex, which hindered the adsorption of H2O2 and thus inhibited the decomposition of H2O2 [141] (Table 4).
In-situ environmental applications of photocatalytic H2O2 production
H2O2 is widely used as a green oxidizer, such as hydrogen production [142, 143], sterilization, degradation of pollutants [144, 145], sewage treatment [146, 147] and so on. Taking into account some other factors, in-situ application has the advantages of no storage and transportation, and has been widely studied. The following mainly introduces its two in-situ environment applications.
As one of the commonly used fungicides, H2O2 can directly change the permeability of cell membranes to achieve bactericidal and bacteriostatic effects. Some photocatalytic nanomaterials have peroxidase-like properties, which can convert the in-situ production of H2O2 into ·OH with a stronger oxidizing ability under sunlight excitation. Typically, photocatalytic H2O2 production is desired to inhibit decomposition to obtain higher yields of H2O2. However, in the environmental field, photocatalytic H2O2 production is desired to rapidly decompose and activate to produce ·OH, which is used for in-situ sterilization or removal of organic pollutants.
Photocatalytic H2O2 production for in-situ disinfection
H2O2 produced by photocatalysis can be converted into oxygen-active substances (ROS) such as ·OH for efficient in-situ sterilization. Ideally, H2O2 is produced efficiently and consumed in time. However, current catalysts have poor H2O2 activation capacity, which limits the in-situ application of H2O2. The construction of heterogeneous structures is favorable for improving the separation efficiency of photogenerated carriers and increasing the synthesis rate of H2O2. In addition, metal sulfides with reducing active sites are co-catalysts for the decomposition of H2O2, which is conducive to the application of H2O2 in the in-situ environment. The photogenerated carrier separation and transfer efficiency of ZnIn2S4/g-C3N4 heterojunction developed by Shao et al. is high, and the H2O2 production rate can reach 798.11 μmol h−1 g−1. At the same time, the in-situ production of H2O2 through the cascade reaction was rapidly activated to ·OH, which can cooperate with ·O2− and h+ produced in the process of e−ORR to efficiently sterilize. Most E. coli and S. aureus were killed after 25 min and 60 min, respectively [148]. Geng et al. used ZnO/g-C3N4 heterostructures prepared by thermal condensation polymerization to generate H2O2 in natural water bodies under simulated sunlight and achieve efficient bacterial inactivation. The heterojunction constructed by ZnO and g-C3N4 effectively promoted charge separation, and the yield of H2O2 was 2.65 times higher than that of g-C3N4. Subsequently, H2O2 was converted to ·OH and ·O2− with strong oxidizing properties, and the molds were completely inactivated after 20 min, and the inactivation rate of bacterial microorganisms in the water body reached 97.4% after 60 min [149]. Wang et al. fixed WSe2 nanosheets on the surface of g-C3N4 by in-situ growth method, and the yield of H2O2 was 11.8 times higher than that of g-C3N4. The introduction of the co-catalyst WSe2 not only promoted the visible light absorption but also the Z-scheme heterojunction constructed by WSe2 and g-C3N4 facilitated the spatial separation of charges, with electrons and holes enriched on the CB of WSe2 and the VB of g-C3N4, respectively. The in-situ produced H2O2 was activated by the addition of Fe(II) to produce ·OH and E. coli was completely inactivated within 150 min [150].
Biomimetic nanase has enzyme-like activity and simulates the cascade catalytic reaction of natural enzymes in living organisms. Bionic nanase with a double function of oxidase and peroxidase can catalyze the production and consumption of H2O2 to produce free radicals to achieve efficient sterilization. Qu et al. developed biomimetic nanoenzymes by combining Cu and a monolayer of graphite carbon nitride (Cu2+–C3N4), which is dual-functional of oxidase and peroxidase. Cu2+ acts as an electron acceptor to inhibit the recombination of charge effectively. In the enzyme-like cascade system, Cu2+–C3N4 firstly simulates glucose oxidase (GOx) coupling glucose oxidation and O2 reduction for the production of H2O2 and then simulates horseradish peroxidase (HRP) catalyzing the decomposition of H2O2 to ·OH, which can efficiently kill Shewanella putrefaciens [151].
Photocatalytic H2O2 production for in-situ degradation of pollutants
Adding additional H2O2 to the system is a common strategy for degrading contaminants such as dyes [152,153,154] and antibiotics [155, 156]. Tong et al. achieved effective degradation of doxycycline (DC) by adding additional H2O2 to the light /BiVO4@Ag2O system, and expanded the suitable degradation pH to 3.5–9.5 [157].
Considering the inconvenience of transportation and storage of H2O2, small-scale on-site production of H2O2 for in-situ degradation of pollutants using photocatalysis is an effective and safe energy-saving strategy. Although the concentration of H2O2 prepared by photocatalysis is low, it is sufficient for environmental remediation. Hu et al. investigated the photocatalytic production of H2O2 and in-situ degradation of tetracycline by ultra-thin g-C3N4 nanosheets prepared with different precursor systems. g-C3N4 nanosheets (DCN) prepared from dicyandiamide were found to be the best, which was attributed to the 2e−ORR of DCN with a large number of unpaired electrons, a suitable energy band structure, and a fast charge separation and transfer efficiency [158]. Chen et al. introduced benzene into the CN backbone along with a molten salt heat treatment to promote electron delocalization. It was found that the co-doping of benzene with K+ could enhance the charge separation of g-C3N4 and increase the yield of H2O2. Then H2O2 was converted to ·OH in situ in the absence of Fe2+, and the degradation rates of RhB and Congo red (CR) were 93.3% and 96.6%, respectively. The development of heterojunction photocatalysts with magnetic properties not only efficiently degrades pollutants, but also facilitates separation and reuse, thus saving treatment costs [159]. Kumar et al. used hydrothermal method and multi-step precipitation to prepare a magnetic Ag/s-(Co3O4/NiFe2O4) heterojunction photocatalyst, which can produce a large amount of H2O2 in pure water under visible light and effectively degrade tetracycline in situ. NiFe2O4 is magnetic, which is conducive to the separation and reuse of the catalyst [160].
To accelerate the hole oxidation kinetics, it is usually necessary to use a high concentration of sacrificial agent (such as ethanol, isopropanol, etc.) to obtain a high H2O2 yield. Electrostatic adsorption of organic pollutants on the surface of the photocatalyst, as an electron donor replacement sacrificial agent, not only helps to accelerate the hole oxidation rate and promote the generation of more H2O2 but also facilitates the partial oxidation of the pollutant molecules themselves. Xu et al. constructed a photocatalytic system coupled with H2O2 generation and activated degradation of pollutants. A novel carbon nitride nanorod (GCN-Rod) was developed by alkali-assisted and functionalized methods. The introduction of the K-NC2 site made the Zeta potential on the catalyst surface negatively charged, and the surface of the carbon nitride nanorods adsorbed a large number of protons. Then, tetracycline was protonated and electrostatically adsorbed on the negatively charged catalyst surface, which accelerated the hole oxidation kinetics. The EPR and energy band structure confirmed that the active oxygen originated from H2O2, and tetracycline was degraded by photocatalytic production of H2O2 and in-situ conversion of oxygen actives formed by ·OH and ·O2− under visible light, and the degradation rate was higher than 95% within 15 min [161].
The conventional Fenton process can effectively degrade pollutants selectively, but the addition of H2O2 and the low utilization efficiency significantly increase the cost, which limits its wide application. As an oxidant of the Fenton reaction, the photocatalytic yield and utilization efficiency of H2O2 directly determine the degradation performance. However, the photocatalytic-self-Fenton cascade reaction not only eliminates the need for additional H2O2 but also efficiently utilizes in-situ produced H2O2 through timely decomposition and accelerates the degradation of pollutants in coordination with iron-based catalysts or Fe2+, which is considered to be one of the most efficient and promising wastewater treatment processes. The construction of heterojunctions is a common modification strategy for the preparation of highly efficient photocatalysts that significantly promote the spatial separation of carriers. Li et al. designed P–C3N4/O–C3N4 heterojunctions, and the materials realized the effective separation of photogenerated carriers under the effect of the built-in electric field, and the synthesis rate of H2O2 was 7.2 times higher than that of C3N4. The addition of Fe2+ degraded 91.6% of metronidazole within 1 h. The photocatalytic and in-situ Fenton cascade mode significantly increased the utilization of H2O2 to 77.7%, which was 9 times higher than that of the conventional homogeneous Fenton reaction [162]. Fe2(MoO4)3 is considered as a material with high Fenton reactivity and visible light trapping ability, which is beneficial for accelerated degradation of pollutants when used in photocatalytic Fenton system. Ma et al. developed Fe2(MoO4)3/Ag/Ag3PO4 Z-scheme heterojunctions, which not only enhanced the visible light absorbing ability but also promoted the charge separation. In addition, the Z-scheme heterostructure retains a high oxidation potential, and the Fe2(MoO4)3 nanoparticles have a high Fenton reactivity. Catalase experiments showed that the produced H2O2 was immediately converted to ·OH in situ on the surface of Fe2(MoO4)3. 96% of RhB was degraded within 5 min, which was about 254 and 7 times higher than that of Fe2(MoO4)3 and Ag3PO4, respectively, and the degradation efficiency of 91.2% was still maintained after 5 cycles [163]. Sun et al. constructed a red mud/cadmium sulfide (RM/CdS) S-scheme heterojunction, loaded RM particles from the waste of an aluminum oxidation plant onto CdS nanospheres, realized in-situ production and activated H2O2 to generate ·OH at the same time, effectively degrading cephalosporin (AMX) antibiotics. The built-in electric field (BIEF) formed at the interface of RM and CdS promotes the spatial separation of charge carriers [164].
The photocatalytic self-Fenton system not only makes H2O2 utilized efficiently but also continuously produces abundant ·OH [165]. Compared with the photocatalytic system and Fenton system, the photocatalytic self-Fenton system significantly improved the degradation and mineralization performance. Ma et al. utilized the in-situ generation of H2O2 from g-C3N4 under visible light irradiation for the degradation and mineralization of 2,4-dichlorophenol. Compared with the C3N4 photocatalytic system and the Fenton system, the degradation performance was improved by 20.5 and 3.7 times, and the mineralization degree was increased by 22.5 and 3.8 times, respectively [166]. Wang et al. constructed a photocatalytic self-Fenton system by oxygen-doped porous g-C3N4 nanosheets and adding Fe3+. Porous nanosheets and oxygen doping accelerated the photogenerated charge transfer and provided more reaction sites for the production of H2O2. The additional Fe3+ was reduced to Fe2+ by photogenerated electrons while inhibiting charge recombination and promoting the cyclic conversion of Fe3+/Fe2+. Finally, H2O2 was efficiently utilized to produce abundant ·OH through heterogeneous Fenton reaction. The degradation rate of 2,4-dichlorophenol was 11.5 and 9.9 times higher than that of the g-C3N4 photocatalytic system and the Fenton system, respectively, and the mineralization rate was 11.4 and 4.2 times higher, respectively [167]. Lu et al. utilized 4-carboxyphenylboronic acid edge covalently modified g-C3N4 (CPBA-CN) for the photocatalytic removal of persistent 4-chlorophenol (4-CP) by a photocatalytic self-Fenton. The photocatalyzed in-situ formation of H2O2 on CPBA-CN, the photogenerated electrons help accelerate the regeneration of Fe2+ to improve the utilization efficiency of H2O2, and the photogenerated holes promote the enhancement of 4-CP mineralization. Under the conjugation of CPBA, the electronic structure of CN was optimized and the molecular dipole was enhanced, leading to the deepening of the VB position, the accelerated separation of electron–hole pairs, and the improved adsorption and activation of O2. Therefore, the increment of the 4-CP degradation rate in the CPBA-CN photocatalytic self-Fenton process was close to 0.099 min−1, which was 3.1 times higher than that of photocatalysis. The parallel mineralization efficiency increased to 74.6%, which was 2.1 and 2.6 times higher than that of photocatalysis and Fenton, respectively [168].
The degradation of pollutants by photocatalytic self-Fenton cascade reactions occurring at natural pH is a sustainable process for wastewater treatment. However, exploring photocatalytic self-Fenton with efficient pollutant degradation performance at natural pH is challenging, which limits its wide application. Jiang et al. developed a photocatalytic self-Fenton system of CdS/rGO/Fe2+, which achieved in-situ generation of H2O2 at natural pH and timely formation of ·OH by combining with Fe2+ in the system. Phenol could be completely degraded in 1 h, and the mineralization rate is 43.66%. The system can work without pH adjustment, which overcomes the limitation of the traditional Fenton process to work in an acidic pH environment [169]. Shi et al. used in-situ deposition to homogeneously anchor amorphous FeOOH QDs on the surface of ultrathin porous g-C3N4 (UPCN), with a yield of 23.91 μmol L−1 of H2O2 under visible light. The ultrathin porous structure promotes the rapid transfer of photogenerated electrons from the UPCN to the amorphous FeOOH QDs, and at the same time facilitates the reduction of Fe3+ to produce Fe2+. The FeOOH QDs converted the H2O2 to ·OH in situ, and the degradation rate of oxytetracycline (OTC) at natural pH reached 86.23% [170]. Li et al. prepared a FeS2-modified resorcinol formaldehyde resin (FeS2-RFR) with dual active sites for the in-situ generation of H2O2 and utilized it for the efficient removal of organic arsenic contaminants (ROX). The resorcinol formaldehyde resin efficiently produced H2O2 under simulated sunlight irradiation with yields up to 500 μmol g−1 h−1. Subsequently, Fe2+ ionized from FeS2 decomposed H2O2 to ·OH in situ, which maintained a high degradation capacity over a wide pH range (pH = 2.8–6.8). The degradation of ROX was as high as 97% in 2 h, and the highly toxic ROX was oxidized to the low toxicity As(V) [171]. Our research group used FeOOH QDs modified resorcinol formaldehyde resin to achieve efficient degradation of Roxarsone. FeOOH QDs acted as activators and electron shuttles to decompose H2O2 produced in situ by light excitation. ·OH, ·O2−, and 1O2 participated in the degradation, of which 1O2 was the dominant reactive species. The system exhibited excellent reusability and stability over a wide pH range, providing a new strategy for the efficient in-situ removal of ROX from water [172] (Fig. 17).

Summary and outlook
Photocatalytic H2O2 production is economical, clean, and sustainable, and has attracted wide attention. In this paper, the basic principles of photocatalytic H2O2 production are reviewed, and there are three main pathways, namely, oxygen reduction (ORR), water oxidation (WOR), and dual-pathway synthesis of H2O2. The advanced photocatalysts for H2O2 production and their modification methods are highlighted. TiO2 and g-C3N4 are the most widely studied advanced photocatalysts, and the modification methods mainly include morphology control, defect control, elemental doping, loading of noble metal nanoparticles, and construction of heterojunction structure. The emerging photocatalysts BiVO4, MOFs, COFs, RFs, and HOFs have attracted attention because of their high activity. Then this paper introduces several more advanced systems and improvement strategies from the reactant and product perspectives. The establishment of a spontaneous system without a sacrificial agent and the construction of a gas–liquid–solid three-phase system is in the direction of greener and more sustainable development, and the utilization of pure water or seawater, as well as the abundant O2 in the atmosphere for photocatalytic production, is an inevitable trend for the future development of this field. In addition to improving the activity of photocatalytic production, inhibiting the decomposition of H2O2 is also important for maximizing yield. Finally, the in-situ environmental applications of photocatalytic H2O2 production are introduced, which mainly include in-situ sterilization and disinfection and in-situ degradation of pollutants. The photocatalytic H2O2 production technology is at a challenging stage of development, and the yield is expected to be further increased, which requires not only exploring more advanced semiconductors but also further optimizing the whole reaction system. Currently, some photocatalytic H2O2 production systems have reached the millimolar level, but they are still a long way away from industrial application in terms of feasibility and economy, and the future production process will be developed in the direction of being more efficient, green, and clean.
References
K. Sato, M. Aoki, R. Noyori, Science 281, 1646 (1998)
G. Luo, Y. Jiao, X. Lv, X. Zhang, X. Gao, Res. Chem. Intermed. 44, 5377 (2018)
L.K. Verna, S.A. Holman, V.C. Lee, J. Hoh, Cell Biol. Toxicol. 16, 303 (2000)
X. Wei, X. Fu, S. Lyu, Res. Chem. Intermed. 48, 4459 (2022)
C. Hao, C. Jun, J. Lu, W. Lei, H. Xu, Chem. Mater. 34, 4259 (2022)
S. Mansoor, M. Tayyab, M. Khan, Z. Akmal, L. Zhou, J. Lei, M. Anpo, J. Zhang. Res. Chem. Intermed. 49, 3723 (2023)
Y. Yamada, M. Yoneda, S. Fukuzumi, Energy Environ. Sci. 8, 1698 (2015)
P. Sun, C. Tyree, C.-H. Huang, Environ. Sci. Technol. 50, 4448 (2016)
J.M. Campos-Martin, G. Blanco-Brieva, J.L.G. Fierro, Angew. Chem. Int. Ed. 45, 6962 (2006)
S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff, I.E.L. Stephens, ACS Catal. 8, 4064 (2018)
R. Bortolo, D. Bianchi, R. D’Aloisio, C. Querci, M. Ricci, J. Mol. Catal. A-Chem. 153, 25 (2000)
Y. Yi, L. Wang, J. Yu, H. Guo, J. Zhang, C. Meng, AICHE J. 64, 981 (2018)
N. Zhang, S. Lin, F. Wang, Y. Liu, J. Zhang, L. Zhou, J. Lei. Res. Chem. Intermed. 47, 3379 (2021)
L.X. Wang, J.J. Zhang, Y. Zhang, H.G. Yu, Y.H. Qu, J.G. Yu, Small 18, 2104561 (2022)
J. Low, J. Yu, M. Jaroniec, S. Wageh, A.A. Al-Ghamdi, Adv. Mater. 29, 1601694 (2017)
X.K. Zeng, Y. Liu, X.Y. Hu, X.W. Zhang, Green Chem. 23, 1466 (2021)
G.J. Rani, M.A.J. Rajan, G.G. Kumar, Res. Chem. Intermed. 43, 2669 (2017)
H. Ye, S. Lu, Res. Chem. Intermed. 41, 139 (2015)
E.M. Goliaei, N. Seriani, J. Phys. Chem. C 123, 2855 (2019)
Y. Xia, J. Yu, Chem 6, 1039 (2020)
X. Li, C. Chen, J. Zhao, Langmuir 17, 4118 (2001)
C. Yang, Y. He, K. Li, Y. Yao, R. Cao, Y. Wang, J. Jia. Res. Chem. Intermed. 41, 5365 (2015)
A.E. Gaidoumi, J.M. Doña-Rodríguez, E.P. Melián, O.M. González-Díaz, B.E. Bali, J. Antonio Navío, A. Kherbeche, Res. Chem. Intermed. 45, 333 (2019)
M. Teranishi, S. Naya, H. Tada, J. Am. Chem. Soc. 132, 7850 (2010)
K. Kim, J. Park, H. Kim, G.Y. Jung, M.-G. Kim, ACS Catal. 9, 9206 (2019)
M.R. Miah, T. Ohsaka, Anal. Chem. 78, 1200 (2006)
D. Tsukamoto, A. Shiro, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka, T. Hirai, ACS Catal. 2, 599 (2012)
C. Chu, D. Huang, Q. Zhu, E. Stavitski, J.A. Spies, Z. Pan, J. Mao, H.L. Xin, C.A. Schmuttenmaer, S. Hu, J.-H. Kim, ACS Catal. 9, 626 (2019)
L. Wang, S. Cao, K. Guo, Z. Wu, Z. Ma, L. Piao, Chin. J. Catal. 40, 470 (2019)
S. Cao, T.-S. Chan, Y.-R. Lu, X. Shi, B. Fu, Z. Wu, H. Li, K. Liu, S. Alzuabi, P. Cheng, M. Liu, T. Li, X. Chen, L. Piao, Nano Energy 67, 104287 (2020)
L.H. Zheng, H.R. Su, J.Z. Zhang, L.S. Walekar, H.V. Molamahmood, B.X. Zhou, M.C. Long, Y.H. Hu, Appl. Catal. B-Environ. 239, 475 (2018)
G.H. Moon, W. Kim, A.D. Bokare, N.E. Sung, W. Choi, Energy Environ. Sci. 7, 4023 (2014)
X. Zeng, Z. Wang, G. Wang, T.R. Gengenbach, D.T. McCarthy, A. Deletic, J. Yu, X. Zhang, Appl. Catal. B-Environ. 218, 163 (2017)
W.-Y. Hu, Q.-Y. Li, G.-Y. Zhai, Y.-X. Lin, D. Li, X.-X. He, X. Lin, D. Xu, L.-H. Sun, S.-N. Zhang, J.-S. Chen, X.-H. Li, Small 18, 2200885 (2022)
W. Hu, Q. Li, D. Xu, G. Zhai, S. Zhang, D. Li, X. He, J. Jia, J. Chen, X. Li, Nano Res. 15, 10142 (2022)
Y. Chen, W. Gu, L. Tan, Z. Ao, T. An, S. Wang, Appl. Catal. A 618, 118127 (2021)
L. Feng, B. Li, Y. Xiao, L. Li, Y. Zhang, Q. Zhao, G. Zuo, X. Meng, V.A.L. Roy, Catal. Commun. 155, 106315 (2021)
A. Behera, P. Babu, K. Parida, Inorg. Chem. Front. 8, 1489 (2021)
Y. Yang, B. Cheng, J.G. Yu, L.X. Wang, W.K. Ho, Nano Res. 16, 4506 (2023)
L. Wang, J. Zhang, H. Yu, I.H. Patir, Y. Li, S. Wageh, A.A. Al-Ghamdi, J. Yu, J. Phys. Chem. Lett. 13, 4695 (2022)
Z. Jiang, Q. Long, B. Cheng, R. He, L. Wang, J. Mater. Sci. Technol. 162, 1 (2023)
E. Salehi Ghalehsefid, Z. Ghorbani Jahani, A. Aliabadi, M. Ghodrati, A. Khamesan, A. Parsaei-Khomami, M. Mousavi, M.A. Hosseini, J.B. Ghasemi, X. Li, J. Environ. Chem. Eng. 11, 110160 (2023)
S. Cao, J. Yu, J. Phys. Chem. Lett. 5, 2101 (2014)
Y. Shiraishi, Y. Kofuji, H. Sakamoto, S. Tanaka, S. Ichikawa, T. Hirai, ACS Catal. 5, 3058 (2015)
H. Ou, P. Yang, L. Lin, M. Anpo, X. Wang, Angew. Chem. Int. Ed. 56, 10905 (2017)
W. Liu, C. Song, M. Kou, Y. Wang, Y. Deng, T. Shimada, L. Ye, Chem. Eng. J. 425, 130615 (2021)
L. Zhou, J. Feng, B. Qiu, Y. Zhou, J. Lei, M. Xing, L. Wang, Y. Zhou, Y. Liu, J. Zhang. Appl. Catal. B-Environ. 267, 118396 (2020)
Y. Xu, W. Tai, Z. Wang, L. Zhang, D. Wang, J. Liao. Sci. China Mater. (Early Access) (2023)
S. Li, G. Dong, R. Hailili, L. Yang, Y. Li, F. Wang, Y. Zeng, C. Wang, Appl. Catal. B-Environ. 190, 26 (2016)
H. Xie, Y.M. Zheng, X.L. Guo, Y.Y. Liu, Z. Zhang, J.J. Zhao, W.J. Zhang, Y.X. Wang, Y. Huang, ACS Sustain. Chem. Eng. 9, 6788 (2021)
J. Lei, B. Chen, W. Lv, L. Zhou, L. Wang, Y. Liu, J. Zhang, ACS Sustain. Chem. Eng. 7, 16467 (2019)
Y. Zheng, Y. Luo, Q. Ruan, J. Yu, X. Guo, W. Zhang, H. Xie, Z. Zhang, J. Zhao, Y. Huang, J. Colloid Interface Sci. 609, 75 (2022)
Y. Wang, D. Meng, X. Zhao, Appl. Catal. B-Environ. 273, 119064 (2020)
X. Zhang, P.J. Ma, C. Wang, L.Y. Gan, X.J. Chen, P. Zhang, Y. Wang, H. Li, L.H. Wang, X.Y. Zhou, K. Zheng, Energy Environ. Sci. 15, 830 (2022)
Y. Cong, S. Zhang, Q. Zheng, X. Li, Y. Zhang, S.-W. Lv, J. Colloid Interface Sci. 650, 1013 (2023)
S. Wu, H. Yu, S. Chen, X. Quan, ACS Catal. 10, 14380 (2020)
L. Chen, C. Chen, Z. Yang, S. Li, C.H. Chu, B.L. Chen, Adv. Funct. Mater. 31, 2105731 (2021)
J. Yuan, N. Tian, Z. Zhu, W. Yu, M. Li, Y. Zhang, H. Huang, Chem. Eng. J. 467, 143379 (2023)
H. Wang, Y. Guan, S. Hu, Y. Pei, W. Ma, Z. Fan, NANO 14, 1950023 (2019)
C. Feng, L. Tang, Y. Deng, J. Wang, J. Luo, Y. Liu, X. Ouyang, H. Yang, J. Yu, J. Wang. Adv. Funct. Mater. 30, 2001922 (2020)
C. Feng, L. Tang, Y. Deng, J. Wang, Y. Liu, X. Ouyang, H. Yang, J. Yu, J. Wang. Appl. Catal. B-Environ. 281, 119539 (2021)
L. Xue, H. Sun, Q. Wu, W. Yao, J. Colloid Interface Sci. 615, 87 (2022)
H. Kim, K. Shim, K.E. Lee, J.W. Han, Y. Zhu, W. Choi, Appl. Catal. B-Environ. 299, 120666 (2021)
C. Zhang, J. Bai, L. Ma, Y. Lv, F. Wang, X. Zhang, X. Yuan, S. Hu, Diam. Relat. Mat. 87, 215 (2018)
H. Che, X. Gao, J. Chen, J. Hou, Y. Ao, P. Wang, Angew. Chem. Int. Ed. 60, 25546 (2021)
J. Zhang, C. Yu, J. Lang, Y. Zhou, B. Zhou, Y.H. Hu, M. Long, Appl. Catal. B-Environ. 277, 119225 (2020)
S. Hu, X. Qu, P. Li, F. Wang, Q. Li, L. Song, Y. Zhao, X. Kang, Chem. Eng. J. 334, 410 (2018)
R. Du, K. Xiao, B. Li, X. Han, C. Zhang, X. Wang, Y. Zuo, P. Guardia, J. Li, J. Chen, J. Arbiol, A. Cabot, Chem. Eng. J. 441, 135999 (2022)
Q. Hu, Y. Huang, X. Yu, S. Gong, Y. Wen, Y. Liu, G. Li, Q. Zhang, R. Ye, X. Chen, ACS Appl. Mater. Interfaces 15, 42611 (2023)
S. Zhao, T. Guo, X. Li, T. Xu, B. Yang, X. Zhao, Appl. Catal. B-Environ. 224, 725 (2018)
Y.W. Shan, Y. Guo, Y. Wang, X.R. Du, J. Yu, H. Luo, H. Wu, B. Boury, H. Xiao, L.L. Huang, L.H. Chen, J. Colloid Interface Sci. 599, 507 (2021)
Y.L. Peng, L. Zhou, L.Z. Wang, J.Y. Lei, Y.D. Liu, S. Daniele, J.L. Zhang, Res. Chem. Intermed. 45, 5907 (2019)
Y. Yang, Z.T. Zeng, G.M. Zeng, D.L. Huang, R. Xiao, C. Zhang, C.Y. Zhou, W.P. Xiong, W.J. Wang, M. Cheng, W.J. Xue, H. Guo, X. Tang, D.H. He, Appl. Catal. B-Environ. 258, 117956 (2019)
S.F. Lin, N. Zhang, F.C. Wang, J.Y. Lei, L. Zhou, Y.D. Liu, J.L. Zhang, ACS Sustain. Chem. Eng. 9, 481 (2021)
C. Chu, Q. Zhu, Z. Pan, S. Gupta, D. Huang, Y. Du, S. Weon, Y. Wu, C. Muhich, E. Stavitski, K. Domen, J.-H. Kim, Proc. Natl. Acad. Sci. 117, 6376 (2020)
X. Zhang, H. Su, P. Cui, Y. Cao, Z. Teng, Q. Zhang, Y. Wang, Y. Feng, R. Feng, J. Hou, X. Zhou, P. Ma, H. Hu, K. Wang, C. Wang, L. Gan, Y. Zhao, Q. Liu, T. Zhang, K. Zheng, Nat. Commun. 14, 7115 (2023)
H. Tan, P. Zhou, M. Liu, Q. Zhang, F. Liu, H. Guo, Y. Zhou, Y. Chen, L. Zeng, L. Gu, Z. Zheng, M. Tong, S. Guo, Nat. Synth. 2, 557 (2023)
H. Hirakawa, S. Shiota, Y. Shiraishi, H. Sakamoto, S. Ichikawa, T. Hirai, ACS Catal. 6, 4976 (2016)
T. Liu, Z. Pan, J.J.M. Vequizo, K. Kato, B. Wu, A. Yamakata, K. Katayama, B. Chen, C. Chu, K. Domen, Nat. Commun. 13, 1034 (2022)
T. Liu, Z. Pan, K. Kato, J.J.M. Vequizo, R. Yanagi, X. Zheng, W. Yu, A. Yamakata, B. Chen, S. Hu, K. Katayama, C. Chu, Nat. Commun. 13, 7783 (2022)
D. Dai, X. Bao, Q. Zhang, Z. Wang, Z. Zheng, Y. Liu, H. Cheng, Y. Dai, B. Huang, P. Wang, Chem. A Eur. J. 29, e202203765 (2023)
Y. Isaka, Y. Kondo, Y. Kawase, Y. Kuwahara, K. Mori, H. Yamashita, Chem. Commun. 54, 9270 (2018)
Y. Isaka, Y. Kawase, Y. Kuwahara, K. Mori, H. Yamashita, Angew. Chem. Int. Ed. 58, 5402 (2019)
Y. Kawase, Y. Isaka, Y. Kuwahara, K. Mori, H. Yamashita, Chem. Commun. 55, 6743 (2019)
C. Xiaolang, Y. Kuwahara, K. Mori, C. Louis, H. Yamashita, J. Mater. Chem. A 8, 1904 (2020)
X. Chen, Y. Kuwahara, K. Mori, C. Louis, H. Yamashita, ACS Appl. Energ. Mater. 4, 4823 (2021)
L. Yujie, M. Fahao, Z. Liren, L. Yuanyuan, W. Zeyan, W. Peng, Z. Zhaoke, C. Hefeng, D. Ying, H. Baibiao, Mater. Horizons 8, 2842 (2021)
J.H. You, Y. Zhao, L. Wang, W.T. Bao, J. Clean. Prod. 291, 125822 (2021)
C. Krishnaraj, H.S. Jena, L. Bourda, A. Laemont, P. Pachfule, J. Roeser, C.V. Chandran, S. Borgmans, S.M.J. Rogge, K. Leus, C.V. Stevens, J.A. Martens, V. Van Speybroeck, E. Breynaert, A. Thomas, P. Van der Voort, J. Am. Chem. Soc. 142, 20107 (2020)
S. Zhou, Y. Shi, G. Chen, W. Kong, H. Hu, H. Xie, C. Li, J. Qin, Z. Zhang, L. Peng, X. Ke, Y. Kong, Chem. Eng. J. 477, 146946 (2023)
X. Yu, B. Viengkeo, Q. He, X. Zhao, Q. Huang, P. Li, W. Huang, Y. Li, Adv. Sustain. Syst. 5, 2100184 (2021)
C. Wu, Z. Teng, C. Yang, F. Chen, H.B. Yang, L. Wang, H. Xu, B. Liu, G. Zheng, Q. Han, Adv. Mater. 34, 2110266 (2022)
H.Z. Wang, C. Yang, F.S. Chen, G.F. Zheng, Q. Han, Angew. Chem. Int. Ed. 61, e202202328 (2022)
L. Chen, L. Wang, Y.Y. Wan, Y. Zhang, Z.M. Qi, X.J. Wu, H.X. Xu, Adv. Mater. 32, 1904433 (2020)
H. Cheng, H.F. Lv, J. Cheng, L. Wang, X.J. Wu, H.X. Xu, Adv. Mater. 34, 2107480 (2022)
M.P. Kou, Y.Y. Wang, Y.X. Xu, L.Q. Ye, Y.P. Huang, B.H. Jia, H. Li, J.Q. Ren, Y. Deng, J.H. Chen, Y. Zhou, K. Lei, L. Wang, W. Liu, H.W. Huang, T.Y. Ma, Angew. Chem. Int. Ed. 61, e202200413 (2022)
Y. Zhang, J. Qiu, B. Zhu, M.V. Fedin, B. Cheng, J. Yu, L. Zhang, Chem. Eng. J. 444, 136584 (2022)
H. Wang, E. Almatrafi, Z. Wang, Y. Yang, T. Xiong, H. Yu, H. Qin, H. Yang, Y. He, C. Zhou, G. Zeng, P. Xu, J. Colloid Interface Sci. 608, 1051 (2022)
Y. Luo, B. Zhang, C. Liu, D. Xia, X. Ou, Y. Cai, Y. Zhou, J. Jiang, B. Han, Angew. Chem. Int. Ed. 62, e202305355 (2023)
Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa, T. Hirai, Nat. Mater. 18, 985 (2019)
Y. Shiraishi, T. Hagi, M. Matsumoto, S. Tanaka, S. Ichikawa, T. Hirai, Comm. Chem. 3, 169 (2020)
Y. Shiraishi, M. Matsumoto, S. Ichikawa, S. Tanaka, T. Hirai, J. Am. Chem. Soc. 143, 12590 (2021)
Y. Shiraishi, K. Miura, M. Jio, S. Tanaka, S. Ichikawa, T. Hirai, ACS Mater. Au 2, 709 (2022)
C. Zhao, X. Wang, Y. Yin, W. Tian, G. Zeng, H. Li, S. Ye, L. Wu, J. Liu. Angew. Chem. Int. Ed. 62, e202218318 (2023)
X. Wang, X. Yang, C. Zhao, Y. Pi, X. Li, Z. Jia, S. Zhou, J. Zhao, L. Wu, J. Liu. Angew. Chem. Int. Ed. 62, e202302829 (2023)
R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou, B. Chen, Chem. Soc. Rev. 48, 1362 (2019)
M.R. di Nunzio, I. Hisaki, A. Douhal, J. Photochem. Photobiol. C Photochem. Rev. 47, 100418 (2021)
P. Wu, X. Yin, Y. Zhao, F. Li, Y. Yang, N. Liu, J. Liao, T. Lan, J. Hazard. Mater. 459, 132179 (2023)
B. Yu, L. Li, S. Liu, H. Wang, H. Liu, C. Lin, C. Liu, H. Wu, W. Zhou, X. Li, T. Wang, B. Chen, J. Jiang. Angew. Chem. Int. Ed. 60, 8983 (2021)
W. Huang, H. Yuan, H. Yang, L. Tong, R. Gao, X. Kou, J. Wang, X. Ma, S. Huang, F. Zhu, G. Chen, G. Ouyang, JACS Au 2, 2048 (2022)
S. Ghosh, S. Prasanthkumar, S. Das, A. Saeki, S. Seki, A. Ajayaghosh, Chem. Commun. 58, 6837 (2022)
M. Hu, C. Wu, S. Feng, J. Hua, Molecules 28, 6850 (2023)
Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata, C. Su, B. Liu, T. Ohno, Nat. Catal. 4, 374 (2021)
L. Xu, Y. Liu, L. Li, Z. Hu, J.C. Yu, ACS Catal. 11, 14480 (2021)
Y. Liu, X. Wang, Y. Zhao, Q. Wu, H. Nie, H. Si, H. Huang, Y. Liu, M. Shao, Z. Kang, Nano Res. 15, 4000 (2022)
C. Chu, Q. Li, W. Miao, H. Qin, X. Liu, D. Yao, S. Mao, Appl. Catal. B-Environ. 314, 121485 (2022)
Q. Wu, J. Cao, X. Wang, Y. Liu, Y. Zhao, H. Wang, Y. Liu, H. Huang, F. Liao, M. Shao, Z. Kang, Nat. Commun. 12, 483 (2021)
A. Gopakumar, P. Ren, J. Chen, B.V.M. Rodrigues, H.Y.V. Ching, A. Jaworski, S. Van Doorslaer, A. Rokicinska, P. Kustrowski, G. Barcaro, S. Monti, A. Slabon, S. Das, J. Am. Chem. Soc. 144, 2603 (2022)
T. Soltani, A. Yamamoto, S. Singh, A. Anzai, E. Fudo, A. Tanaka, H. Kominami, H. Yoshida, ACS Appl. Energ. Mater. 4, 6500 (2021)
Z. Liu, X. Sheng, D. Wang, X. Feng, Iscience 17, 67 (2019)
W. Xu, X. Sheng, H. Zhou, D. Wang, Z. Ding, X. Feng, Chem. Eng. J. 410, 128342 (2021)
L. Chen, S. Li, Z. Yang, C. Chen, C. Chu, B. Chen, Appl. Catal. B-Environ. 305, 121066 (2022)
L. Li, L. Xu, Z. Hu, J.C.Y. Yu, Adv. Funct. Mater. 31, 2106120 (2021)
M. Sun, X. Wang, Y. Li, H. Pan, M. Murugananthan, Y. Han, J. Wu, M. Zhang, Y. Zhang, Z. Kang, ACS Catal. 12, 2138 (2022)
Z. Chen, H. Chen, K. Wang, J. Chen, M. Li, Y. Wang, P. Tsiakaras, S. Song, ACS Catal. 13, 6497 (2023)
V. Maurino, C. Minero, E. Pelizzetti, G. Mariella, A. Arbezzano, F. Rubertelli, Res. Chem. Intermed. 33, 319 (2007)
R. Cai, Y. Kubota, A. Fujishima, J. Catal. 219, 214 (2003)
V. Maurino, C. Minero, G. Mariella, E. Pelizzetti, Chem. Commun. 20, 2627 (2005)
G. Zuo, B. Li, Z. Guo, L. Wang, F. Yang, W. Hou, S. Zhang, P. Zong, S. Liu, X. Meng, Y. Du, T. Wang, V.A.L. Roy, Catalysts 9, 623 (2019)
L. Zheng, J. Zhang, Y.H. Hu, M. Long, J. Phys. Chem. C 123, 13693 (2019)
L. Li, B. Li, L. Feng, X. Zhang, Y. Zhang, Q. Zhao, G. Zuo, X. Meng, Molecules 26, 3844 (2021)
T. Lee, H. Tran, J. Yoo, M. Ra, S.H. Han, W. Kim, W. Kwon, ACS Appl. Mater. Interfaces 11, 41196 (2019)
R.Y. Ma, L. Wang, H. Wang, Z.Y. Liu, M.Y. Xing, L.F. Zhu, X.J. Meng, F.S. Xiao, Appl. Catal. B-Environ. 244, 594 (2019)
J. Zhang, J. Lang, Y. Wei, Q. Zheng, L. Liu, Y.-H. Hu, B. Zhou, C. Yuan, M. Long, Appl. Catal. B-Environ. 298, 120522 (2021)
L. Zhou, J.Y. Lei, F.C. Wang, L.Z. Wang, M.R. Hoffmann, Y.D. Liu, S.I. In, J.L. Zhang, Appl. Catal. B-Environ. 288, 119993 (2021)
H. Yerin, C. Yongjoon, G. Eun Min, P. Sharma, C. Hyeonjin, L. Byongkyu, L. Sang Myeon, S.O. Park, K. Myohwa, K. Sang Kyu, Y. Changduk, J. Ji-Wook, Chem. Eng. J. 418, 129346 (2021)
J.H. Lee, H. Cho, S.O. Park, J.M. Hwang, Y. Hong, P. Sharma, W.C. Jeon, Y. Cho, C. Yang, S.K. Kwak, H.R. Moon, J.-W. Jang, Appl. Catal. B-Environ. 284, 119690 (2021)
J. Sun, H. Sekhar Jena, C. Krishnaraj, K. Singh Rawat, S. Abednatanzi, J. Chakraborty, A. Laemont, W. Liu, H. Chen, Y.-Y. Liu, K. Leus, H. Vrielinck, V. Van Speybroeck, P. Van Der Voort, Angew. Chem. Int. Ed. 62, e202216719 (2023)
B.O. Burek, D.W. Bahnemann, J.Z. Bloh, ACS Catal. 9, 25 (2019)
M. Teranishi, S.-I. Naya, H. Tada, J. Phys. Chem. C 120, 1083 (2016)
J. Zhang, L. Zheng, F. Wang, C. Chen, H. Wu, S.A.K. Leghari, M. Long, Appl. Catal. B Environ. 269, 118770 (2020)
P. Chen, H.Y. He, Res. Chem. Intermed. 40, 1947 (2014)
H.Y. He, Res. Chem. Intermed. 37, 1057 (2011)
M.A. Rahman, M. Muneer, D. Bahnemann, Res. Chem. Intermed. 29, 35 (2003)
H. Zhang, D. Liu, S. Ren, H. Zhang, Res. Chem. Intermed. 43, 1529 (2017)
C. Zhan, F. Chen, J. Yang, M. Zhong, J. Song, X. Jiang, Res. Chem. Intermed. 44, 2425 (2018)
S. Rahimi, B. Ayati, A. Rezaee, Res. Chem. Intermed. 43, 1935 (2017)
Y. Shao, J. Hu, T. Yang, X. Yang, J. Qu, Q. Xu, C.M. Li, Carbon 190, 337 (2022)
X.L. Geng, L. Wang, L. Zhang, H. Wang, Y.Y. Peng, Z.Y. Bian, Chem. Eng. J. 420, 129722 (2021)
W. Wang, W. Gu, G. Li, H. Xie, P.K. Wong, T. An, Environ. Sci.-Nano 7, 3877 (2020)
L. Qu, X. Fang, T. Xie, H. Xu, G. Yang, W. Liu, Sens. Actuator B-Chem. 353, 131156 (2022)
A. Chakib, K. Mohamed, A. Elaziouti, W. Touati, I.K. Allah, A. Benhamed, A. Bekka, Res. Chem. Intermed. 49, 1213 (2023)
V.B. Khajone, P.R. Bhagat, Res. Chem. Intermed. 46, 783 (2020)
W. Tan, J. Ai, Y. Fan, X. Liu, Y. Xu, H. Zhang, Y. Huang, Res. Chem. Intermed. 46, 4423 (2020)
E. Ouahiba, M. Chabani, A.A. Assadi, A. Abdeltif, F. Florence, B. Souad, Res. Chem. Intermed. 49, 1 (2023)
F. Pan, J. Yang, J. Cai, L. Liu, Res. Chem. Intermed. 47, 4595 (2021)
Y. Tong, Z. Chen, J. Kang, J. Deng, L. Sun, H. Liu, Res. Chem. Intermed. 49, 5471 (2023)
J. Hu, P. Zhang, T. Yang, Y. Cai, J. Qu, X. Yang, Appl. Surf. Sci. 576, 151841 (2022)
Y. Chen, X. Yan, H. Lin, C. Wang, J. Xu, J. Taiwan Inst. Chem. Eng. 131, 104179 (2022)
U. Kumar, J. Kuntail, A. Kumar, R. Prakash, M.R. Pai, I. Sinha, Appl. Surf. Sci. 589, 153013 (2022)
Z. Xu, S. Gong, W. Ji, S. Zhang, Z. Bao, Z. Zhao, Z. Wei, X. Zhong, Z.-T. Hu, J. Wang. Chem. Eng. J. 446, 137009 (2022)
J. Li, Y. Mei, S. Ma, Q. Yang, B. Jiang, B. Xin, T. Yao, J. Wu, J. Colloid Interface Sci. 608, 2075 (2022)
S. Ma, Y. Yang, J. Li, Y. Mei, Y. Zhu, J. Wu, L. Liu, T. Yao, Q. Yang, J. Colloid Interface Sci. 606, 1800 (2022)
X. Sun, K. He, Z. Chen, H. Yuan, F. Guo, W. Shi, Sep. Purif. Technol. 324, 124600 (2023)
S.A. El-Molla, Res. Chem. Intermed. 41, 4635 (2015)
J. Ma, K. Wang, C. Wang, X. Chen, W. Zhu, G. Zhu, W. Yao, Y. Zhu, Appl. Catal. B-Environ. 276, 119150 (2020)
F. Wang, J. Xu, Z. Wang, Y. Lou, C. Pan, Y. Zhu, Appl. Catal. B-Environ. 312, 121438 (2022)
T. Lu, H. Zhao, L. Jian, R. Ji, C. Pan, G. Wang, Y. Dong, Y. Zhu, Environ. Res. 222, 115361 (2023)
Z. Jiang, L. Wang, J. Lei, Y. Liu, J. Zhang. Appl. Catal. B-Environ. 241, 367 (2019)
W. Shi, W. Sun, Y. Liu, K. Zhang, H. Sun, X. Lin, Y. Hong, F. Guo, J. Hazard. Mater. 436, 129141 (2022)
X. Li, J. He, J. Lu, Y. Zhou, Y. Zhou, J. Hazard. Mater. 424, 127650 (2022)
W. Shen, L. Zhou, Y. Liu, J. Zhang, J. Lei. Res. Chem. Intermed. 49, 2569 (2023)
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22076046, 22176061), the Science and Technology Commission of Shanghai Municipality (21230712000), the Shanghai Pujiang Program (21PJD016), and the Fundamental Research Funds for the Central Universities (SLB13233301).
Author information
Authors and Affiliations
Contributions
Song Huang and Xingzi Yang Wrote original Draft; Liang Zhou and Yongdi Liu reviewed and revised the manuscript; Juying Lei and Jinlong Zhang reviewed and revised the manuscript and provided funding.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, S., Yang, X., Zhou, L. et al. Photocatalytic production of H2O2 and its in-situ environmental applications. Res Chem Intermed 50, 2917–2969 (2024). https://doi.org/10.1007/s11164-024-05301-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-024-05301-w