
Abstract
In the present work, we have fabricated a novel mesoporous TiO2–rGO nanocomposite by a facile one-step solvothermal method using titanic sulfate as the TiO2 source. The as-prepared composites were characterized by transmission electron microscopy, X-ray diffraction; UV–Vis diffuse reflectance spectra, X-ray photoelectron spectroscopy and photoluminence spectra. In situ nucleation and anchoring of TiO2 nanoparticles onto a graphene sheet is favorable fpr forming an intimate interfacial contact, and the chemically bonded TiO2–rGO nanocomposites commendably enhanced their photocatalytic activity in the photodegradation of rhodamine B and phenol. The high photocatalytic activity of the as-synthesized nanocomposites are primarily ascribed to the mesoporous structure, efficient charge transportation and separation with enhanced visible light absorption, which come from the appealing nanoarchitecture, for instance, ultra-dispersed and ultra-small TiO2 nanocrystals along with intimate and absolute interfacial contact between the TiO2 nanocrystals and the graphene sheet.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the recent years, semiconductor nanocrystals have drawn considerable interest on account of their exceptional physicochemical features and strong prospects for applications in solar energy conversion, supercapacitors, and the photodegradation of hazardous pollutants [1,2,3]. TiO2 photocatalysts have been regarded as one of the most efficient and reliable materials due to its superior photocatalytic performance, non-toxicity, low cost, biological and chemical inertness and long-term physical and chemical stability [4,5,6]. Undesirably, the fast recombination rate of photogenerated charge carriers within TiO2 results in its low quantum productivity, consequently deterring its real-world applications. In previous years, numerous approaches have been devised in an endeavor to improve the photocatalytic process and to enhance the photocatalytic performance of TiO2 photocatalysts [7]. In particular, carbon–titania nanocomposites have been attaining considerable responsiveness as a state-of-the-art class of photocatalysts [8, 9], which can potentially offer an appropriate ability for sorting out electron–hole pairs.
Recently, graphene, a single layer of carbon atoms, has enthralled researchers because of its extraordinarily specific surface area, thermal and mechanical properties, and better electron mobility, and many efforts have been made to use graphene as a substrate for producing very large numbers of functional nanohybrids with improved catalytic performance [10,11,12,13,14,15]. Although many exhilarating advancements have been accomplished in the synthesis of TiO2-reduced graphene nanocomposites, there still exist numerous complications hampering the further improvement of their photoactivity. The aggregated or large TiO2 nanoparticles are a big concern, which meaningfully reduced the surface–volume ratio of TiO2 and lengthens the diffusion distance of radiative electrons and holes, consequently resulting in a decline in photocatalytic activity [16]. Additionally, the random distribution of TiO2 nanoparticles, with a notable degree of exposed and undecorated graphene sheet or irreversible aggregation of graphene sheets encouraged by means of robust p–p interactions, lead to a substantial shrinkage of the available surface area and substrate approachability, hence losing material efficiency over a brief time frame [17,18,19]. Furthermore, most of the existing approaches for the preparation of TiO2 nanocrystals on graphene sheets involve environmentally detrimental surfactants (e.g., P123 [20], SDS [21], SDBS [22], CTAB [23], and PVP [24]), and complicated synthetic steps such as a two-step solution process, gas phase synthesis pathway, direct deposition and sol–gel methods [25,26,27,28,29,30,31,32] also profoundly restrict the industrialized expansion of the nanohybrids. The ability to control the size, morphology, dispersion density and strong interaction of inorganic nanocrystals on graphene sheets, permitting one to tailor nanocomposites toward a specific application, is still an imperative challenge, and demands more grit and determination, because it is extremely challenging to match their compatibilities and interactions as well as regulating the reduction of graphene oxide (GO) simultaneously in the synthetic process, which could thwart the transfer of electrons and thus impede applications in energy storage and catalysis [33, 34]. Compared to physically blending two compounds together, in which the pathetic interaction between TiO2 and graphene sheets is a foremost shortcoming, in situ anchoring of TiO2 nanocrystals onto graphene sheets through chemical bonds improves the stability of the nanocomposite and encourages charge separation and mutual electron transport between TiO2 and graphene [35]. It has been established that additives like polyaniline can be used as the face-controlling and/or linker agent to accomplish in situ growth of ultra-dispersed TiO2 nanoparticles on the surface of TiO2-graphene aerogels; however, the loading amount of TiO2 nanoparticles was small and difficult to control [36]. Therefore, it is still a challenge and immensely desired to accomplish in situ growth of TiO2 nanocrystals on a graphene sheet with sturdy binding interactions and well-regulated loading concentrations.
Here, we report a simple surfactant-free facile one-step solvothermal method for the synthesis of mesoporous TiO2 nanocrystals on graphene. In the present work, the TiO2 nanoparticles nucleated, grew and anchored on GO sheets using an inexpensive and stable titanium sulfate as a TiO2 source in a water and ethylene glycol system with a trivial amount of ammonia. The resultant mesoporous TiO2–rGO nanocomposites not only possess highly dispersed small-size anatase nanoparticles (~5–10 nm) but also exhibits enhanced visible light photocatalytic performance for the degradation of rhodamine B dye and phenol.
Experimental
Materials used
Graphite powder (<20 μm, synthetic), titanium sulfate (Ti (SO4)2), ethylene glycol, NaNO3 (99.0%), H2SO4 (95.0%), KMnO4 (99.3%), H2O2 (30%) and NH4OH were purchased from Sinopharm Chemical Regent (Shanghai). Degussa supplied the commercial P25. Deionized water was used in all the experiments.
Synthesis of graphene oxide (GO)
GO was prepared by a modified Hummer method [10, 37]. In a typical preparation protocol, 2 g graphite (500 mesh), 1 g NaNO3 and 50 mL concentrated H2SO4 were put into a 250-mL three-necked flask in an ice bath. After continuous stirring for 2 h, 7.3 g KMnO4, 7 mL hydrogen peroxide (30 wt%) and 150 mL DI water were added into the above reaction mixture. The resulting yellow–brown product was obtained after centrifugation and washed multiple times with 200 mL warm (nearly 40 °C) dilute HCl (3 wt%) and finally dried at 60 °C for 24 h in a vacuum oven. The as-prepared GO was then dispersed ultrasonically for 1 h in 500 mL deionized water at room temperature. The unexfoliated GO in the suspension was extracted by successive centrifugation at 4500 rpm for 30 min. The as-obtained solid was dried in vacuum at 60 °C overnight and finally pulverized into powdered GO.
Synthesis of TiO2–rGO nanocomposites
The synthesis of mesoporous TiO2–rGO composites is illustrated in Scheme 1. In a typical synthesis, different amounts of GO were dispersed in a mixed equimolar 60 mL solution of water and ethylene glycol (EG), followed by ultrasonication for 1 h. Then, Ti (SO4)2 (2 mmol) was added to the GO dispersion under magnetic stirring and pursued by the addition of a small amount of ammonia solution (100 µL). After stirring for 30 min, the resultant suspension was transferred to a 100-mL Teflon-lined stainless steel autoclave for solvothermal treatment for 12 h at 180 °C. In this case, the EG was used as both reducing agent and solvent. The ammonia solution adjusts the alkalinity to assist the reduction of GO and help in the subsequent deposit of TiO2 nanoparticles on the graphene. After the autoclave was cooled to room temperature, the resulting products were collected and thoroughly washed with DI water and ethanol for several times to eliminate undecorated TiO2 nanoparticles, unreacted chemicals, and residual EG. Finally, the as-prepared nanocomposite was dried at 60 °C under vacuum condition overnight before characterization. The proposed mass ratio of GO to TiO2 was maintained as 0, 2.5, 5 and 10.0 wt% and the resultant final products are represented as TG0, TG2.5, TG5 and TG10%, respectively.

The synthesis of TiO2–rGO nanocomposites
Material characterization
XRD investigation of the prepared photocatalysts was conducted at room temperature with a Rigaku D/MAX 2550 VB/PC apparatus using Cu Kα radiation (λ = 1.5406 Å) and a graphite monochromator, operated at 40 kV and 30 mA. Diffraction patterns were recorded in the angular range of 5°–80° with a step width of 0.02°. The ultraviolet–visible diffuse reflectance spectra (UV–Vis DRS) were obtained using a scan UV–Vis spectrophotometer (Varian Cary 500) equipped with an integrating sphere assembly, using BaSO4 as the reflectance sample. The spectra were obtained at STP within the range of 200–800 nm. The surface morphologies and particle sizes were examined by transmission electron microscopy (TEM; JEM2000EX), using an accelerating voltage of 200 kV. The chemical states of the nanocomposite were inspected by using X-ray photoelectron spectroscopy (XPS) with a Perkin-Elmer PHI 5000 C ESCA System with Al Kα radiation functioning at 250 W. The shift of binding energy owing to relative surface charging was corrected using the C 1s level at 284.6 eV as an internal standard. The Fourier transform infrared (FT-IR) spectra were executed with KBr disks holding the powder sample with an FT-IR spectrometer (Nicolet Magna 550). Raman spectra were obtained using a micro-Raman spectrometer (Renishaw In Via) with 514 nm excitation light. The recombination of electron-holes in the samples were studied with a spectrofluorophotometer (Shimadzu, RF–5301) by using an Xe lamp as the excitation source at room temperature with the excitation wavelength of 320 nm, and the slit widths at the excitation and emission of the spectrofluorimeter were 5 and 2 nm, respectively. Brunauer–Emmett–Teller (BET) surface area measurements were conducted by N2 adsorption at 77 K using an ASAP2020 instrument. All the samples were degassed at 180 °C prior to analysis and the BET surface area was verified by adsorption data in the relative pressure (p/p 0) range of 0.06–0.2.
Photocatalytic activity measurement
The photocatalytic performance was assessed by photodegradation of RhB and phenol solution under visible light irradiation. The photodegradation of RhB was carried out using a homemade photo-reactor furnished with 500-W halogen lamp and 420-nm cut-off glass optical filter, which used as a light source to provide visible light irradiation. In a typical photocatalytic activity experiment, 50 mg of the photocatalyst was added into 50 mL of the above stated 10 mg L−1 RhB solution. Before starting the light source, the reaction mixture was stirred for 30 min in the dark with the purpose of adsorption/desorption equilibrium between the dye and the photocatalyst. At a given time interval of visible light irradiation, a sample of 3 mL was withdrawn and then centrifuged to essentially separate all the photocatalyst. The concentrations of the remaining dye were spectrophotometrically examined by using a UV–Vis spectrophotometer (Shimadzu UV-2450) and evaluating the absorbance of the solutions at 554 nm in the course of the photodegradation process. The photocatalytic activity of phenol was evaluated using a 300-W Xe lamp under simulated solar light (with an AM1.5 air mass filter). Typically, the as-prepared photocatalyst (50 mg) was placed in a cylindrical Pyrex glass tube having 50 mL phenol aqueous solution (10 mg L−1), which was first magnetically stirred in the dark for 30 min in order to accomplish adsorption/desorption equilibrium and then exposed to visible light. At given time intervals, 3-mL aliquots of the irradiated solution were sampled, centrifuged, and analyzed by using high-performance liquid chromatography.
Results and discussion
Characterization of TiO2–rGO nanocomposites
Transmission electron microscopy (TEM) examined the morphological and structural features of the synthesized composite. Figure 1a discloses that a large number of TiO2 nanocrystals are highly dispersed on the rGO sheet and the majority of TiO2 nanocrystals range from 5 to 10 nm. The size distribution curve of the TiO2 nanocrystals demonstrates a mean diameter centered at ~8 nm (Fig. 1a, inset). Notably, intra-particle aggregation constitutes a mesoporous structure, which is further proven by the nitrogen adsorption–desorption isotherm. The HRTEM image (Fig. 1b) portrays a distinct crystalline lattice with a d spacing of 0.35 nm corresponding to the (101) plane of anatase TiO2 [38].

a TEM images of the TiO2–rGO nanocomposite (TG5%). Inset in (a) is the corresponding particle size distribution of the loaded TiO2 nanoparticles derived from 100 of TiO2 particles shown in (a). b HRTEM image showing nanosized TiO2
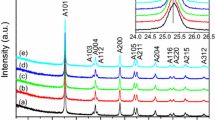
Figure 2a demonstrates the XRD patterns of the bare TiO2 and TiO2–rGO nanocomposites, As depicted from the figure, only anatase phase TiO2 (JCPDS No. 21-1272) was observed in the bare TiO2 sample (TG0%) and TiO2–rGO nanocomposites photocatalyst (samples TG2.5, TG5 and TG10%). Above all, no distinctive diffraction peaks of GO or graphene were detected in all the TiO2–rGO nanocomposites, signifying that the aggregation of graphene sheets through the reduction route can be efficiently barred. This can be attributed to the idea that the systematic stacking pattern of GO was shattered under the solvothermal reduction conditions and could not be resolved using the XRD pattern.

XRD pattern (a), UV–Visible absorption spectra (b) of samples TG0% (a), TG2.5% (b), TG5% (c), TG10% (d)
UV–Vis DRS has been recognized as an efficient optical characterization approach to investigate the probable modification in the band gap and interaction between TiO2 and rGO as shown in Fig. 2b. The TiO2–rGO nanocomposites show an enhanced red-shift in the absorption edge as compared to its bare TiO2 nanoparticle (TG0%) counterparts which show typical and intense absorption in the UV region, resulting in electron transition from the valence band (VB) to the conduction band (CB). This broad circumstantial absorption of TiO2–rGO in visible light could be attributed to the existence of reduced GO in the as-prepared titania nanocomposites and this red-shift is helpful for the degradation of RhB dye and phenol. Nevertheless, any specific tendency or quantifiable variation in the shift of the absorption edge has not been seen. Zhang et al. also [39] affirmed similar results. According to the above results and the shift of the absorption edge of TiO2–rGO nanocomposites was probably attributed to the interaction between the TiO2 nanocrystals and the reduced GO, and it is anticipated that the Ti–O–C bond between TiO2 and rGO was formed which was also soundly established by XPS examination.
X-ray photoelectron spectroscopy (XPS) was used to investigate the interactions and successful functionalization of TiO2 nanocrystals on the graphene sheet. Figure 3a, b demonstrate the high-resolution C 1s XPS spectra of GO and the TiO2–rGO nanocomposite (sample TG 5%), respectively. The C 1s XPS spectrum of the GO nanosheets clearly reveals the existence of two kinds of carbon bonds. The peak positioned at 285 eV is typically assigned to the Sp2 hybridized carbon of the graphene sheet while an other peak at 287.4 eV represented the carbonaceous band (C–OH), proposing a substantial degree of oxidation for the GO nanosheets and providing plenty of active sites for direct linking of the TiO2 nanoparticles. After solvothermal treatment of the GO nanosheet in the TiO2–rGO nanocomposite, there is a decrease in the XPS peak intensity, suggesting the effective deoxygenation of the GO nanosheet and emphasizing that the GO nanosheet in the TiO2–GO scheme can be in situ reduced to produce TiO2–rGO nanocomposites.

XPS spectra of C 1s for GO (a), the TG5% (b) samples
Raman spectroscopy is an excellent non-destructive technique to describe the crystalline feature of carbon and provide an additional insight into the local order of the products. The substantial structural changes that occurred during the solvothermal reaction route from GO to graphene are also echoed in the Raman spectra. Figure 4a demonstrates a comparison of the Raman spectra of GO and the TiO2–rGO nanocomposite (sample TG5%). In the case of TG5% (Fig. 4a, spectrum b), numerous characteristic bands at 146, 397, 516 and 637 cm−1 correspond to the Eg(1), B1 g (1), A1 g + B1 g(2) and Eg(2) modes of anatase, respectively There are two typical Raman bands assigned for GO (Fig. 4a, spectrum a). The band at about 1352 cm−1 normally called the D-band and is assigned to disordered Sp2 carbon. While the other band near 1587 cm−1 is adjacent to the well-ordered graphite domain and is frequently called the G-band [40, 41]. For the TiO2–rGO (TG5%) composite, with all Raman bands for anatase along with two distinctive peaks at nearly 1360 and 1604 cm−1 for the D- and G-bands, respectively, are also found, indicating the successful incorporation of graphene into the hybrids. The D/G intensity ratio of GO is 0.996, but this value rises to 1.06 in TiO2–rGO (TG5%). This rise in the intensity of D/G ratio results in a reduction in the average size of the sp2 domains prepared through the solvothermal course of reaction [41].

(a) Raman spectra of the GO (a) and (b) TG5% samples, (b) nitrogen adsorption–desorption isotherms and the corresponding pore-size distribution curves (inset) of the TG5%
The specific surface area and porous structure of the prepared TiO2–rGO and TiO2 nanocomposite were determined by the multi-point BET techniques. Figure 4b demonstrates the nitrogen adsorption–desorption isotherms and the corresponding pore size distribution curve (inset in Fig. 4b) for the TG5% sample, which can be regarded as type IV isotherm with distinct mesopores and H3 type hysteresis loop relating to the IUPAC classification [42]. The predominant average pore size exists between 2 and 10 nm and all prepared TiO2–rGO nanocomposites possess a mesoporous texture. Table 1 displays that all TiO2–rGO nanocomposite samples have larger specific surface areas than pure TiO2 and P25. The introduction of rGO in TiO2 not only itself proliferates the BET surface area, but also the existing rGO decreases the crystal size of TiO2, which ultimately results in the augmentation of surface area. This is due to the existence of graphene in the nanocomposites, which has an exceptionally high surface area (theoretical value of 2600 m2 g−1). But at a certain limit with rising contents of GO in the nanocomposite, an increasing trend of specific surface area became apparent and the graphene content reaching 10% then declining in the specific surface area were observed. This may possibly be caused by aggregation of GO, which happens at higher concentrations or TiO2 nanoparticles, are coated by a graphene nanosheet. Consequently, when the surface area of TiO2–rGO nanocomposites is calculated, its size is largely governed by surface properties of anatase TiO2 nanoparticles.
FT-IR spectra of the as-prepared nanocomposites were recorded, as demonstrated in Fig. 5a, spectrum a, GO was found to display numerous typical absorption bands of oxygen-containing groups. The absorption peaks were at 1052 cm−1 for C–O stretching, 1220 cm−1 for phenolic C–OH stretching [42], 1625 cm−1 for the -OH groups of molecular H2O [43] and 1729 cm−1 for C=O stretching vibration of residual COOH groups [44]. The minor peaks positioned at 2856 and 2920 cm−1 in the spectrum correspond to the CH2 stretching vibration mode. The FT-IR results for the TiO2–rGO nanocomposite (TG5%) are shown in Fig. 5 (spectrum b). It is apparent that the intensities of oxygen-containing functional groups, for instance, C–O (1052 cm−1), vividly diminished. The phenolic and carbonyl C=O bands at 1220 and 1729 cm−1, respectively, also disappeared in the TiO2–rGO (TG5%) nanocomposite. The spectra also display sturdy absorption bands at 400–900 cm−1, signifying the existence of the Ti–O–Ti stretching vibration mode in TiO2 [45, 46].

a FTIR spectra of the GO (a) and (b) TG5% samples, (b) comparison of the photoluminescence spectra of samples TG0% (a), TG2.5% (b), TG10% (c), TG5% (d)
In a photocatalytic process, the photogenerated electron and hole recombination is one of the many traits mainly liable for the low photocatalytic efficiency. It is well documented that graphene is a viable candidate for the electron acceptor material owing to its two-dimensional p-conjugation edifice [47], and has been efficiently applied to the TiO2–graphene scheme that retards the recombination of photogenerated electrons in TiO2 [48, 49]. The photoluminescence (PL) emission spectra have been broadly used to explore the efficacy of radiative charge carrier recombination, migration and transfer, and to understand the lifespan of photogenerated charge carriers in the semiconductors [50]. Figure 5b exhibits the PL for pure TiO2 (TG0%) and well-adorned TiO2-nanocrystals on reduced GO nanocomposites with different mass ratios of GO. It is detected that the PL intensity of TG0% (bare TiO2) diminished significantly after successful introduction of rGO. This noteworthy PL quenching is due to the effectual electron transfer from TiO2 to graphene sheets. In addition, the uppermost PL quenching happens in TG5%, which suggests that there is an optimum amount of GO in order to obtain the paramount and efficient electron transportation. However, if the amount of GO is in excess up to 10% (TG10%), the PL quenching intensity increased instead of decreasing, because the number of active sites capturing the photo-induced electrons declined. Moreover, excessive graphene can wrap the surface of TiO2 nanoparticles, leading to a drop-off in the concentration of photogenerated charge carriers and the photocatalytic activity of the photocatalyst.
Photocatalytic measurement of TiO2–rGO nanocomposites
Finally, we tested the photocatalytic performance of our mesoporous TiO2–rGO nanocomposites in the photodegradation of rhodamine B (RhB) and phenol under visible light irradiation and compared it with free TiO2 nanocrystals synthesized by the same method in the absence of GO and P25 (a well-known commercial TiO2 photocatalyst). It is well recognized that RhB and phenol are important chemicals and have been extensively used in industrial sectors, and so are often found as notorious environmental contaminants. Therefore, in the current work, we selected them as model organic substances to assess the photocatalytic performance of the as-prepared TiO2–rGO nanocomposites photocatalyst. Prior to photodegradation experiment, the dye and phenol solution with the photocatalyst were magnetically stirred in the dark for 30 min to ensure the absorption/desorption equilibrium of the pollutants with the photocatalyst. Figure 6 exhibits the photodegradation efficiencies and temporal evaluation of the RhB and phenol concentration in the presence of different catalysts under visible light irradiation. The photocatalytic activity of commercial TiO2 (P25) was investigated for the comparison purpose in the study of the degradation of RhB under the same reaction conditions. Interestingly, even though the photocatalytic activity of pure TiO2 nanoparticles (TG0%) was lower than P25, all the three TiO2–rGO nanocomposites still exhibited the remarkably higher photocatalytic activity as compared to P25 and its bare TiO2 counterparts. The bare TiO2 also showed some photocatalytic activity, which is possibly due to the mesoporous structure with a surface area of 111 m2 g−1.

Photodegradation of RhB under visible light irradiation (a); photodegradation of phenol under simulated solar light irradiation (b); fractional removal of RhB under visible light illumination for 90 min (c); fractional removal of phenol under simulated solar light illumination for 150 min (d)
As is depicted from Fig. 6, the GO contents have a substantial effect on the photocatalytic activity of TiO2 for the degradation of RhB and phenol. The sample TG0% (bare TiO2) displays a low photocatalytic activity due to the rapid recombination between CB electrons and VB holes in pure TiO2. However, photodegradation was enhanced remarkably after the introduction of graphene sheet in TiO2–rGO nanocomposite photocatalysts. The photocatalytic activity increased with increasing contents of GO. However, at a certain limit increase in GO contents favored the enhanced photocatalytic activity and a further increase in GO contents especially resulted in the decline of the photocatalytic activity, in the present system when the graphene contents reached to 10.0 wt%, a slight decrease in photocatalytic activity has been observed. The interpretation for the reduced GO contents dependency of photocatalytic performance of the TiO2–rGO nanocomposites and the relative decrease in photocatalytic activity is perhaps attributable to the intensification in the opaqueness and light scattering of TG10%. The loading of a higher concentration of graphene resulted in shielding the TiO2 from absorbing visible light and leading to rapid diminution of irradiation through the reaction suspension. Moreover, certain active sites of the photocatalyst (TiO2) was also detrimentally occupied by reduced GO, which leads to a lessening in the number of potential reaction sites of the photocatalyst, resulting in a subsequent decrease in photocatalytic activity. The photocatalytic efficiency is at a maximum at the optimum reduced GO contents in the photocatalyst. Therefore, the nanocomposite of TG5% demonstrates the most excellent photocatalytic activity for the degradation of RhB. After 90 min of visible light exposure, 85% of RhB is photocatalytically degraded by the TiO2–rGO photocatalyst. Additionally, photodegradation of phenol has also been achieved to further uncover the photocatalytic activity and universality of the as-prepared TiO2–rGO nanocomposites (Fig. 6b). In comparison with bare TiO2, the TiO2–rGO nanocomposites reveal a remarkable photodegradation rate of phenol and, after 150 min exposure under the simulated solar light irradiation, 100% degradation of phenol took place, signifying that the as-synthesized TiO2–rGO nanocomposites embrace not only an enhanced photocatalytic activity but also a relative generality for the photodegradation of various pollutants. This experimental observation not only understandably reveal that phenol was degraded by the catalyst but also further strengthen the finding that degradation of RhB happens because of the excitation of TiO2-reduced GO rather than merely the photoexcitation of the dye [39, 51].
The enhanced photocatalytic activity could be largely credited to the synergistic coupling outcomes in the nanocomposites. The ultra-small TiO2 nanocrystals can curtail the radial length for the diffusion of radiative electrons and holes to the TiO2 surface, and the ultra-dispersed nature of TiO2 nanocrystals on graphene not only improve the intimate interfacial contact but also expedite the charge separation and transfer of electrons from TiO2 to the graphene nanosheet. Graphene has been described as a viable and efficient acceptor material owing to its two-dimensional π-conjugation structure, and the excited electrons of TiO2 could transfer from the conduction band to the graphene. Therefore, the swift transport of charge carriers could be attained and, subsequently, improved charge transportation and separation of photogenerated charge carriers is accomplished. Besides the efficient charge separation and transportation, the enhanced light absorption intensity and light absorption range of the nanocomposites resulted in the more proficient utilization of the solar spectrum and exhibited noteworthy improvements in the photodegradation of the pollutant. This visible light photocatalytic activity of the catalyst is probably helpful to expedite its use in practical environmental remediation. Another possible contributory factor is that the hybrid material has a mesoporous structure with good absorptivity for pollutants and enlarged BET surface area (147 m2 g−1) that offer more potential reaction sites for RhB dye and phenol adsorption and successive redox reactions. It is well acknowledged that samples with enlarged surface areas readily exhibit enhanced photocatalytic competency, predominantly owing to the abundant exposed active sites and large light harvesting.
Figure 7 demonstrates the anticipated mechanism of RhB dye degradation. As the photocatalyst becomes excited in the visible light irradiation, the transfer of electrons from the valance band to the conduction band of TiO2 takes place where reduced GO can behave as an electron trap and photogenerated electrons are then transported to rGO. These electrons then react with the surface oxygen resulting in the formation of successive active oxygen radicals such as O2 −, which lead to a decreased rate of electrons and holes recombination. A reduced GO matrix improves the charge separation and hence the holes are made available for the formation of hydroxyl (·OH) radicals. The consequential free radicals generated then play a significant role in the degradation of the dye molecule. The main reactions involved in all this process are shown below:

Schematic of proposed mechanism of rhodamine B dye degradation
Conclusions
In summary, we have devised a simple, scalable and environmentally benign one-step solvothermal method for producing highly dispersed TiO2 nanocrystals on graphene with a small particle size (~5–10 nm). Titanium sulfate is used as TiO2 source with the assistance of ultra-low content of ammonia to accomplish in situ growth of mesoporous TiO2 nanocrystals on graphene sheets. Graphene has proven to be a dynamic cocatalyst to improve the photocatalytic activity of TiO2 nanoparticles, and its contents have a substantial impact on the photoactivity of TiO2. Notably, the as-synthesized nanocomposites with an appealing nanoarchitecture have revealed a mesoporous structure and effective charge separation with enhanced visible light absorption, which have bestowed the nanocomposites with admirable photocatalytic performance for RhB dye and phenol degradation. We anticipate that the work presented here will open up a scalable and cost-effective approach to prepare robust materials for organic dye degradation and deleterious phenol oxidation for the purification of polluted water resources.
References
J.M. Szeifert, J.M. Feckl, D. Fattakhova-Rohlfing, Y. Liu, V. Kalousek, J. Rathousky, T. Bein, J. Am. Chem. Soc. 132, 36 (2010)
P. Wang, J. Lei, M. Xing, L. Wang, Y. Liu, J. Zhang, J. Environ. Chem. Eng. 961, 3 (2015)
H. Li, L. Zhou, L. Wang, Y. Liu, J. Lei, J. Zhang. Phys. Chem. Chem. Phys. 17406, 17 (2015)
J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo, D.W. Bahnemann, Chem. Rev. 9919, 114 (2014)
L. Zhou, L. Wang, J. Zhang, J. Lei, Y. Liu, Res. Chem. Intermed. (2016). doi:10.1007/s11164-016-2748-8
B. Qiu, Y. Zhou, Y. Ma, X. Yang, W. Sheng, M. Xing, J. Zhang, Sci. Rep. 5, 8591 (2015)
J. Zhang, Y. Wu, M. Xing, S.A.K. Leghari, S. Sajjad, Energy Environ. Sci. 715, 3 (2010)
M. Xing, X. Li, J. Zhang, Sci. Rep. 4, 5493 (2014)
C. Cheng, D. Lu, B. Shen, Y. Liu, J. Lei, L. Wang, J. Zhang, M. Matsuoka, Micropor. Mesopor. Mater. 79, 226 (2016)
M. Moshari, M. Rabbani, R. Rahimi, Res. Chem. Intermed. 5441, 42 (2016)
X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey, H. Zhang, Small 7, 14 (2011)
S. Abdolhosseinzadeh, H. Asgharzadeh, S. Sadighikia, A. Khataee, Res. Chem. Intermed. 4479, 42 (2016)
W. Chang, H. Kim, G.Y. Lee, B.J. Ahn, Res. Chem. Intermed. 71, 42 (2016)
M. Xing, W. Fang, X. Yang, B. Tian, J. Zhang, Chem. Commun. 50, 50 (2014)
G. Qi, W. Zhang, Y. Dai, Res. Chem. Intermed. 1149, 41 (2015)
H.-I. Kim, G.-H. Moon, D. Monllor-Satoca, Y. Park, W. Choi, J. Phys. Chem. C 116, 1 (2011)
H. Liu, K. Cao, X. Xu, L. Jiao, Y. Wang, H. Yuan, ACS Appl. Mater. 7, 21 (2015)
P.M. Sudeep, T.N. Narayanan, A. Ganesan, M.M. Shaijumon, H. Yang, S. Ozden, P.K. Patra, M. Pasquali, R. Vajtai, S. Ganguli, ACS Nano 7, 8 (2013)
Y. Jiang, W.-N. Wang, P. Biswas, J.D. Fortner, ACS Appl. Mater. 6, 14 (2014)
K.M. Cho, K.H. Kim, H.O. Choi, H.-T. Jung, Green Chem. 17, 7 (2015)
X. Xin, X. Zhou, J. Wu, X. Yao, Z. Liu, ACS Nano 6, 12 (2012)
D. Wang, D. Choi, J. Li, Z. Yang, Z. Nie, R. Kou, D. Hu, C. Wang, L.V. Saraf, J. Zhang, ACS Nano 3, 4 (2009)
R. Mo, Z. Lei, K. Sun, D. Rooney, Adv. Mater. 26, 13 (2014)
C. Zhu, S. Guo, P. Wang, L. Xing, Y. Fang, Y. Zhai, S. Dong, Chem. Commun. 38 (2010)
Y. Liang, H. Wang, H.S. Casalongue, Z. Chen, H. Dai, Nano Res. 3, 701–705 (2010)
K.K. Manga, Y. Zhou, Y. Yan, K.P. Loh, Adv. Funct. Mater. 19, 22 (2009)
Y. Gu, M. Xing, J. Zhang, Appl. Surf. Sci. 319 (2014)
W. Li, F. Wang, Y. Liu, J. Wang, J. Yang, L. Zhang, A.A. Elzatahry, D. Al-Dahyan, Y. Xia, D. Zhao, Nano Lett. 15, 2186–2193 (2015)
X. Huang, X. Qi, F. Boey, H. Zhang, Chem. Soc. Rev. 41, 2 (2012)
H. Wang, H. Dai, Chem. Soc. Rev. 42, 7 (2013)
M. Xing, F. Shen, B. Qiu, J. Zhang, Sci Rep. 4 (2014)
S. Guo, S. Dong, Chem. Soc. Rev. 40, 2644–2672 (2011)
Y. Liang, Y. Li, H. Wang, H. Dai, J. Am. Chem. Soc. 135, 6 (2013)
F. Sakhaei, E. Salahi, M.E. Olya, I. Mobasherpour, Res. Chem. Intermed. (2017). doi:10.1007/s11164-017-2860-4
Z. Mou, Y. Wu, J. Sun, P. Yang, Y. Du, C. Lu, ACS Appl. Mater. 6, 16 (2014)
W. Ma, D. Han, S. Gan, N. Zhang, S. Liu, T. Wu, Q. Zhang, X. Dong, L. Niu, Chem. Commun. 49, 71 (2013)
W.S. Hummers Jr., R.E. Offeman, J. Am. Chem. Soc. 80, 6 (1958)
J. Chen, S. Qin, Y. Liu, F. Xin, X. Yin, Res. Chem. Intermed. 637, 40 (2014)
Y. Zhang, Z.-R. Tang, X. Fu, Y.-J. Xu, ACS Nano 4, 12 (2010)
Q. Xiang, J. Yu, M. Jaroniec, J. Phys. Chem. C 115, 15 (2011)
Y. Zhou, C. Guo, S. Xi, C. Sun, H. Wu, F. Shang, Res. Chem. Intermed. 1495, 43 (2017)
Z. Wang, B. Huang, Y. Dai, Y. Liu, X. Zhang, X. Qin, J. Wang, Z. Zheng, H. Cheng, CrystEngComm 14, 5 (2012)
L. Pan, J.-J. Zou, S. Wang, X.-Y. Liu, X. Zhang, L. Wang, ACS Appl. Mater. 4, 3 (2012)
J. Shen, B. Yan, M. Shi, H. Ma, N. Li, M. Ye, J. Mater. Chem. 21, 10 (2011)
N. Yang, J. Zhai, D. Wang, Y. Chen, L. Jiang, ACS Nano 4, 2 (2010)
B. Zhang, W. Zou, J. Zhang, Res. Chem. Intermed. 3157, 41 (2015)
C. Ran, M. Wang, W. Gao, J. Ding, Y. Shi, X. Song, H. Chen, Z. Ren, J. Phys. Chem. C 116, 43 (2012)
X.-Y. Zhang, H.-P. Li, X.-L. Cui, Y. Lin, J. Mater. Chem. 20, 14 (2010)
J. Liu, H. Bai, Y. Wang, Z. Liu, X. Zhang, D.D. Sun, Adv. Funct. Mater. 20, 23 (2010)
H. Yamashita, Y. Ichihashi, M. Harada, G. Stewart, M.A. Fox, M. Anpo, J. Catal. 158, 1 (1996)
J. Strunk, W.C. Vining, A.T. Bell, J. Phys. Chem. C 114, 40 (2010)
Acknowledgements
National Nature Science Foundation of China (21237003 and 21377038), the National Basic Research Program of China (973 Program, 2013CB632403), the Science and Technology Commission of Shanghai Municipality (16JC1401400), the Science and Technology Commission of Jiangsu Province (BC2015135), the Fundamental Research Funds for the Central Universities (222201717003) have supported this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue of the 1st International Symposium on Photocatalysis at Fuzhou University.
Rights and permissions
About this article

Cite this article
Iqbal, W., Tian, B., Anpo, M. et al. Single-step solvothermal synthesis of mesoporous anatase TiO2-reduced graphene oxide nanocomposites for the abatement of organic pollutants. Res Chem Intermed 43, 5187–5201 (2017). https://doi.org/10.1007/s11164-017-3049-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-017-3049-6