
Abstract
A highly efficient, green protocol has been developed for the synthesis of various structurally diverse 2,2′-arylmethylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) and 2-substituted-1H-benzimidazole derivatives. The reaction was performed in water under ultrasound irradiation, using BiOCl nanoparticles as a catalyst. The nanocatalyst was found to be reusable for seven subsequent reactions without much loss in activity. Simple methodology with short reaction times and mild reaction conditions with easy work-up procedure are the salient features of this method.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The development of rapid and selective synthetic routes for functionalized heterocyclic building blocks is of great importance to both human health and the environment. During the last few years, increasing environmental consciousness in chemical research and industry has meant that organic researchers are now directed to the finding of methods that largely take into account the criterion of sustainable chemistry [1]. Indeed, due to the demands of clean technology, many recyclable nano-catalysts have been reported [2–5].
Tetraketones, with four carbonyl functionalities, is an important intermediate in the preparation of xanthendione, acridinedione [6] and 4H-1-benzopyran derivatives [7]. 2,2′-aryl-methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) or tetraketones exhibit a broad spectrum of biological activities, such as lipoxygenase inhibition, anti-oxidant [8] antibacterial and antiviral [9], and the prevention and treatment of thrombosis [10]. Because of their biological and chemical importance, synthesis of tetraketones is under considerable attention of many organic chemists. Tetraketones have been mainly synthesized through Knoevenagel condensations and Michael additions of aldehydes with cyclohexane-1,3-diones, dimedones or various 1,3-cyclic diketones [11]. Various synthetic methodologies have been developed, involving the use of pyridine [12], metal hydroxides [13, 14], l-proline [15], HClO4-SiO2 [16], choline chloride [17], and nano-Fe/NaY zeolite [18] as catalysts, cetyltrimethyl ammonium bromide as surfactant [19], or without any catalyst in water [11].
Benzimidazole is one of the important analogues generally found in drugs and agrochemicals. Hence, its derivatives are being targeted in medicinal chemistry research due to their pharmacological interest. Benzimidazole shows antibacterial [20], anti-inflammatory [21], antiviral [22], antiprotozoal [23], anticancer [24] and antimalarial activity [25].Various strategies employed in the synthesis of substituted benzimidazoles include the cyclo-condensation reaction of o-phenylenediamines with carboxylic acids, nitriles and orthoesters [26]. Another protocol is the direct condensation of 1,2-phenylenediamines with aryl aldehydes using a variety of catalysts, such as heteropoly acid containing ionic liquids [27], bismuth chloride [28], polyaniline-sulfate [29], Zn2+-K10-clay [30], (bromodimethyl)sulfonium bromide [31], TiCl3OTf [32], NaY-zeolite [33], nano-In2O3 [34] Fe(HSO4)3 [35] and MgCl2·6H2O [36]. However, these methods suffer from limitations such as longer reaction times (generally 2–24 h), and the use of expensive catalysts and organic solvents such as MeCN, CH2Cl2, CHCl3, DMF, etc. Moreover, non-reusability of catalysts such as MgCl2·6H2O and harsh reaction conditions decreasea the efficacy of the reported methods. Therefore, it is important to develop more efficient catalysts and alternate synthetic methodologies for the preparation of these compounds. Ultrasound-promoted organic synthesis [37, 38] has attracted considerable attention as it places emphasis on minimizing energy requirements, the avoidance of toxic reagents, the reduction of waste and increasing reaction efficiency. Bismuth oxychloride (BiOCl) nano-particles have been successfully employed as semiconductor photocatalysts [39–44]. The high reactivity of BiOCl nanoparticles is attributed to their large surface area which enables their use in the synthesis of organic compounds.
Materials and methods
All the chemicals were purchased from S.D. Fine Chemicals (India) and were used without further purification. All melting points were measured on a Gallenkamp melting point apparatus and are uncorrected. 1H NMR spectra were recorded on a Bruker Avance-400 MHz spectrometer in CDCl3 in the presence of tetramethylsilane as an internal standard. IR spectra were recorded using a Perkin-Elmer 843 spectrometer with KBr plates. Sonochemical synthesis was performed with the help of an ultrasonic instrument (ACE, USA Horn-type). Conditions: diameter of stainless steel tip of horn: 1.3 × 10−2 m, rated output power: 750 W, operating frequency: 22 kHz, surface area of ultrasound irradiating face: 1.32 × 10−4 m2 and intensity: 3.4 × 105 W/m2.
The temperature of the process was maintained at 35 ± 2 °C by means of a supply of water to the jacketed reactor used for the synthesis. The reactions were monitored by TLC on silicagel PolyGram SILG/UV 254 plates (Merck). All products are known compounds and were identified by comparison with those reported in the literature.
Synthesis and characterization of BiOCl nanoparticles
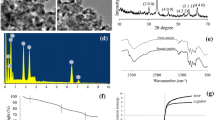
The BiOCl nanoparticles were prepared by the reported method using bismuth nitrate pentahydrate Bi(NO3)3·5H2O as the precursor [45, 46]. Synthesized BiOCl nanoparticles were characterized by XRD and Fourier transform infrared (FT-IR).The crystal structure of BiOCl was determined by an X-ray diffractometer (Bruker D8 Advance Diffractometer) with CuKα radiations (λ = 1.5416 Å) in the range of 20°–80° as shown in Fig. 1a. Figure 1b represents the XRD patterns of reused BiOCl nanoparticles after seven cycles. The prepared BiOCl sample is in good agreement with the tetragonal structure and matched with the JCPDS data (01-075-1533). The crystallite size (D) was calculated from the XRD peak using the Debye–Scherrer formula.
where K is the shape factor, which is a constant taken as 0.9, λ is the wavelength of the X-ray radiation (λ = 1.5416 Å), β is the full-width at half-maximum (FWHM) in radians, and θ is the Bragg’s angle in degree. The crystallite size of the sample obtained from Eq. (1) has a grain size between 23 and 29 nm. The FT-IR spectrum in Fig. 2a shows the characteristic absorption peaks of the as-synthesized sample. The absorption band at 524 cm−1 is assigned to the stretching vibration of Fourier transform infrared Bi-O bond in BiOCl. [47] Figure 2b represents Fourier transform infrared IR spectra of recycled BiOCl nanoparticles after Fourier transform infrared seventh reuse.

XRD patterns of BiOCl: (a) initial; (b) after seven cycles of reuse

FT-IR of BiOCl: (a) initial; (b) after seven cycles of reuse
Optimization of the reaction conditions
The reaction conditions were optimized for the synthesis of 2,2′-(4-methoxylphenyl)methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) (3a) and 2-(4-nitrophenyl)-1H-benzo[d]imidazole (5b) under ultrasound irradiation (Table 1). The effects of various solvents on the model reactions were explored. Several organic solvents such as EtOH, CH3CN, toluene, CHCl3 and DMF were examined (Table 1, entries 1–5 and 16–20). It is exciting to find that water affords the product in good yield even better than other solvents for (3a) and (5b) synthesis (Table 1, entries 15 and 29). Water is highly polar, which possibly creates the necessary conditions for the formation of intermediates and their conversion to final products on the catalyst surface.
The catalytic activity of BiOCl was also studied. When the reaction was performed at room temperature in the presence of BiOCl nanoparticles and in the absence of ultrasonic irradiation, 3a was obtained in 45% yield after 150 min (Table 1, entry 6) and 5b was obtained in 50% yield after 120 min (Table 1, entry 21). In the presence of the catalyst (30 mg) and ultrasonic irradiation (400 W), the yield of 3a increased to 80% and the reaction time was reduced to 40 min (Table 1, entry 11) and in the presence of (Table 1, entry 6)catalyst (12 mg) and ultrasonic irradiation (400 W), the yield of 5b increased to 71% and the reaction time was reduced to 25 min (Table 1, entry 25). It was verified that the combination method (ultrasound irradiation and BiOCl nanoparticles) can be regarded as having an important role in the product yield and reaction time.
General procedure for synthesis of 2, 2′-arylmethylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one)
A mixture of aldehyde (1 mmol) and dimedone (2 mmol) in the presence of BiOCl (30 mg) nanoparticles was added to water (5 mL) as solvent. The reaction mixture was then placed under sonication using an ultrasonic horn (22 kHz frequency) for the required time with a 5-s ON and 5-s OFF cycle from time t = 0 h. The progress of the reaction was monitored by TLC (eluent: 7:3 n-hexane–ethyl acetate) for the time period as indicated in Table 2. After completion of the reaction, the mixture was diluted by 1:1 H2O: EtOAc (10 mL) stirred at ambient temperature (20 min) and centrifuged to separate the solid catalyst. The organic layer of the solution was separated, dried over sodium sulfate, and the organic solvent and other residues were stripped in a vacuum evaporator. The residue obtained was purified by column chromatography (silica gel, 60–120 mesh; pet ether) to afford the desired products. All the products (3a–m) are known compounds and are confirmed by comparison of their spectroscopic data with literature data.
Spectral data for 2,2′-arylmethylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one)
2,2′-(4-Methoxylphenyl)methylenebis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3a )
IR (KBr, cm−1) υ max = 3050, 2960, 2870, 1725, 1601. 1H NMR (CDCl3, 400 MHz) δ: 11.93 (s, 1H, OH), 11.56 (brs, 1H, OH), 7.02 (d, J = 8.3 Hz, 2H, Ph-H), 6.82 (d, J = 8.3 Hz, 2H, Ph-H), 5.50 (s, 1H, CH), 3.79 (s, 3H, CH3O), 2.17–2.49 (m, 8H, CH2), 1.24 (s, 6H, CH3), 1.12 (s, 6H, CH3). 13C NMR (400 MHz, DMSO-d 6 ) δ: 27.3, 29.6, 31.4, 32.0, 46.4, 55.2, 113.6, 115.8, 127.8, 129.8, 157.6, 189.3, 190.0
2,2′-(4-Nitrophenyl)methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3b )
IR (KBr, cm−1) υ max = 3062, 2955, 1590, 1510, 1384, 1345.1H NMR (CDCl3, 400 MHz) δ: 11.80 (s, 1H, OH), 11.26 (brs, 1H, OH), 8.10 (d, J = 8.3 Hz, 2H, Ph-H), 7.22 (d, J = 8.3 Hz, 2H, Ph-H), 5.53 (s, 1H, CH), 2.30–2.49 (m, 8H, CH2), 1.21 (s, 6H, CH3), 1.09 (s, 6H, CH3). 13C NMR (400 MHz, DMSO-d 6 ) δ: 27.4, 29.4, 31.4, 33.2, 46.4, 114.8, 123.4, 127.6, 146.0, 146.6, 189.5, 191.0
2,2′-(3-Nitrophenyl)methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3c )
IR (KBr, cm−1) υ max = 3055, 2961, 1592, 1514, 1391, 1340. 1H NMR (CDCl3, 400 MHz) δ: 11.83 (s, 1H, OH), 11.21(brs, 1H, OH), 8.26(m, 2H, Ph-H), 8.01 (s, 1H, Ph-H), 7.37(d, J = 8.6 Hz, 1H, Ph-H) 5.50 (s, 1H, CH), 2.37–2.47 (m, 8H, CH2), 1.22 (s, 6H, CH3), 1.14 (s, 6H, CH3). 13C NMR (400 MHz, DMSO-d 6 ) δ: 27.3, 29.6, 31.3, 32.8, 46.3, 46.9, 114.7, 120.9, 122.1, 129.0, 132.8, 140.6, 148.3, 189.5, 191.0
2,2′-(2-Nitrophenyl)methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3d )
IR (KBr, cm−1) υ max = 3052, 2960, 1594, 1510, 1396, 1347. 1H NMR (CDCl3, 400 MHz) δ:11.80 (s, 1H, OH), 11.23(brs, 1H, OH), 7.57 (d, J = 7.9 Hz, 1H, Ph-H), 7.43 (t, J = 7.9 Hz, 1H, Ph-H), 7.31 (t, J = 7.9 Hz, 1H, Ph-H), 7.20 (d, J = 7.9 Hz, 1H, Ph-H), 5.82 (s, 1H, CH), 2.22–2.50 (m, 8H, CH2), 1.14 (s, 6H, CH3), 1.07 (s, 6H, CH3).
2, 2′-(4-Chlorophenyl)methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3e )
IR (KBr, cm−1) υ max = 3465, 3010, 2975, 1733, 1594. 1H NMR (CDCl3, 400 MHz) δ: 11.86 (s, 1H, OH), 11.47 (brs, 1H, OH), 7.21 (d, J = 8.1 Hz, 2H, Ph-H), 7.00 (d, J = 8.1 Hz, 2H, Ph-H), 5.46 (s, 1H, CH), 2.28–2.47 (m, 8H, CH2), 1.21 (s, 6H, CH3), 1.09 (s, 6H, CH3). 13C NMR (400 MHz, DMSO-d 6 ) δ: 27.4, 29.6, 31.4, 32.4, 46.4, 47.0, 47.04, 115.3, 128.2, 128.3, 131.5, 136.7, 189.4, 190.6
2, 2′-(3-Chlorophenyl)methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3f )
IR (KBr, cm−1) υ max = 3469, 3017, 2970, 1735, 1591. 1H NMR (CDCl3, 400 MHz) δ: 11.91(s, 1H, OH), 11.50 (brs, 1H, OH), 7.19–7.31 (m, 2H, Ph-H), 7.04 (s, 1H, Ph-H), 6.95 (d, J = 7.8 Hz, 1H, Ph-H), 5.41 (s, 1H, CH), 2.21–2.36 (m, 8H, CH2), 1.22 (s, 6H, CH3), 1.10 (s, 6H, CH3). 13C NMR (400 MHz, DMSO-d 6 ) δ: 27.2, 29.4, 31.3, 32.5, 46.2, 47.1, 115.2, 124.8, 126.1, 127.0, 129.3, 134.0, 140.3, 189.3, 190.5.
2, 2′-(2-Chlorophenyl)methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3g )
IR (KBr, cm−1) υ max = 3461, 3022, 2972, 1729, 1596. 1H NMR (CDCl3, 400 MHz) δ: 11.84(s, 1H, OH), 11.38 (brs, 1H, OH), 7.31 (d, J = 7.9 Hz, 1H, Ph-H), 7.27 (d, J = 7.9 Hz, 1H, Ph-H), 7.20 (t, J = 7.9 Hz, 1H, Ph-H), 7.13 (t, J = 7.9 Hz, 1H, Ph-H), 5.50 (s, 1H, CH), 2.23–2.43 (m, 8H, CH2), 1.18 (s, 6H, CH3), 1.09 (s, 6H, CH3).
2,2′-(4-Hydroxyphenyl)methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3h )
1H NMR (CDCl3, 400 MHz) δ: 11.81 (s, 1H, OH), 11.49 (brs, 1H, OH), 7.33 (d, J = 8.4 Hz, 2H, Ph-H), 7.24 (d, J = 8.4 Hz, 2H, Ph-H), 5.50 (s, 1H, OH), 5.42 (s, 1H, CH), 2.21–2.44 (m, 8H, CH2), 1.22 (s, 6H, CH3), 1.11 (s, 6H, CH3).
2,2′-(4-Hydroxy-3-methoxyphenyl)methylenebis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3i )
1H NMR (CDCl3, 400 MHz) δ: 11.77 (s, 1H, OH), 11.26 (brs, 1H, OH), 6.98 (d, J = 8.2 Hz, 1H, Ph-H), 6.67 (s, 1H, Ph-H), 6.59 (d, J = 8.2 Hz, 1H, Ph-H), 5.47 (s, 1H, OH), 5.44 (s, 1H, CH), 3.81 (s, 3H, CH3O), 2.31–2.44 (m, 8H, CH2), 1.24 (s, 6H, CH3), 1.12 (s, 6H, CH3).
2,2′-(2,4-Dichlorophenyl)methylenebis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3j )
1H NMR (CDCl3, 400 MHz) δ: 11.81 (brs, 1H, OH), 10.67 (s, 1H, OH). 7.34–7.31(m, 2H, Ph-H), 7.20–7.18 (dd, J = 7.6 Hz, J = 2.5 Hz, 1H, Ph-H), 5.51 (s, 1H, CH), 2.24–2.41 (m, 8H, CH2), 1.12 (s, 6H, CH3), 1.08 (s, 6H, CH3).
2,2′-(4-Methylphenyl)methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3k )
1H NMR (CDCl3, 400 MHz) δ: 11.56 (brs, 1H, OH), 10.67 (s, 1H, OH). 7.11 (d, J = 8.2 Hz, 2H, Ph-H), 6.99 (d, J = 8.2 Hz, 2H, Ph-H), 5.49 (s, 1H, CH), 2.36–2.44 (m, 8H, CH2), 2.31 (s, 3H, CH3), 1.14 (s, 6H, CH3), 1.07 (s, 6H, CH3).
2,2′-Phenylmethylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3l )
1H NMR (CDCl3, 400 MHz) δ: 11.66 (brs, 1H, OH), 11.07 (s, 1H, OH) 7.27–7.45 (m, 5H, Ph-H), 5.44 (s, 1H, CH), 2.30–2.41 (m, 8H, CH2), 1.15 (s, 6H, CH3), 1.10 (s, 6H, CH3).
2,2′-(3-Bromophenyl)methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) ( 3m )
1H NMR (CDCl3, 400 MHz) δ: 11.87(s, 1H, OH), 11.22 (brs, 1H, OH), 7.17–7.30 (m, 2H, Ph-1.21 (s, 6H, CH3), 1.16 (s, 6H, CH3).
General procedure for synthesis of 2-substituted-1H-benzimidazoles
1, 2-phenylenediamine derivative (1 mmol), aryl aldehyde (1 mmol) and BiOCl nanoparticles (12 mg) were added to water (5 mL) as solvent. The reaction mixture was then placed under sonication using an ultrasonic horn (22 kHz frequency) for the required time with a 5-s ON and 5-s OFF cycle from time t = 0 h. The progress of the reaction was monitored by TLC (eluent: 7:3 n-hexane–ethyl acetate) for the time period as indicated in Table 3. After completion of the reaction, the mixture was diluted by 1:1 H2O: EtOAc (10 mL) stirred at ambient temperature (20 min) and centrifuged to separate the solid catalyst. The organic layer of the solution was separated, dried over sodium sulfate, and the organic solvent and other residues were stripped in a vacuum evaporator. The residue obtained was recrystallized using the appropriate solvent. The products (5a–l) are known compounds and their 1H NMR and melting points compared with those were reported in literature.
Spectral data for 2-substituted-1H-benzimidazoles
2-Phenyl-1H-benzimidazole ( 5a )
1H NMR (DMSO-d 6 , 400 MHz) δ: 13.01 (br, 1H, NH), 8.11–8.23 (m, 2H, Ph-H), 7.48–7.56 (m, 5H, Ph-H), 7.15–7.21 (m, 2H, Ph-H). 13C NMR (400 MHz, DMSO-d 6 ) δ: 111.84, 119.39, 122.1, 122.9, 126.8, 129.3, 130.2, 130.5, 135.4, 143.9, 151.6
2-(4-Nitrophenyl)-1H-benzo[d]imidazole ( 5b )
1H NMR (CDCl3, 400 MHz) δ: 13.04 (br, 1H, NH), 8.30 (d, J = 8.6 Hz, 2H, Ph-H), 8.03 (d, J = 8.6 Hz, 2H, Ph-H), 7.33–7.38 (m, 2H, Ph-H), 7.27–7.30(m, 2H, Ph-H). 13C NMR (400 MHz, DMSO-d 6 ) δ: 112.41, 120.07, 123.09, 124.90, 128.03, 136.71, 148.45, 149.66
2-(3-Nitrophenyl)-1H-benzo[d]imidazole ( 5c )
1H NMR (DMSO-d 6 , 400 MHz) δ: 12.81 (br, 1H, NH), 8.22 (m, 2H, Ph-H), 8.10 (s, 1H, Ph-H), 7.34 (d, J = 8.5 Hz, 1H, Ph-H), 7.31–7.37 (m, 2H, Ph-H), 7.20–7.28 (m, 2H, Ph-H). 13C NMR (400 MHz, DMSO-d 6 ) δ: 115.7, 116.9, 118.4, 123.0, 125.2, 128.8, 129.7, 134.0, 135.6, 138.2, 142.9, 148.7, 153.8
2-(2-Nitrophenyl)-1H-benzo[d]imidazole ( 5d )
1H NMR (DMSO-d 6 , 400 MHz) δ: 12.89 (br, 1H, NH), 7.77 (d, J = 8.1 Hz, 1H, Ph-H), 7.40 (t, J = 8.0 Hz, 1H, Ph-H), 7.33 (t, J = 8.0 Hz, 1H, Ph-H), 7.21–7.27 (m, 2H, Ph-H), 7.11 (d, J = 8.1 Hz, 1H, Ph-H), 6.85–6.95 (m, 2H, Ph-H).
2-(4-Methoxyphenyl)-1H-benzo[d]imidazole ( 5e )
1H NMR (DMSO-d 6 , 400 MHz) δ: 13.14 (br, 1H, NH), 8.26 (d, J = 8.1 Hz, 2H, Ph-H), 7.50–7.59 (m, 4H, Ph-H), 7.30 (d, J = 8.1 Hz, 2H, Ph-H), 3.82 (s, 3H, CH3O). 13C NMR (400 MHz, DMSO-d 6 ) δ: 55.84, 114.0, 121.9, 132.1, 128.4, 129.4, 151.7, 161.0.
2-(4-Chlorophenyl)-1H-benzo[d]imidazole ( 5f )
1H NMR (DMSO-d 6 , 400 MHz) δ: 13.04 (br, 1H, NH), 8.19 (d, J = 8.1 Hz, 2H, Ph-H), 7.58–7.63 (m, 4H, Ph-H), 7.33 (d, J = 8.1 Hz, 2H, Ph-H). 13C NMR (400 MHz, DMSO-d 6 ) δ: 111.0, 118.5, 121.8, 126.1, 128.5, 129.4, 129.1, 134.7, 143.4, 151.0
2-(4-Cyanophenyl)-1H-benzimidazole ( 5g )
1H NMR (CDCl3, 400 MHz) δ: 12.92 (br, 1H, NH), 8.36 (d, J = 8.5 Hz, 2H, Ph-H), 8.11 (d, J = 8.5 Hz, 2H, Ph-H), 7.35–7.37 (m, 2H, Ph-H), 7.21–7.27 (m, 2H, Ph-H).
2-(4-N,N-dimethylbenzenamine)-1H-benzimidazole ( 5h )
1H NMR (CDCl3, 400 MHz) δ: 11.89 (br, 1H, NH), 8.36 (d, J = 8.4 Hz, 2H, Ph-H), 8.23 (m, 2H, Ph-H), 7.30–7.35 (d, J = 8.4 Hz, 2H, Ph-H), 7.22–7.26(m, 2H, Ph-H), 2.13 (s, 6H, N(CH3)2. 13C NMR (400 MHz, DMSO-d 6 ) δ: 21.40, 40.38, 111.79, 116.11, 170.02,119.41, 127.19, 130.43, 136.11, 136.58, 142.01, 152.53, 156.91
2-(4-Tolyl)-1H-benzimidazole ( 5i )
1H NMR (CDCl3, 400 MHz) δ: 11.91 (br, 1H, NH), 8.15 (d, 2H, J = 8.0 Hz, Ph-H), 7.56 (m, 2H, Ph-H), 7.30 (d, 2H, J = 8.0 Hz, Ph-H), 7.12 (m, 2H, Ph-H), 2.31 (s, 3H, CH3).
2-(2-Pyridinyl)-1H-benzoimidazole ( 5j )
1H NMR (CDCl3, 400 MHz) δ: 12.33 (br, 1H, NH), 7.80–7.84(m, 2H, Ph-H), 7.53–7.57 (m, 2H, Ph-H), 7.12 (m, 2H, Ph-H), 6.92–6.96 (m, 2H, Ph-H).
2-(2-Furanyl)-1H-benzoimidazole ( 5k )
1H NMR (CDCl3, 400 MHz) δ: 12.18 (br, 1H, NH), 7.87(d, J = 4.4 Hz, 1H,Ph-H), 7.53–7.50 (m, 2H, Ph-H), 7.25 (dd, 1H, J = 4.4 Hz &J = 2.0 Hz, Ph-H), 6.63 (d, 1H, J = 2.0 Hz, Ph-H), 6.36–6.40 (m, 2H, Ph-H).
2-(2-Flurophenyl)-1H-benzo[d]imidazole ( 5l )
1H NMR (CDCl3, 400 MHz) δ: 12.41 (br, 1H, NH), 8.32 (d, J = 8.2 Hz, 2H, Ph-H), 8.21 (d, J = 8.2 Hz, 2H, Ph-H), 7.36–7.38 (m, 2H, Ph-H), 7.27–7.30 (m, 2H, Ph-H).
Results and discussion
To explore the scope and limitations of the protocol under optimized conditions (Table 1, entry 15), the reaction of dimedone (1) with various aromatic aldehydes 3(a–m) bearing electron-donating and electron-withdrawing groups gives rise to the corresponding 2,2′-arylmethylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) 3a–m in good to excellent yields (83–95%) at 35 °C within 12–17 min (Table 2). As is indicated in Table 2, both aromatic aldehydes bearing electrondonating and ithdrawing groups react at a faster rate with dimedone.
Encouraged by these excellent results, we then decided to test the generality and versatility of optimized reaction conditions (Table 1, entry 29) in the synthesis of substituted benzimidazoles, and several substituted aromatic aldehydes and heteroaromatic aldehydes 5(a–l) were reacted with o-phenylenediamine (4). These results are shown in Table 3. Aromatic aldehydes bearing electron-donating and -withdrawing groups undergo smooth conversion to afford various benzimidazoles (5a–i, l) in good to excellent yields. When heteroaromatic aldehydes such as pyridine-2-aldehyde and 2-furfural (Table 3, entries 10, 11) were reacted with o-phenylenediamine under similar reaction conditions they gave corresponding benzimidazoles (5j, 5k) in lower yield compared to benzimidazole derivatives obtained from aromatic aldehydes.
Reusability of BiOCl nano-catalyst
Ther catalytic efficiency of the BiOCl nano-catalyst was estimated by a reusability study. We have conducted the experiments using the recycled BiOCl catalyst for the synthesis of 2,2′-arylmethylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) and 2-substituted-1H-benzimidazoles. The recovered catalyst was purified after each run by filtration, washed with acetone and dried at 100 °C for 2 h in an oven before use in the next catalytic cycle. The BiOCl catalyst was found to be reusable for seven times without any significant loss in activity (Fig. 3). After each subsequent run, more than 95% of the catalyst was easily recovered from the reaction mixture.

Reusability study of the catalyst
Conclusion
In summary, BiOCl nanoparticles (NPs) provide an alternative and efficient pathway for the production of heterocyclic intermediates. We have developed a practical and convenient synthetic method in aqueous media by using BiOCl NPs as the catalyst for the facile synthesis of 2,2′-arylmethylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) and 2-substituted-1H-benzimidazoles under ultrasound irradiation. Milder conditions, easy recovery and reusability of the BiOCl NPs catalyst make the method highly efficient.
References
M. Doble, A. Kumar, Green Chemistry and Engineering (Elsevier, Amsterdam, 2007)
J. Safaei-Ghomi, A. Ziarati, S. Zahedi, J. Chem. Sci. 124, 933–939 (2012)
J. Safaei-Ghomi, A. Ziarati, J. Iran. Chem. Soc. 10, 135–139 (2013)
J. Safaei-Ghomi, M.A. Ghasemzadeh, Acta Chim. Slov. 59, 697–702 (2012)
M.A. Ghasemzadeh, J. Safaei-Ghomi, H. Molaei, Compt. Rend. 15, 969–974 (2012)
P. Shanmugasundaram, K.J. Prabahar, V.T. Ramakrishnan, J. Heterocycl. Chem. 30, 1003 (1993)
B.A. Marco, T.A. Manuel, P.M. Itzia, M.M. Francisco, E. Georgina, M. Elies, E. Enrique, J. Chem. Crystallogr. 29, 759 (1999)
G.M. Maharvi, S. Ali, N. Riaz, N. Afza, A. Malik, M. Ashraf, L. Iqbal, M. Lateef, J. Enzyme Inhib. Med. Chem. 23, 62–69 (2008)
R.W. Lambert, J.A. Martin, J.H. Merrett, K.E.B. Parkes, G.J. Thomas, PCT Int. Appl. WO 9706178 (1997) Chem. Abstr. 126, 212377Y (1997)
J. Lehmann, Lancet 241, 611 (1943)
Yu. Jian-Jun, Li-Min Wang, Jin-Qian Liu, Feng-Lou Guo, Ying Liu, Ning Jiao, Green Chem. 12, 216–219 (2010)
K.M. Khan, G.M. Maharvi, M.T.H. Khan, A.J. Shaikh, S. Perveen, S. Begun, M.I. Choudhary, Bioorg. Med. Chem. 14, 344 (2006)
G. Cravotto, A. Demetri, G.M. Nano, G. Palmmisano, A. Penoni, S. Tagliapietra, Eur. J. Org. Chem. 22, 4438 (2003)
B.A. Marco, T.A. Manuel, P.M. Itzia, M.M. Francisco, E. Georgina, M. Elies, E. Enrique, J. Chem. Crystallogr. 29, 759 (1999)
D.B. Ramachary, M. Kishor, J. Org. Chem. 72, 5056 (2007)
S. Kantevari, R. Bantu, L. Nagarapu, J. Mol. Catal. A Chem. 53, 269 (2007)
N. Azizi, S. Dezfooli, M.M. Hashemi, Compt. Rend. Chim. 16, 997 (2013)
M. Tajbakhsh, M. Heidary, R. Hosseinzadeh, Res. Chem. Intermed. 42, 1425–1439 (2016)
Z.J. Ren, W.G. Cao, W.Q. Tong, X.P. Jing, Synth. Commun. 32, 1947 (2002)
S. Sharma, S. Gangal, A. Rauf, Eur. J. Med. Chem. 44, 1751–1757 (2009)
M. Pedini, B.G. Alunni, A. Ricci, L. Bastianini, E. Lepri, Farmaco 49, 823–827 (1994)
B. Simone, K. Mariola, G. Agata, K. Zygmunt, E. Henning, C. Paolo, S. Frank, Arkivoc 3, 225–250 (2009)
V.G. Navarette, R. Cedilla, C.A. Hernandez, A. Yepez, L.F. Hernandez, J. Valdez, R. Morales, R. Cortes, M. Hernandez, R. Castillo, Bioorg. Med. Chem. 11, 187 (2001)
J.S. Kim, B. Gatto, C. Yu, A. Liu, L.F. Liu, E.J. LaVoie, J. Med. Chem. 39, 992 (1996)
P. Toro, A.H. Klahn, B. Pradines, F. Lahoz, A. Pascual, C. Biot, R. Arancibia, Inorg. Chem. Commun. 35, 126–129 (2013)
T.A. Fairley, R.R. Tidwell, I. Donkor, N.A. Naiman, K.A. Ohemeng, R.J. Lombardy, J.A. Bentley, M. Cory, J. Med. Chem. 36, 1746–1753 (1993)
H.G.O. Alvim, H.C.B. de Oliveira, G.A. Bataglion, M.N. Eberlin, L.M. Ramos, W.A. Silva, RSC Adv. 5, 69418 (2015)
Y.-S. Su, C.-M. Sun, Synlett 8, 1243 (2005)
U. Srinivas, C. Srinivas, P. Narender, V.J. Rao, S. Palaniappan, Cat. Commun. 8, 107 (2007)
A. Dhakshinamoorthy, K. Kanagaraj, K. Pitchumani, Tetrahedron Lett. 52, 69–73 (2011)
B. Das, H. Holla, Y. Srinivas, Tetrahedron Lett. 48, 61 (2007)
J. Azizian, P. Torabi, J. Noei, Tetrahedron Lett. 2, 185–188 (2016)
A. Mobinikhaledi, N. Forughifar, M. Zendehdel, M. Jabbarpour, Synth. React. Inorg. Met. Org. Nano-Metal Chem. 38, 390 (2008)
S. Santra, A. Majee, A. Hajra, Tetrahedron Lett. 53, 1974 (2012)
H. Eshghi, M. Rahimizadeh, A. Shiri, P. Sedaghat, Bull. Korean Chem. Soc. 33, 515 (2012)
P. Ghosh, R. Subba, Tetrahedron Lett. 21, 2691–2694 (2015)
T.J. Mason, D. Peters, Practical Sonochemistry (Ellis Horwood, New York, 1991)
D. Habibi, H. Sahebekhtiari, M. Nasrollahzadeh, A. Taghipour, Lett. Org. Chem. 10, 209 (2013)
K. Norihito, M. Koichi, S. Masao, O. Tomo, K. Tomohiro, Y. Hiroyuki, Appl. Cat. A 206, 237–244 (2001)
N. Kijima, K. Matano, M. Saito, T. Oikawa, T. Konishi, H. Yasuda, T. Sato, Y. Yoshimura, Appl. Catal. A 206, 237–244 (2006)
C. Wang, C. Shao, Y. Liuc, L. Zhang, Scripta Mater. 59, 332–335 (2008)
C. Yang, F. Li, T. Li, CrystEngComm 17, 7676–7683 (2015)
C. Yang, F. Li, T. Li, W. Cao, J. Mol. Catal. A Chem. 418, 132–137 (2016)
C. Yang, F. Li, M. Zhang, T. Li, W. Cao, J. Mol. Catal. A Chem. 423, 1–11 (2016)
H.Y. Hao, Y.Y. Xu, P. Liu, G.Y. Zhang, Chin. Chem. Lett. 26, 133–136 (2015)
W.J. Kim, D. Pradhan, B.K. Min, Y. Sohn, Appl. Cat. B Environ. 147, 711–725 (2014)
F.D. Gao, D.W. Zeng, Q.W. Huang, S.Q. Tian, C.S. Xie, Phys. Chem. Chem. Phys. 14, 10572–10578 (2012)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sapkal, B.M., Labhane, P.K. & Satam, J.R. In water–ultrasound-promoted synthesis of tetraketones and 2-substituted-1H-benzimidazoles catalyzed by BiOCl nanoparticles. Res Chem Intermed 43, 4967–4979 (2017). https://doi.org/10.1007/s11164-017-2924-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-017-2924-5