
Abstract
Conceptual density functional theory, including chemical hardness, electronic chemical potential, global and local electrophilicity index and Fukui functions, is used to predict reactivity and regioselectivity of 1,3-dipolar cycloadditions (13DCs) between five aryl azides (1–5) and an electron-deficient alkyne at the B3LYP/6-31G(d,p) level. Two reaction paths (a) and (b) are considered which result in the corresponding regioisomeric 1,2,3-triazoles P(1-5)a and P(1-5)b, respectively. All the 13DCs proceed via rather asynchronous TSs and the path (b) is clearly more synchronous than the path (a). All the reactions are high exoergic [∆Gº = −45.1 to −51.4 kcal/mol for path (a) and −47.7 to −55.9 kcal/mol for path (b)] with the moderate and nearly similar activation barriers (E a = 15.4–16.7 kcal/mol) referring a relatively low regioselectivity. All reactivity descriptors but one clearly suggest that path (a) is somewhat preferred over path (b). FMO interactions occur between HOMO13DP and LUMODPh due to the corresponding lower energy gap. All the reactions considered in this work classified as polar 13DCs with NED character. Our theoretical results are in good agreement with those reported experimentally.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
1,3-Dipolar cycloadditions (13DCs) constitute one of the most important classes of organic reactions and are among the most versatile and powerful preparative methods for the synthesis of five-membered heterocyclic compounds [1]. A 1,3-dipole (13DP) can add onto asymmetric dipolarophiles (DPhs) in two different pathways; however, in many cases, the regiochemistry proceeds mainly through one reaction path, which is lower in energy [2–4]. The controversies between the concerted 4π s + 2π s mechanism in which two new σ bonds are formed rather simultaneously of Huisgen [5] and the stepwise biradical mechanism of Firestone [6] for 13DC have been solved helping numerous experimental and computational investigations. The concerted mechanism is favored for the reactions of the unsubstituted 13DPs with alkenes or alkynes while the other mechanism becomes likely when the 13DPs and DPhs are substituted by radical stabilizing groups [7].
The first organic azide, phenyl azide, as an explosive and hazardous compound [8] is one of the most important 13DPs applied in the regioselective 13DCs [9, 10]. Phenyl azide and its analogues undergo the azide-alkyne Huisgen cycloaddition, a classic example of click chemistry. Although azides are not the most reactive 13DPs available for reaction, they are preferred for their relative lack of side reactions and stability in typical synthetic conditions. Several 1,4,5-trisubstituted 1,2,3-triazoles were prepared by 13DC reactions of some 2-substituted phenyl azides to dimethylacetylene dicarboxylate under regular stirring at room temperature or under microwave irradiation [11].
Houk et al. [12] applied frontier molecular orbital (FMO) perturbation theory for interpretation of regioselectivity in the concerted 13DCs. Based on FMO, three types of concerted 13DCs can occur. One is the FMO interaction between the highest occupied molecular orbital (HOMO) of the 13DP and the lowest unoccupied molecular orbital (LUMO) of the DPh (normal electron demand, NED). A 13DP of this class is referred to as a HOMO-controlled dipole or a nucleophilic dipole, which includes azomethine ylide, carbonyl ylide, nitrile ylide, azomethine imine, carbonyl imine and diazoalkane [13]. A second type is dominated by the reaction between the HOMO of the DPh and the LUMO of the 13DP (inverse electron demand, IED). A dipole of this class is referred to as a LUMO-controlled dipole or an electrophilic dipole, which includes nitrous oxide and ozone [14]. Finally, in the third type of 13DC, the HOMO of the 13DP can interact with LUMO of the DPh; alternatively, the HOMO of the DPh can interact with the LUMO of the 13DP. This two-way interaction arises because the energy gap in either direction is similar. A dipole of this class is referred to as a HOMO–LUMO-controlled dipole or an ambiphilic dipole, which includes azides [15]. Any substituent on the DPh would accelerate the reaction by lowering the energy gap between the two interacting orbitals; i.e., an electron-withdrawing substituent (EWG) would lower the LUMO while an electron-donating substituent (EDG) would raise the HOMO. Despite some exceptions such as ozonolysis of olefins and addition of nitrile oxides to alkynes [16], many more substrate combinations result in no cycloadduct formation when 13DP and DPh are simply mixed. To accelerate these types of reactions, two complementary strategies focus on modulating the reactivity of the DPh. In the first strategy, Lewis acids are included to lower the LUMO of the DPh. Under such conditions the dipole reacts through its HOMO to generate the cycloaddition product [17]. Alternatively, additives that increase the electron density of the DPh can accelerate cycloaddition via an inverse demand cycloaddition interaction. This approach is less common than the Lewis acid-based methods [18] but is most notably operative in the copper-catalyzed synthesis of triazoles from alkynes and azides [19]. In general, the regioselectivity is mainly dependant on the functional groups on the alkyne, and the reactivity of the 13DC reaction is dominantly controlled by HOMO13DP–LUMOalkyne interactions. So, an EWG on the alkyne will accelerate the reaction with diazo compounds usually under very mild conditions. However, the balance of factors, strong electrostatic effects and closed shell repulsions may result in some complications which make the regioselectivity to be not straightforward at all times [20].
Nevertheless, this is a fact that, unlike within the molecular orbital (MO) theory, molecular orbitals do not exist entirely in the DFT, and MOs used as an approximation to the resolution of Schrödinger’s equation have no physical existence. Consequently, DFT-based MOs employed to explain the reactivity in organic chemistry within FMO theory should be used with care [21].
Thus, it should be useful to find out the utility of several reactivity descriptors for the elucidation of the regioselectivity, concurrently. To the best of our knowledge, theoretical investigations on the 13DCs of unsymmetrical alkynes with substituted phenyl azides have not been addressed [22]. Hence, the main goal of this study is examination of various reactivity descriptors based on density functional theory (DFT) on this reaction to validate their applicability in such important organic reactions (Fig. 1). The comparison of our calculated results with those of experimentally observed would be decisive in this work.

The experimentally reported 13DC of aryl azides with methyl 2-perfluoroalkynoates to perfluoroalkylated 1,2,3-triazoles [22]
Methodology
All gas phase Gaussian 03 [23] quantum chemical calculations are done using the B3LYP exchange–correlation functional [24, 25] together with the standard 6-31G* basis set [26]. This level of theory had been confirmed by numerous theoretical studies on the related cycloaddition reactions [27–32]. Geometry optimizations for the reactant, transition state, and product of all the studied reactions are carried out by the Berny analytical gradient method [33]. To verify the nature of the stationary species and evaluate the activation energy barriers, frequency calculations are performed at 298 K and 1 atm. For minimum state structures, only real frequency values and for the TSs, only a single imaginary frequency value is accepted. In the calculation of the condensed Fukui function, the atomic electronic populations of neutral, cationic and anionic systems are analyzed by natural population analysis (NPA) at the optimized geometries of the corresponding neutral systems [34] with the use of the NBO program version 3.1(link 607, Gaussian 03).
Results and discussion
13DC reactions of methyl 2-trifluoromethylalkynoate with 13DPs 1–5 can take place along two regioisomeric pathways (a) and (b), leading to the formation of methyl 5-(trifluoromethyl)-3-aryl-3H-1,2,3-triazole-4-carboxylates P(1-5)a and methyl 5-(trifluoromethyl)-1-aryl-1H-1,2,3-triazole-4-carboxylates P(1-5)b, respectively, after passing through their corresponding TSs (Fig. 2).

Two possible reaction pathways (a, b) along with the atom numbering for five 13DCs (1–5) under study
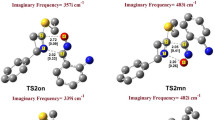
The B3LYP/6-31G*-optimized structures of the TSs associated with these reactions along two regioisomeric pathways are given in Fig. 3.

The B3LYP/6-31G*-optimized structures of transition states corresponding to the 13DCs between alkyne and 13-DPs 1–5 (bond lengths are given in Å)
In general, the linearity of both alkyne and azide moieties are diminished going from the reactants to TSs which denote the product-like nature of TSs based on the Hammond’s postulate. The most important geometrical parameter considered in these two reaction paths is the difference between the lengths of the two newly formed sigma bonds: Δr a = |rN1–C1 − rN3–C2| and Δr b = |rN1–C2 − rN3–C1|. These calculated Δr values provide a simple index to assess the synchronicity of the process. Meanwhile, Δr is zero for fully synchronous processes and the deviation from zero is reflected in the asynchronicity of the processes. Accordingly, our calculated Δr values indicate that all the 13DCs proceed via rather asynchronous TSs (Δr a = 0.175–0.194 Å, ca. 8 %, and Δr b = 0.021–0.05 Å, ca. 1 %) and the path (b) is clearly more synchronous than the path (a) and the highest asynchronous nature is observed for reaction 4 (Δra = 0.194 Å). Another interesting point on the geometry of TS structures is the regularity in the path (a) for having larger bond lengths of C1–N1 compared to the C2–N3 ones. On the other side, such regularity is not observed in the TS structures of path (b) since TSs 4b and 5b are different from the TSs 1b–3b for having larger lengths of N1–C2 compared to the N3–C1 ones.
The potential energy surface (PES) involving activation energies (Ea) as well as the standard Gibbs free energy changes (∆G0) of all 13DC reactions along two regioisomeric pathways are presented in Fig. 4.

The B3LYP/6-31G*-calculated PES (kcal/mol) corresponding to all 13DC reactions along two regioisomeric pathways (a, b)
Our data show that all the reactions are high exoergic [∆G0 = −45.1 to −51.4 kcal/mol, for path (a) and −47.7 to −55.9 kcal/mol, for path (b)]. Moreover, all the 13DC reactions along path (b) are more exoergic than their corresponding reactions along path (a) by 1–2 kcal/mol. On the other hand, calculated activation barriers of path (a) are insignificantly smaller than those of path (b). These thermodynamic and kinetic data besides the similarity in the steric effects of all reactions denote a low regioselectivity for such 13DC reactions between aryl azides with alkyne [22].
In parallel to the energetic data, chemical selectivity and reactivity can be provided based on conceptual DFT in terms of global electronic descriptors such as electronic chemical potential (μ), hardness (η) and softness (S) [17, 35]. Since DFT is becoming an efficient and computationally cost effective tool for the study of electronic structures, it is worthwhile to find out how DFT-based reactivity concepts can be used for the interpretation of reaction mechanisms and especially the regiochemistry in the present 13DC reactions [36]. The conceptual DFT has become a powerful tool to study polar organic reactions [37]. Hence, a brief review of employed descriptors is presented hereafter. For an N-electron system with total energy E and external potential v, electronic chemical potential is defined as the first-order derivatives of \(E(\mu = (\partial E/\partial N)_{v} )\) [38, 39]. Moreover, hardness and softness are second-order derivatives of \(E\left( {2\eta = S^{ - 1} = (\partial^{2} E/\partial N^{2} )_{v} = (\partial \mu /\partial N)_{v} } \right)\) [40, 41]. Furthermore, following Koopmans’ theorem [42], the chemical potential and hardness can be rewritten as: \(\mu = \frac{1}{2}\left( {\varepsilon {\text{HOMO}} + \varepsilon {\text{LUMO}}} \right)\) and \(\eta = \frac{1}{2}\left( {\varepsilon {\text{LUMO}} - \varepsilon {\text{HOMO}}} \right)\), where εHOMO and εLUMO are the highest occupied and lowest unoccupied molecular orbital energies, respectively. Pearson showed that many electronic systems arrange themselves, in their ground or valence states, to be as hard as possible (the maximum hardness principle, MHP) [43]. In connection with the MHP, Chattaraj and Sengupta proposed a minimum polarizability principle (MPP), which states that the natural direction of evolution of any system is toward a state of minimum polarizability, α [44]. The static electric dipole polarizability is the relative propensity of a charge distribution, like the electron density of a molecule, to be distorted from its normal shape in the presence of an electric field and, for this work, is calculated under the equation of \(\alpha = \frac{1}{3}(\alpha_{xx} + \alpha_{yy} + \alpha_{zz} )\) where αxx, αyy, and αzz are the diagonal elements of the polarizability tensor. Another global index is the electrophilicity power of a system in terms of its electronic chemical potential and hardness. The electrophilicity index was introduced by Parr et al. [45] and can be expressed as \(\omega = \frac{{\mu^{2} }}{2\eta }\). This property is a measure of energy lowering due to the maximal electron flow between donor and acceptor. Together with the global electrophilicity ω index, the global nucleophilicity N index is also used to study reactivity [46]. Finally, Noorizadeh et al. [47] showed that in the Diels–Alder cycloadditions, the major product has always less electrophilicity than the minor product and the electrophilicity index can be used as an indicator for regioselectivity which is named the minimum electrophilicity principle (MEP).
Recently, a number of reports were published on the mechanism of 13DC reactions by Domingo et al. For instance, they established good correlations between the pseudodiradical (pr) character, the hardness η, and the nucleophilicity N index of the tri-atom-component's TACs with the feasibility of these nonpolar reactions through analysis of 12 TACs participating in 13DC reactions towards ethylene and acetylene [48]. This study made it possible to establish a useful classification of 13DC reactions into zwitterionic (zw)-type reactions involving TACs with a high zw character, and pr-type reactions involving TACs with a high pseudodiradical character. Moreover, a set of seven non-substituted TACs participating in 13DC reactions has been studied using the reactivity indices defined within the conceptual DFT at the B3LYP/6-31G(d) level of theory and it was concluded that the substitution is required in both, TACs and the ethylene species, in order to experimentally perform these zw-type 13DC reactions under mild conditions [49]. Finally, the participation of azomethine ylides (AYs) and carbonyl ylides (CYs) in 13DC reactions has been analysed at the DFT B3LYP/6-31G(d) level where the asymmetric substitution broke the pseudodiradical character of the simplest TACs, modifying their electrophilic and nucleophilic behaviors [50].
Therefore, in this study, the obtained frontier molecular orbital energies, polarizability, hardness and electrophilicity values for all the studied regioisomers are presented in Table 1.
These data show that while MHP and MEP rules predict the regioisomers of path (a) as the major cycloadducts, the MPP predicts an entirely inverse situation in favor of path (b). Again, these global reactivity indexes denote a low regioselectivity on the 13DC reactions of aryl azides with alkynes under study.
Another method for explanation of the regiochemistry of concerted reactions proposed by Gazquez and Mendez based on a local version of the hard-soft acid–base theory (HSAB principle) [51, 52]. The Gazquez–Mendez rule indicates that the interaction between any two chemical species will occur through the centers with nearly equal softness. To predict the regioselectivity using this rule, it is necessary to determine the type of electron demand character of the reactions. To gain this objective, four properties are considered: HOMO–LUMO gaps between the two reactants, calculated μ and ω of reactants, and charge transfer (CT) analysis in the transition states (Table 2).
It is important to know that for calculation of these parameters, the actual HOMOs and LUMOs are used (relevant HOMO and LUMO). Inspecting the reactant FMOs reveals that in our 13DCs, the theoretical and actual molecular orbitals are the same since both the reactants should have one node in their FMOs which is apparent in the pictures (Fig. 5).

The visualized FMOs of reactants in this study
Considering the visualized molecular orbitals of the reactants and their FMO energies, two possible interactions consisting of HOMO13DP–LUMODPh and/or HOMODPh–LUMO13DP can take place, both of which are symmetrically allowed. The energy differences between the two possible HOMO/LUMO combinations show that for all the cycloadditions, the main interaction occurs between HOMO13DP and LUMODPh due to the lower energy gap (Fig. 6). However, the differences among five interactions appear unimportant and range from 4.31 (13DP1) to 4.90 eV (13DP5). Consequently, based on the HOMO–LUMO gaps, the 13DC reactions considered in this work have the NED character.

Possible FMO interactions in the 13DCs between alkyne (DPh) and 13DPs 1–5
On the other hand, calculated μ values of all 13DPs 1–5 (ranging from −0.1331 to −0.1593 au) are more positive than that of DPh (–0.1853 au) which is indicative of electron flow from the 13DPs to the dipolarophile DPh (Table 2). This finding is referring again to the NED character of these reactions. The largest μ pertains to the 13DP4 with a para-CN substituent. Besides the moderate electrophilicity values encountered for all reactants, 13DP4 is also the most electrophilic (ω = 0.0713 au) and the softest dipole among all. The polar character of the studied reactions can be measured by the difference in the absolute electrophilicity power (Table 2). Hence, 13DC reactions of 13DPs 1–5 with the alkyne have a rather low polar character and the reaction of 13DP1 has the highest polarity with ∆ω = 0.641 eV.
The last method for determining the electronic nature of the scrutinized reactions is charge transfer (CT) analysis in the transition states. CT analysis with the NPA method shows that a relatively low net charge transfer takes place from the DPs to the DPh which, again, is in agreement with the NED character of the 13DCs (Table 3).
These data show that values of the CT are between 0.0504e and 0.0817e for all studied TSs in both paths (a) and (b). The values of CT are clearly smaller than those of previously reported Diels–Alder reactions [53]. These results indicate that the reactions can be classified as polar 13DCs, with a more polar nature in the path (a).
In the last step, calculation of Fukui functions, f(r), in the reaction sites of reagents is presented. The Fukui function is the most fundamental and useful local reactivity indicator in chemical DFT. It defines the reactivity and selectivity of a specific site in a molecule as the differential change in electron density due to an infinitesimal change in the number of electrons at a constant external potential v(r) [54]. An other definition of Fukui function is the response of the chemical potential μ of a system to a change in the external potential v(r) [55]. \(f(r) = \left[ {\frac{\partial \rho (r)}{\partial N}} \right]_{v(r)} = \left[ {\frac{\delta \mu }{\delta v(r)}} \right]_{N}\). The condensed forms of Fukui functions of an atom, say k, in a molecule with N electrons, depending on the type of electron transfer, have been proposed by Yang and Mortier [56]: \(f_{k}^{ + } = \left[ {\rho_{k} (N + 1) - \rho_{k} (N)} \right]\) for nucleophilic attack; \(f_{k}^{ - } = \left[ {\rho_{k} (N) - \rho_{k} (N - 1)} \right]\) for electrophilic attack, and \(f_{k}^{o} = \frac{1}{2}\left[ {\rho_{k} (N + 1) - \rho_{k} (N - 1)} \right]\) for radical attack, where \(\rho_{k} (N)\), \(\rho_{k} (N - 1)\), \(\rho_{k} (N + 1)\) are the gross electronic population of the site k in neutral, cationic and anionic systems, respectively. Since the electronic mechanism of the studied reactions is NED, Fukui functions for electrophilic (\(f_{k}^{ - }\)) and nucleophilic (\(f_{k}^{ + }\)) attack at the reaction sites (k) are calculated for 13DPs and DPh, respectively, and collected in Table 4.
The local softness, \(s_{k}^{\alpha }\), and local philicity index, \(\omega_{k}^{\alpha }\), condensed to reaction sites are easily obtained by their global quantities and condensed Fukui functions. Three different types of \(s_{k}^{\alpha }\) and \(\omega_{k}^{\alpha }\) can be readily defined as [57]: \(s_{k}^{\alpha } = Sf_{k}^{\alpha }\) and \(\omega_{k}^{\alpha } = \omega f_{k}^{\alpha }\) where α = + and − refer to nucleophilic and electrophilic attack, respectively. Now, using local softness s(r) values and the Gazquez–Mendez rule concept, the Δs values for both pathways are computed according to the formula: \(\Delta s_{a} = \left( {s_{{{\text{N}}1}}^{ - } - s_{{{\text{C}}1}}^{ + } } \right)^{2} + \left( {s_{{{\text{N}}3}}^{ - } - s_{{{\text{C}}2}}^{ + } } \right)^{2}\) and \(\Delta s_{b} = \left( {s_{{{\text{N}}1}}^{ - } - s_{{{\text{C}}2}}^{ + } } \right)^{2} + \left( {s_{{{\text{N}}3}}^{ - } - s_{{{\text{C}}1}}^{ + } } \right)^{2}\)(Fig. 2). It is recommended that the reaction channel with lower activation energy corresponding to the predominant product is associated with a smaller \(\Delta s\) value. The values of \(\Delta s_{a}\) and \(\Delta s_{b}\) are given in Table 5.
It is clear that values of \(\Delta s_{a}\) are smaller than those of \(\Delta s_{b}\) for all the studied reactions. Therefore, the Gazquez–Mendez rule based on NPA charges predicts that path (a) would be chosen over the alternative path (b).
According to Chattaraj’s polar model [44], prediction of the regioselectivity using local descriptors is possible. In this model, the most favored interactions take place between highest values of local electrophilicity powers between reaction sites (Fig. 7). The values of the local nucleophilicity powers, ω−, for atoms N1 and N3 of 13DPs 1–5 and the local electrophilicity powers, ω+, for atoms C1 and C2 of DPh, are shown. This figure evidently shows that Chattaraj’s model also predicts regioselectivity of these 13DCs in favor of path (a).

Calculated local nucleophilicites, ω−, for 13DPs 1–5, and local electrophilicites, ω+, for DPh centres
It is worth mentioning that local reactivity in polar reactions can also be studied within the conceptual DFT through the analysis of the electrophilic and nucleophilic Parr functions based on the spin density distribution at the radical anion and at the radical cation of a neutral molecule which allow for the characterization of the most electrophilic and nucleophilic centers of molecules, and for the establishment of the regio- and chemoselectivity in polar reactions [58].
In summary, all the conceptual DFT reactivity indexes (except for MPP) denote relatively more reactivity in path (a) of the 13DCs which results in the formation of methyl-5-(trifluoromethyl)-3-aryl-3H-1,2,3-triazole-4-carboxylates, P1–5a. The regioselectivities in the 13DCs discussed here could be determined from the relative energies of the two TSs associated with the two modes of cycloadditions. Due to the similarity of Arrhenius pre-exponential factor and entropy values, the relative amounts of the two cycloadducts can be assumed to be proportional to the Arrhenius exponential factor [\(\exp \left( {\Delta E_{a}^{\# } } \right)/RT\)]. Consequently, the calculated product ratios, P1–5a/P1–5b, appear to be insignificant just by 2.6 times in favor of P1–5a. Low regioselectivity pertaining to these 13DCs are in good agreement with the results of X-ray crystallography and 19F nuclear magnetic resonance (NMR) spectra reported by Zhang et al. [22] where they found that all the path (a) regioisomers P1-5a are preferred by 2.1–2.4 times over the corresponding path (b) regioisomers P1-5b.
Interestingly, the fact that the MPP index fails to explain the observed regiochemistry of 13DC reaction is in line with some earlier reports in the literature. In 2006, Noorizadeh and Maihami argued that that, in many cases, the MHP and MPP cannot predict the major regioisomer of a Diels–Alder reaction [47], and, in 2013, Blair and Thakkar reported that careful scrutiny of 2386 experimental and theoretical polarizabilities revealed 86 violations and suggested that the MPP is violated approximately two to three per cent of the time [59]. Moreover, Torrent-Sucarrat et al. carried out diagonalizations of the Hessian matrix of the polarizability to determine the molecular distortions with a more marked MPP or anti-MPP character and, subsequently, they could derive a set of simple rules that allow predicting a priori without calculations the existence of vibrational modes that break the MPP [60].
Conclusions
The mechanism, reactivity and regioselectivity of 13DC five aryl azides and an electron-deficient alkyne leading to two regioisomeric series (a) and (b) are studied at the B3LYP/6-31G(d,p) level of theory using different approaches (the maximum hardness principle, minimum polarizability principle, minimum electrophilicity principle, activation energies, FMO energies, Gazquez–Mendez rule and Chattaraj’s polar model). All the 13DCs are high exoergic, have a NED character, HOMO13DP–LUMODPh interaction and are rather asynchronous. Except for polarizability values, the remaining methods predict the path (a) regioisomers as the major product. Interestingly, these finding are in good agreement with those experimentally reported for the titled reactions.
References
A. Padwa, W.H. Pearson (eds.), Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products (Wiley, Hoboken, 2003)
M. Regitz, Phosphaalkynes: new building blocks in synthetic chemistry. Chem. Rev. 90, 191–213 (1990)
M.T. Nguyen, An analysis of reactant approach in concerted 1, 3-dipolar cycloadditions by the second moment of localized orbitals. J. Mol. Struct. (THEOCHEM) 105, 343–349 (1983)
L. Nyulaszi, P. Varnai, W. Eisfeld, M. Regitz, Regioselectivity in cycloaddition reaction between phosphaacetylene and diazomethane: an ab initio study. J. Comput. Chem. 18, 609–616 (1997)
R. Huisgen, 1, 3-dipolar cycloadditions: past and future. Angew. Chem. Int. Ed. 2, 565–632 (1963)
R.A. Firestone, Orientation in 1,3-dipolar cycloadditions according to the diradical mechanism: partial formal charges in the linnet structures of the diradical intermediates. J. Org. Chem. 37, 2181–2191 (1972)
S.A. Siadati, An example of a stepwise mechanism for the catalyst-free 1, 3-dipolar cycloaddition between a nitrile oxide and an electron rich alkene. Tetrahedron Lett. 56, 4857–4863 (2015)
P. Griess, Ueber diazocyanbenzol. Ber. Dtsch. Chem. Ges. 2, 369–370 (1869)
E.F.V. Scriven, K. Turnbull, Azides: their preparation and synthetic uses. Chem. Rev. 88, 297–368 (1988)
W.H. Pearson, P. S. Ramamoorthy in Encyclopedia of Reagents for Organic Synthesis, ed. by L. Paquette L. (Wiley, New York, 2004)
B. Souad, C.E. Fatmi, T. Mabrouk, Synthesis of some 1,4,5-trisubstituted 1,2,3-triazoles by 1,3-dipolarcycloaddition of 2-substituted phenyl azides to dimethyl acetylene dicarboxylate (DMAD), regular versus microwave irradiation: a comparative study. Rasayan. J. Chem. 4, 806–809 (2011)
K.N. Houk, J. Sims, C.R. Watts, L.J. Luskus, Origin of reactivity, regioselectivity, and periselectivity in 1, 3-dipolar cycloadditions. J. Am. Chem. Soc. 95, 7301–7315 (1973)
J. Geittner, R. Huisgen, R. Sustmann, Kinetics of 1, 3-dipolar cycloaddition reactions of diazomethane; a correlation with homo-lumo energies. Tetrahedron Lett. 18, 881–884 (1977)
D.G. Williamson, R.J. Cvetanovic, Rates of ozone-olefin reactions in carbon tetrachloride solutions. J. Am. Chem. Soc. 90, 3668–3672 (1968)
R. Huisgen, G. Szeimies, L. Mobius, 1.3-Dipolare cycloadditionen, XXXII. Kinetik der additionen organischer azide an CC-Mehrfachbindungen. Chem. Ber. 100, 2494–2507 (1967)
T.M.V.D. Pinho e Melo, Recent advances on the synthesis and reactivity of isoxazoles. Curr. Org. Chem. 9, 925–958 (2005)
K.N. Houk, Frontier molecular orbital theory of cycloaddition reactions. Acc. Chem. Res. 8, 361–369 (1975)
J. Barluenga, C. Valdes, G. Beltran, M. Escribano, F. Aznar, Developments in Pd catalysis: synthesis of 1H1,2,3-triazoles from sodium azide and alkenyl bromides. Angew. Chem. Int. Ed. 45, 6893–6896 (2006)
F. Himo, T. Lovell, R. Hilgraf, V.V. Rostovtsev, L. Noodleman, K.B. Sharpless, V.V. Fokin, Copper (I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 127, 210–216 (2005)
R.A. Firestone, Orientation in the 1, 3-dipolar cycloaddition of diazomethane and ethyl vinyl ether. J. Org. Chem. 41, 2212–2214 (1976)
L.R. Domingo, A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4, 32415–32428 (2014)
J. Wei, J. Chen, J. Xu, L. Cao, H. Deng, W. Sheng, H. Zhang, W. Cao, Scope and regioselectivity of the 1,3-dipolar cycloaddition of azides with methyl 2-perfluoroalkynoates for an easy, metal-free route to perfluoroalkylated 1,2,3-triazoles. J. Fluor. Chem. 133, 146–154 (2012)
M.J. Frisch et al., Gaussian 03, Revision B.03 (Gaussian, Pittsburgh, 2003), p. 9
A.D. Becke, Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993)
C. Lee, W. Yang, R.G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988)
R. Ditchfield, W.J. Hehre, J.A. Pople, Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 54, 724–728 (1971)
E. Paredes, R. Brasca, M. Kneeteman, P.M.E. Mancini, A novel application of the Diels–Alder reaction: nitronaphthalenes as normal electron demand dienophiles. Tetrahedron 63, 3790–3799 (2007)
C.N. Alves, A.S. Carneiro, J. Andres, L.R. Domingo, A DFT study of the Diels-Alder reaction between methyl acrolein derivatives and cyclopentadiene. Understanding the effects of Lewis acids catalysts based on sulfur containing boron heterocycles. Tetrahedron 62, 5502–5509 (2006)
L.R. Domingo, A density functional theory study for the Diels–Alder reaction between N-acyl-1-aza-1, 3-butadienes and vinylamines. Lewis acid catalyst and solvent effects. Tetrahedron 58, 3765–3774 (2002)
C. Della Rosa, C. Ormachea, M.N. Kneeteman, C. Adam, P.M.E. Mancini, Diels-Alder reactions of N-tosylpirroles developed in protic ionic liquids. Theoretical studies using DFT methods. Tetrahedron Lett. 52, 6754–6757 (2011)
P.M.E. Mancini, C.M. Ormachea, C.D. Della Rosa, M.N. Kneeteman, A.G. Suarez, L.R. Domingo, Ionic liquids and microwave irradiation as synergistic combination for polar Diels-Alder reactions using properly substituted heterocycles as dienophiles. A DFT study related. Tetrahedron Lett. 53, 6508–6511 (2012)
S. Bouacha, A.K. Nacereddine, A. Djerourou, A theoretical study of the mechanism, stereoselectivity and Lewis acid catalyst on the Diels–Alder cycloaddition between furan and activated alkenes. Tetrahedron Lett. 54, 4030–4033 (2013)
H.B. Schlegel, Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 3, 214218 (1982)
A.E. Reed, F. Weinhold, Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Phys. Chem. 78, 4066–4073 (1983)
W. Kohn, A.D. Becke, R.G. Parr, Density functional theory of electronic structure. J. Phys. Chem. 100, 12974–12980 (1996)
P.K. Chattaraj, U. Sarker, D. Ranjan Roy, Electrophilicity index. Chem. Rev. 106, 2065–2091 (2006)
L.R. Domingo, E. Chamorro, P. Perez, Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 73, 4615–4624 (2008)
P. Geerlings, F. De Proft, W. Langenaeker, Conceptual density functional theory. Chem. Rev. 103, 1793–1874 (2003)
R.G. Parr, R.A. Donnelly, M. Levy, W.E. Palke, Electronegativity: the density functional viewpoint. J. Chem. Phys. 68, 3801–3807 (1978)
B. Gmez, P.K. Chattaraj, E. Chamorro, R. Contreras, P. Fuentealba, A density functional study of the claisen rearrangement of allyl aryl ether, allyl arylamine, allyl aryl thio ether, and a series of meta-substituted molecules through reactivity and selectivity profiles. J. Phys. Chem. A 106, 11227–11233 (2002)
R.G. Parr, R.G. Pearson, Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 105, 7512–7516 (1983)
T.A. Koopmans, Uber die zuordnung von wellenfunk tionen und eigenwerten zu den einzelenen electronen eines atoms. Physica 1, 104–113 (1934)
R.G. Pearson, Chemical hardness: applications from molecules to solids (Wiley-VCH Verlag GMBH, Weinheim, 1997)
P.K. Chattaraj, S. Sengupta, Popular electronic structure principles in a dynamical context. J. Phys. Chem. 100, 16126–16130 (1996)
R.G. Parr, L.V. Szentpaly, S. Liu, Electrophilicity index. J. Am. Chem. Soc. 121, 1922–1924 (1999)
L.R. Domingo, P. Perez, The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 9, 7168–7175 (2011)
S. Noorizadeh, H. Maihami, A theoretical study on the regioselectivity of Diels–Alder reactions using electrophilicity index. J. Mol. Struct. (THEOCHEM) 763, 133–144 (2006)
L.R. Domingo, S.R. Emamian, Understanding the mechanisms of [32] cycloaddition reactions. The pseudoradical versus the zwitterionic mechanism. Tetrahedron 70, 1267–1273 (2014)
L.R. Domingo, M.J. Aurell, P. Perez, A DFT analysis of the participation of zwitterionic TACs in polar [32] cycloaddition reactions. Tetrahedron 70, 1–7 (2014)
L.R. Domingo, M.J. Aurell, P. Perez, A mechanistic study of the participation of azomethine ylides and carbonyl ylides in [32] cycloaddition reactions. Tetrahedron 71, 1050–1057 (2015)
F. Mendez, J.L. Gazquez, Chemical reactivity of enolate ions: the local hard and soft acids and bases principle viewpoint. J. Am. Chem. Soc. 116, 9298–9301 (1994)
J.L. Gazquez, A. Martinez, F. Mendez, Relationship between energy and hardness differences. J. Phys. Chem. 97, 4059–4063 (1993)
A. Nouri, E. Zahedi, F.J. Jafari, A. Nouri, Diels–Alder reactions of α-cyano α, β-unsaturated ketones with 2-methyl-1, 3-butadiene: DFT study of mechanism, reactivity and regioselectivity. Prog. React. Kinet. Mech. 40, 177–189 (2015)
A.K. Chandra, M.T. Nguyen, Density functional approach to regiochemistry, activation energy, and hardness profile in 1, 3-dipolar cycloadditions. J. Phys. Chem. A 102, 6181–6185 (1998)
A.K. Chandra, M.T. Nguyen, Use of local softness for the interpretation of reaction mechanisms. Int. J. Mol. Sci. 3, 310–323 (2002)
W. Yang, W.J. Mortier, The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 108, 5708–5711 (1986)
H. Chemouri, S.M. Mekelleche, Elucidation of the substitutent effects on the reaction pathway of the cycloaddition of 1, 3-diazabuta-1, 3-dienes with ketenes using DFT-based reactivity indexes. J. Mol. Struct. (THEOCHEM) 813, 67–72 (2007)
L.R. Domingo, P. Perez, J.A. Saez, Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 3, 1486–1494 (2013)
S.A. Blair, A.J. Thakkar, How often is the minimum polarizability principle violated? Chem. Phys. Lett. 556, 346–349 (2013)
M. Torrent-Sucarrat, J.M. Luis, M. Duran, M. Sola, Are the maximum hardness and minimum polarizability principles always obeyed in nontotally symmetric vibrations? J. Chem. Phys. 117, 10561–10570 (2002)
Acknowledgments
The authors gratefully acknowledge The Research Council of the Islamic Azad University Shahr-e-Qods Branch.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dizaji, N.J., Nouri, A., Zahedi, E. et al. Regioselectivity of 1,3-dipolar cycloadditions between aryl azides and an electron-deficient alkyne through DFT reactivity descriptors. Res Chem Intermed 43, 767–782 (2017). https://doi.org/10.1007/s11164-016-2663-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-016-2663-z