
The effect of the formation of sintered TiC–ZrC solid solutions in mullite–WC–TiC–ZrC and mullite– c-BN–TiC–ZrC samples during spark plasma sintering of compositions at a compression load of 60 MPa in the range of 1200–1600°C on their phase composition, microstructure, relative density, open porosity, linear shrinkage, physicomechanical properties, and linear correlation of elastic modulus and fracture toughness is shown. The synthesized WC, c-BN powders, and solid solutions of TiC–ZrC prepared by spark plasma sintering at 1800°C are characterized by intensive crystallization of WC, c-BN and (Ti, Zr)C, respectively, with the formation of a uniform, densely sintered crystalline microstructure. Sintered samples containing 80/20 and 90/10 mol.% solid solutions of TiC and ZrC show intensive development of mullite and (Ti, Zr)C, a gradual increase in c-BN and (Zr, Ti)C, as well as a decrease in the intensity of the formation of the WC phase in the range of 1200 – 1600°C. The microstructure of more evenly and densely sintered samples containing c-BN and solid solutions of TiC–ZrC (in various proportions) is fine-grained with a small number of pores and most strengthened at the boundaries of the regions of oxide and oxide-free crystalline phases. As a result, such samples show higher values of the relative density, linear shrinkage and physicomechanical properties in the range of 1200 – 1600°C, high crack resistance at 1500°C and larger linear correlation of the elastic modulus and fracture toughness in the range of 1200 – 1600°C.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The problem of compaction and structural strengthening of the oxide–oxide-free crystalline phase boundaries is relevant and requires a comprehensive and diverse approach to its practical solution [1, 2]. This problem arises during the sintering of mixtures of oxide and oxide-free powder due to differences in the diffusion coefficients in the powders [1 – 4], and is also caused by phase transformation, for example, c-BN → h-BN, during which a brittle, intermediate layer of h-BN forms at the contact boundaries of particles of dissimilar powders with increasing content of c-BN in sintered powder mixtures, increasing temperature and compression load [5, 6]. This leads to uneven and incomplete sintering of the powders, the formation of dislocation regions, the formation of microcracks in the h-BN boundary layer, embrittlement of the boundaries of the crystalline phase regions and a deterioration of the physicomechanical properties of the material [2 – 4, 6, 7].
One of the traditional and most effective ways to solve this problem is to use additives of oxide powders containing rare-earth metals (for example, Y2O3, Dy2 O3) that intensively form low-melting eutectics with oxide and oxide-free powders, which stimulate diffusion of the substance through the eutectic melt and compensate for the difference in diffusion coefficients in the sintered powders [8 – 10]. These additives cause oxidation of the oxide-free powder with a change in its composition, forming particles of oxide powder, which otherwise affects the sintering of mixtures, grain growth of the oxide-free component and the formation of glass phase of various compositions with an increase in the content of the additive oxide component in sintering mixtures [8 – 10].
Traditionally, ZrB2 and c-ZrO2 additives are added to sintered mixtures of oxide and oxide-free powders, which compact and strengthen the structure of the boundary layers by sintering additive particles of the highest hardness with particles of the initial mixture components at the contact boundaries of dissimilar powders [11, 12]. These additives cause uneven and incomplete solid-phase sintering with increasing temperature and compression load; therefore, it is necessary to carefully select the amount and ratio of additives with the initial components of the mixtures [8 – 12].
At the same time, a method of compaction and strengthening of the structure of boundary layers of materials is introduced in practice by obtaining a TiC–ZrC solid solutionduring spark plasma sintering of a mixture of oxide and oxide- free powders at temperatures above 1500°C [13]. As a result, sintering and the formation of a solid solution develop simultaneously, which contributes to a more intensive and complete incorporation of the crystalline phase of the solid solution into the structure of the boundary layers and a significant improvement in the physicomechanical properties of the material [13]. During this sintering, compaction and strengthening of the structure of the boundary layers is realized through solid-phase sintering and depends on the percentage and structure of the components in the solid solution [13].
The purpose of the present work is to study the effect of the formation of TiC–ZrC solid solutions during spark plasma sintering at a compression load of 60 MPa in the range of 1200 – 1600°C on phase composition, microstructure, relative density, open porosity, linear shrinkage, physicomechanical properties, and linear correlation of the elastic modulus and fracture toughness of mullite–WC-, mullite–c-BN samples.
EXPERIMENTAL METHODS
Al2O3 and SiO2 powders were mixed (Table 1) according to the procedure described in [3]. The synthesis of WC and c-BN powders was carried out in a plasma-chemical unit under vacuum at 1600°C for 1 h (Table 2) according to the reactions:
Solid solutions of TiC–ZrC were obtained by spark plasma sintering under vacuum at 1800°C and a compression load of 70 MPa for 5 min (Table 3). The samples were then ground up in a planetary mill (RETSCH PM 400) for 30 min to obtain powders with particle sizes from 5 to 10 microns. Powders of WC, c-BN and TiC–ZrC solid solutions (see Table 3) were mixed in a planetary mill for 10 min. The resulting mixture of Al2O3 and SiO2 powders was mixed with the prepared groups of WC/80 TiC–20 ZrC, WC/90 TiC–10 ZrC, c-BN/80 TiC–20 ZrC and c-BN/90 TiC–10 ZrC powder mixtures in a planetary mill for 10 min. The component mixtures were poured into a graphite mold with a diameter of 30 mm and prepared by spark plasma sintering (SPS on a Summimoto unit, model SPS 825. CE, Dr. Sinter, Japan) under vacuum (6 Pa) with a compression force of 60 MPa, 2 min holding time in the range of 1200 – 1600°C with a heating rate of 100°C/min.
Phase composition of the synthesized powders, as well as microstructure, open porosity φ, relative density ρrel, linear shrinkage ∆l, elastic modulus E, Vickers hardness HV, and imprint surface area S of each sample (see Table 1) was calculated by the method described in [3]. Theoretical density of powder components was as follows, g/cm3: mullite 3.17, c-BN 3.49, WC 15.6, TiC 4.93, ZrC 6.73. The impact strength KIc of the samples was determined by the method described in [2].
Results And Discussion
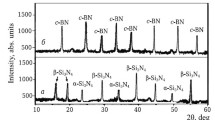
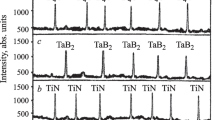
Phase composition of the synthesized WC and c-BN powders is mainly represented by intense diffraction maxima of WC and c-BN with a small amount of tungsten oxycarbide (Fig. 1). This phase is a non-stoichiometric composition of tungsten carbide containing an unreacted amount of WO3 and C. The phase composition of sintered TiC–ZrC solid solutions with different ratios of TiC and ZrC is shown in x-ray diffraction patterns (Fig. 2). The microstructure of sintered TiC–ZrC solid solutions depending on the ratio of TiC and ZrC is shown in Fig. 3.

Phase composition of WC (a) and c-BN (b) powders synthesized by spark plasma sintering at 1600°C: WC) tungsten carbide; WCxOy) tungsten oxycarbide; c-BN) cubic boron nitride.

Phase composition of TiC–ZrC solid solutions synthesized at 1800°C by spark-plasma sintering with a ratio of 80TiC–20ZrC(a) and 90 TiC-10 ZrC (b): (Ti, Zr)C) solid solution of zirconium carbide; (Zr, Ti)C) solid solution of titanium carbide.

Microstructure of TiC–ZrC solid solutions prepared by spark-plasma sintering at 1800°C with 80 TiC–20 ZrC (a) and 90 TiC–10 ZrC (b) component ratios.
X-ray diffraction patterns of sintered TiC–ZrC solid solutions are represented by less intense (Zr, Ti)C and developed (Ti, Zr)C phases (see Fig. 2). The lower intensity of the (Zr, Ti)C phase is explained by TiC content significantly exceeding the content of ZrC (see Table 2), which causes partial incorporation (intercalation) of Ti4+ cations into the ZrC structure, with (Zr, Ti)C developing less actively in the solid phase. In turn, the lowest ZrC content (see Table 2) facilitates the complete incorporation of Zr4+ cations into the TiC structure, developing (Ti, Zr)C in the solid phase more intensively. As a result, the difference between the cationic radii of Ti4+ and Zr4+ is inverse to the development of solid solutions at various ratios of TiC and ZrC (see Table 2). The microstructure of sintered TiC–ZrC solid solutions is crystalline, uniformly and densely sintered, consisting of evenly spaced (Ti, Zr)C regions in (Zr, Ti)C with an insignificant number of individual fine pores (see Fig. 3). The formation of dense microstructures is associated with incomplete development of (Zr, Ti)C in the solid phase, (Ti, Zr)C has a significant compaction and strengthening effect on the forming (Zr, Ti)C microstructure; TiC is mainly spent on solid phase sintering of these compositions at a compression load of 70 MPa.
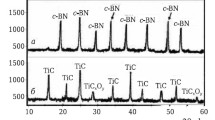
The phase composition of samples prepared from mixtures of the starting components by spark plasma sintering in the range of 1200 – 1600°C is shown in Fig. 4. Samples containing solid solutions with different ratios of TiC and ZrC show intense mullitization in the range of 1200 – 1600°C, which is due to the intensive structuring of mullite during the interaction of Al2O3 and SiO2 and the formation of its stoichiometric composition. (Ti, Zr)C develops more intensively in comparison with (Zr, Ti)C in the range of 1200 – 1600°C. This is explained by a more complete structuring of (Ti, Zr)C in comparison with (Zr, Ti)C in the solid phase in the range of 1200 – 1600°C. As a result, intense and narrow diffraction maxima of (Ti, Zr)C are formed in comparison with weak and individual wide diffraction maxima of (Zr, Ti)C. Crystalline TiC is observed in samples with WC; it is especially intensively developed in the range of 1400 – 1600°C (see Fig. 4a, b ). This is due to the partial decomposition of WC with the formation of crystalline W2C and amorphous carbon by the reaction:

Phase composition of samples sintered in the 1200 – 1600°C range with the following compositions: M3WC97 (80 TiC–20 ZrC) (a), M3WC97 (90 TiC–10 ZrC) (b), M3BN97 (80 TiC–20 ZrC) (c) and M3BN97 (90 TiC–10 ZrC) (d): M) mullite (3Al2O3·2SiO2); WC, W2C) tungsten carbides; c-BN) cubic boron nitride; (Zr, Ti)C) solid solution of titanium carbide; (Ti, Zr)C) solid solution of zirconium carbide.
Amorphous carbon actively diffuses into a less densely structured (Zr, Ti)C with its intense carbon saturation. As a result, (Zr, Ti)C becomes less stable and partially decomposes into a solid solution of non-stoichiometric composition and TiC in the range of 1400 – 1600°C by the reaction:
In the samples, there is an active increase in WC to 1300°C and c-BN in the range of 1200 – 1600°C (see Fig. 4), which is due to intense diffusion and structuring of these components in the solid phase. The intensity of these processes in a less dense (hexagonal) WC structure is higher than in a dense (cubic) c-BN structure. On x-ray diffraction patterns of samples in the range of 1300 – 1600°C, a decrease in the intensity of WC diffraction maxima is observed with the formation of an incidental crystalline W2C phase (see Fig. 4a, b ). This is due to the partial decomposition of WC (reaction (3)), which is insignificant in the range of 1300 – 1400°C and more active in the range of 1400 – 1600°C (see Fig. 4a, b ). These results correspond to the phase diagram of the equilibrium system W–C [14].
In contrast to the partial decomposition of WC, cubic boron nitride is not susceptible to decomposition in conditions of spark plasma sintering of the corresponding compositions (see Fig. 4c, d ). This is explained by the cubic structure of c-BN more resistant to the compression load with increasing temperature compared to the less stable hexagonal structure of WC. At the same time, the formation of h-BN in the range of 1200 – 1600°C is not observed in the samples (see Fig. 4c, d ). This indicates the absence of the c-BN → h-BN phase transformation with increasing temperature. Mullite does not interact with (Zr, Ti)C, (Ti, Zr)C, WC, and c-BN, since there are no products of decomposition of mullite and oxidation of solid solutions, WC and c-BN in the range of 1200 – 1600°C (see Fig. 4).
The microstructure of samples prepared by spark plasma sintering at 1500°C is shown in Fig. 5.

Microstructure of samples sintered at 1500 (Ñ with the composition M3WC97 (80 TiC–20 ZrC) (a), M3WC97 (90 TiC–10 ZrC) (b), M3BN97 (80 TiC–20 ZrC) (c) and M3BN97 (90 TiC–10 ZrC) (d).
The microstructure of samples with WC is crystalline, unevenly and least densely sintered, with weakly sintered areas and many pores of various sizes (see Fig. 5a, b ). This is explained by the partial decomposition of WC with the formation of crystallineW2C and amorphous carbon by reaction (3). A decrease in the content of WC in sintered compositions with increasing temperature (see Fig. 4a, b ) leads to a decrease in structure strengthening, and amorphous carbon slows down sintering at the boundaries of Al2O3, SiO2 and WC, Al2O3, SiO2 and (Zr, Ti)C, (Ti, Zr)C particles the least, and at the boundaries of WC and (Zr, Ti)C, (Ti, Zr)C particles — the most. In the first case, this is due to the viscous flow and activation of diffusion of oxide components, which stimulate more uniform and complete sintering of dissimilar particles. In the second case, this is explained by limited and incomplete solid-phase diffusion at the boundaries of oxide- free sinter particles; as a result, amorphous carbon as the finely dispersed component is more actively embedded and accumulates at the boundary regions of these particles. These processes correspond to the microstructure of the boundaries of crystalline phase regions (Fig. 6a – a4). Densely sintered regions of different sizes are observed in the microstructure of the samples formed during active diffusion between particles of oxide components and WC, (Zr, Ti)C, (Ti, Zr)C.

Microstructure of sintered samples of compositions M3WC97 (90 TiC–10 ZrC) (a – a4-0) with a distorted portion of the boundary between regions (a4) and dislocations (a4-0) and M3BN97 (90 TiC–10 ZrC) (b – b2) at 1300 °C (a – a2, b) and at 1500°C (a3 – a4-0, b1, b2).
The microstructure of c-BN samples is crystalline, most uniformly and densely sintered with an insignificant number of pores and weakly sintered areas (see Fig. 5c, d ). This is due to diffusion between oxide components and (Zr, Ti)C, (Ti, Zr)C particles due to the viscous flow, activation of diffusion of Al2O3 and SiO2, which stimulates mainly intensive and uniform solid-phase sintering of c-BN particles at 1500°C. An insignificant number of pores and weakly sintered areas in the samples is explained by less developed diffusion processes at the boundaries of sintered particles (Zr, Ti)C, (Ti, Zr)C and c-BN; as a result, an uneven, incomplete solid-phase sintering of these oxide-free particles develops.
Changes in ρrel, φ, ∆l, microstructure, E, KIc, HV and a photo of Vickers hardness imprints of samples with different ratios of c-BN and cubic ZrO2, respectively, sintered in the range of 1200 – 1600 and at 1500°C are shown in Figs. 6 – 9.
An increase in ρrel and ∆l with a decrease in φ of samples containing WC and c-BN is approximately similar for samples sintered in the range of 1200 – 1400°C. This is explained by the uniform filling of pores during mullite diffusion, the initiation of structuring of (Zr, Ti)C and (Ti, Zr)C (see Fig. 4). However, above 1400°C, significant sintering differences are observed depending on the oxide-free component found in the sintering composition. The decrease in ρrel and ∆l of the samples containing WC is explained by the partial decomposition of WC (see Fig. 4a, b) with the initiation of diffusion and uneven distribution (content) of W2C, carbon at the boundaries of sintered particles of Al2O3, SiO2 and WC, Al2O3, SiO2, WC and (Zr, Ti)C, (Ti, Zr)C, WC and (Zr, Ti)C, (Ti, Zr)C, as well as with intense diffusion of carbon through the pores in the range of 1400 – 1600°C. This correlates with the microstructure of the boundaries of crystalline phase regions (see Fig. 6a3). As a result, the diffusion of matter between these particles slows down, inhomogeneous sintering of particles develops, and pore filling slows down (see Fig. 5a, b ). At the same time, a slight increase in the φ of the samples is noticeable in the range of 1500 – 1600°C, caused by pore coalescence.
Samples containing c-BN show a larger and more stable increase in ρ and ∆l with a corresponding decrease in φ in the range of 1200 – 1600°C. This is due to the structuring of (Zr, Ti)C and (Ti, Zr)C, active and uniform sintering of c-BN particles in the solid phase (see Fig. 4c, d ) with insignificant formation of pores and weakly sintered areas (see Fig. 5c, d ). The sample with the 90/10 mol.% ratio of TiC/ZrC shows large values of these properties compared with the properties of the sample with the 80/20 mol.% ratio. This is due to a more developed structuring of (Ti, Zr)C in the sample in the 1200 – 1600°C range (see Fig. 4d ).
The increase in KIc and HV of samples with WC up to 1400°C is due to compaction and strengthening of the structure at the boundaries of the mullite–WC (see Fig. 6a ) and mullite–(Zr, Ti)C–(Ti, Zr)C regions (see Fig. 6, a1). At the same time, a gradual increase in the physicomechanical properties is observed in the range of 1200 – 1400°C. This is explained by weak elastic properties at the boundary of the WC–(Zr, Ti)C–(Ti, Zr)C regions (see Fig. 6a2), which cause incomplete compaction and strengthening of the structure at this boundary section. In this case, the sample is characterized by high crack resistance with a minimum number and a small length of microcracks propagating along a relatively rectilinear path (see Fig. 9a ) along the boundary of the less strengthened regions WC–(Zr, Ti)C–(Ti, Zr)C. A decrease in the physicomechanical properties of these samples is observed above 1400°C, and most intensively in the range of 1500 – 1600°C (see Fig. 8). This is explained by the partial decomposition of WC and the corresponding destabilization (decay) of the incompletely structured (Zr, Ti)C with the formation of a less dense crystalline TiC structure in the range of 1400 – 1600°C (see Fig. 4a, b). These processes lead to an uneven distribution of crystalline W2C and amorphous carbon at the boundary section of the mullite–WC regions (see Fig. 6, a3), the formation of a dislocation region at the mullite–WC–(Zr, Ti)C–(Ti, Zr)C boundary (see Fig. 6a4, a4-0) during the incorporation of crystalline TiC into the boundary section WC–(Zr, Ti)C–(Ti, Zr)C (see Fig. 6, a4) through its embrittlement with the non-stoichiometric composition (Zr, Tix–1)Cy–1. As a result, brittleness increases, the compaction and strengthening of the structure decrease, the sliding of particles relative to each other under the action of the external applied load at the boundary sections of the mullite–WC, mullite–WC–(Zr, Ti)C–(Ti, Zr)C regions increases. Finely dispersed (up to 1 μm) amorphous carbon located closer to the boundary of the mullite–WC regions affects these processes to a greater extent, in contrast to the smaller influence of coarse (~2.5 μm) crystalline W2C (see Fig. 6a3). Thus, elastic properties (E, KIc, and HV) decrease and brittle properties increase at these boundary sections of the samples (see Fig. 8), which is caused by the formation of an uneven and less densely sintered microstructure of the samples (see Fig. 5a, b ). Consequently, crack resistance of the sample significantly decreases with a maximum number and greater length of microcracks propagating along a winding path (see Fig. 9a1) along the boundary sections of the least strengthened areas of mullite–WC and mullite– WC–(Zr, Ti)C– (Ti, Zr)C.
A more intense increase in KIc and HV of samples with c-BN up to 1400°C is due to developed elastic properties and, as a result, compaction and strengthening of the structure at the boundaries of the mullite–(Zr, Ti)C–(Ti, Zr)C regions (see Fig. 6b ). Such a sample has high crack resistance and no microcracks (see Fig. 9b ). A further rapid increase in the physicomechanical properties of the samples in the range of 1400 – 1600°C (see Fig. 8) is associated with the absence of decomposition of (Zr, Ti)C, more advanced structuring of (Ti, Zr)C, and a slight development of c-BN (see Fig. 4c, d ), the formation of the most uniformly and densely sintered microstructure (see Fig. 5c, d ), greater compaction and strengthening at the boundaries of the mullite–c-BN and c-BN–(Zr, Ti)C–(Ti, Zr)C regions (see Fig. 6b1, b2) and the most developed elastic properties at the boundary sections of the regions of these crystalline phases of the sample. At the same time, such a sample is characterized by slightly lower crack resistance with a minimum number and length of microcracks propagating along a relatively winding path at 1500°C (see Fig. 9b1). This is explained by the formation of separate unevenly and incompletely sintered sections during solid phase sintering of (Ti, Zr)C and c-BN particles (see Fig. 5c, d ) with microcracks propagating along their boundaries.
The results of linear correlation of E and KIc of sintered samples are shown in Fig. 10. The value of R2 is slightly higher for the M3WC97 (90 TiC–10 ZrC) sample than for M3WC97 (80 TiC–20 ZrC). In this case, the deviation of linear straight lines with respect to the points (values) E and KIc, as well as their location relative to each other, are identical in the range of 1200 – 1600°C. These results correlate with a change in phase composition (see Fig. 4a, b ), microstructure (see Fig. 5a, b ), processes at the boundaries of the regions of oxide – oxide-free and oxide-free crystalline phases (see Fig. 7a – a4-0), physicomechanical properties (see Fig. 8) and crack resistance (see Fig. 9a, a1) of the samples.

Change in ρrel, φ, and ∆l of samples in the range of 1200 – 1600°C depending on the ratio of TiC and ZrC in solid solutions: □) M3WC97 (80 TiC–20 ZrC); ■) M3WC97 (90 TiC–10 ZrC);◊) M3BN97 (80 TiC–20 ZrC); ♦) M3BN97 (90 TiC–10 ZrC).

Change in E,KIc and HVof samples sintered in the range of 1200 – 1600°C depending on the ratio of TiC and ZrC in solid solutions: □) M3WC97 (80 TiC–20 ZrC); ■) M3WC97 (90 TiC–10 ZrC); ◊) M3BN97 (80 TiC–20 ZrC); ♦) M3BN97 (90 TiC–10 ZrC).

Vickers hardness HV imprints of samples sintered at 1300 and 1500°C with compositions M3WC97 (90 TiC–10 ZrC) (a, a1) and M3BN97 (90 TiC–10 ZrC) (b, b1).

Linear correlation of E and KIc of samples sintered in the range of 1200 – 1600°C.
A slight difference of about 0.003, for maximum values, is evident in the R2 value for the compositions M3BN97 (80 TiC–20 ZrC) and M3BN97 (90 TiC–10 ZrC). There is practically no deviation of linear straight lines relative to the points (values) E and KIc, and their position relative to each other in the range of 1200 – 1600°C is approximately the same. The greatest correlation of the property values of samples containing c-BN, compared with samples containing WC, is due to more intensive structuring of (Ti, Zr)C and slightly better development while maintaining the integrity of the composition (Zr, Ti)C (see Fig. 4c, d ), the formation of uniform and most densely sintered microstructures (see Fig. 5c, d ), a large strengthening of the structure with barely noticeable boundaries of the areas of oxide – oxide-free and oxide-free crystalline phases (see Fig. 6b – b2), active development of elastic properties (see Fig. 8) and greater crack resistance (with. Fig. 9b, b1).
Conclusion
The effect of the formation of sintered TiC–ZrC solid solutions in samples of mullite–WC–TiC–ZrC and mullite– c-BN–TiC–ZrC during spark plasma sintering with a compression load of 60 MPa in the range of 1200 – 1600°C on their phase composition, microstructure, ρrel, φ, ∆l, physicomechanical properties and linear correlation of E and KIc is shown.
The synthesized powders of WC, c-BN and solid solutions of TiC–ZrC sintered at 1800°C by spark plasma sintering are characterized by intensive crystallization of WC, c-BN, and (Ti, Zr)C, respectively, with a uniformly, densely sintered crystalline microstructure.
Sintered samples containing 80/20 and 90/10 mol.% solid solutions of TiC and ZrC are characterized by intense mullitization and development of (Ti, Zr)C. However, a gradual increase in c-BN and (Zr, Ti)C and a decrease in the intensity of WC is observed in the samples in the range of 1200 – 1600°C. The microstructure of samples containing c-BN and solid solutions of TiC–ZrC in various proportions, is more uniformly and densely sintered, crystalline, fine-grained with few pores, and most strengthened at the boundaries of the regions of oxide and oxide-free crystalline phases. Such samples show large values of ρ, ∆l and physicomechanical properties in the range of 1200 – 1600°C, high crack resistance with insignificant microcracks at 1500°C, and a significant linear correlation of E and KIc in the range of 1200 – 1600°C.
Change history
08 August 2020
To the article ���Strengthening Oxide���Oxide-Free Materials by Incorporation of TiC���ZrC Solid Solutions into Their Structure during Spark Plasma Sintering of Initial Powder Mixtures under High Compression Loads,��� by A. V. Hmelov, Vol. 60, No. 5, pp. 486 ��� 494, January, 2020
References
S. Chuan, L. Yunkai, W. Yunfei, and Z. Lingbo, “Effect of alumina addition on the densification of boron carbide ceramics prepared by spark plasma sintering technique,” Ceram. Int., 40(8), 12723 – 12728 (2014).
A. V. Hmelov, “Mullite–TiC–c-BN–c-ZrO2 materials produced by spark-plasma sintering and their properties,” Refract. Indust. Ceram., 60(1), 86 – 91 (2019).
A. V. Hmelov, “Preparation of mullite–TiC–TiN materials by a plasma spark method and their properties,” Refract. Indust. Ceram., 58(4), 418 – 425 (2017).
F. Yan, F. Chen, Q. Shen, and L. Zhang, “Spark plasma sintering of α-Si3N4 ceramics with MgO–Al2O3 as sintering additives,” Eng. Mat., 351(2), 176 – 179 (2007).
M. Hotta and T. Goto, “Densification and microstructure of Al2O3–c-BN composites prepared spark plasma sintering,” J. Ceram. Soc. Jap., 116(6), 744 – 748 (2008).
F. Ye, Z. Hou, H. Zhang, and L. Liu, “Spark plasma sintering of c-BN / β-sialon composites,” Mat. Sci. Eng. A, 527(18), 4723 – 4726 (2010).
S. Meir, S. Kalabukhov, and S. Hayun, “Low temperature spark plasma sintering of Al2O3–TiC composites,” Ceram. Int., 40(8), 12187 – 12192 (2014).
X. Zhang, X. Li, J. Han, and W. Han, “Effect of Y2O3 on microstructure and mechanical properties of ZrB2–SiC,” J. All. Comp., 465(1/2), 506 – 511 (2008).
S. Guo and Y. Kagawa, “High-strength zirconium diboridebased ceramic composites consolidated by low temperature hot pressing,” Sci. Techn. Adv. Mat., 13(4), 1 – 6 (2012).
A.-K. Wolfrum, B. Matthey, A. Michaelis, and M. Herrmann, “On the stability of c-BN reinforcing particles in ceramic matrix materials,” Materials, 255(11), 1 – 17 (2018).
A. V. Hmelov, “Producing and properties of mullite–sialon– ZrB2 materials obtained using a spark-plasma technique,” Refract. Indust. Ceram., 59(6), 633 – 641 (2019).
D. Chakravarty and G. Sundararajan, “Microstructure, mechanical properties and machining performance of spark plasma sintered Al2O3–ZrO2–TiCN nanocomposites,” J. Eur. Ceram. Soc., 33(13/14), 2597 – 2607 (2013).
A. V. Hmelov, “Preparation of mullite–TiC–ZrC ceramic materials by a plasma-ARC method and their properties,” Refract. Indust. Ceram., 57(6), 645 – 650 (2017).
A. S. Kurlov and A. I. Gusev, “Tungsten carbides and W–C phase diagram,” Inorg. Mat., 42(2), 121 – 127 (2006).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Novye Ogneupory, No. 10, pp. 18 – 26, October 2019.
Rights and permissions
About this article

Cite this article
Hmelov, A.V. Strengthening Oxide–Oxide-Free Materials by Incorporation of Tic–Zrc Solid Solutions into their Structure during Spark Plasma Sintering of Initial Powder Mixtures under High Compression Loads. Refract Ind Ceram 60, 486–494 (2020). https://doi.org/10.1007/s11148-020-00391-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11148-020-00391-6