
Different c-BN/c-ZrO2 ratios are shown to affect the phase composition, microstructure, relative density, open porosity, linear shrinkage, physicomechanical properties, and linear correlation of the elastic modulus and toughness of mullite–TiC–c-BN–c-ZrO2 samples during spark-plasma sintering at pressing load 70 MPa and 1200 – 1600°C. The synthesized TiC and c-BN powders and c-ZrO2 spark-plasma sintered at 1400°C are characterized by extensive phase crystallization. Mullite and TiC develop profusely in sintered samples with different c-BN/c-ZrO2 ratios. Increasing the c-BN/c-ZrO2 ratio promotes ingrowth of more c-BN than c-ZrO2 at 1200 – 1600°C and causes a less uniformly and densely sintered crystalline microstructure with many pores to form at 1500°C. This sample has lower physicomechanical properties, a poorer linear correlation of elasticity modulus and toughness at 1200 – 1600°C, and lower crack resistance at 1500°C.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The compatibility of oxide and non-oxide powders for spark-plasma sintering deserves special attention [1, 2] because of the different diffusion coefficients in the sintered powders [1,2,3]. It is especially critical for phase transformations, e.g., in boron nitride (BN) as its content in the sintered powder mixture and the temperature increase at low compression loads [1, 4]. This leads to uneven and incomplete sintering of powder mixtures in the transverse and/or longitudinal direction with formation of nonuniformly sintered microstructure and brittleness in boundary areas of oxide and non-oxide crystalline phases. As a result, the crack resistance decreases and the physicochemical properties of the materials are poorer [1,2,3,4].
The solution to this problem is to add rare-earth metals (e.g., Y2O3, Sm2O3, Dy2O3) that readily form low-melting eutectics with the oxide and non-oxide powders and solid solutions in the liquid phase because of the small cationic radii of the metals in them [5, 6]. The resulting low-melting eutectics promote nucleation of crystalline phases, stimulate diffusion of matter through the eutectic melt, and increase the contact area of the sintered particles. As a result, the powder mixture is more evenly and completely sintered [5,6,7]. The solid solutions compact the structure and strengthen it on microscopic and macroscopic levels at boundary areas of oxide and non-oxide crystalline phases and grain contacts, respectively. As a result, elastic properties are generated, the crack resistance increases, and the physicomechanical properties of the materials improve [7, 8]. On the other hand, these additives oxidize the non-oxide powder, change its composition, and diminish its content. The oxide powder particles have a different effect on sintering of mixtures of oxide and non-oxide powders, grain growth of non-oxide powder, and formation of a different glass-phase composition that increases the brittleness as the additive (oxide component) content, temperature, and compression load increase [8, 9]. As a result, the physicomechanical properties of the materials decrease significantly [8, 9].
ZrO2 [10] and ZrB2 powders [11] were added to the mixture of oxide and non-oxide powders to replace the used oxide powder additives and to improve the physicomechanical properties of the materials. However, these components in the powder mixtures caused uneven and incomplete solid-phase sintering. The optimal amount and ratio of additives had to be found to reduce their effects [10, 11].
The goal of the work was to study the effect of different c-BN/c-ZrO2 ratios during spark-plasma sintering with compression load 70 MPa at 1200 – 1600°C on the phase composition, microstructure, relative density, open porosity, linear shrinkage, physicomechanical properties, and linear correlation of the elasticity modulus and toughness of mullite–TiC–c-BN–c-ZrO2 samples.
Experimental
Preparation of mixtures of Al2O3 and SiO2, TiC and c-BN powders, and sintered c-ZrO2, preparation of mixtures of oxide and non-oxide powders
Al2O3 and SiO2 powders were mixed (Table 1) according to the published method [12]. TiC and c-BN powders were synthesized in a plasma-chemical unit in vacuo at 1600°C for 1 h using TiO2 (98.0%, Aldrich, Belgium), C (97.5%, Merck, Germany), and B2O3 powders (97.5%, Merck, Germany) and N2 (99.5%, Aldrich, Belgium) and the reactions TiO2 + 2C → TiC + CO2 and 2 B2O3 + 3.5 N2 → 4 c-BN + 3 NO2. Cubic zirconia c-ZrO2 was spark-plasma sintered in vacuo at compression load 35 MPa for 2 min at 1400°C. Starting ZrO2 (97.5%, Merck, Germany) and Y2O3 powders (99.5%, Aldrich, Belgium) were used in a 17.58/1 ratio, which corresponded to a biphasic system on the ZrO2–Y2O3 equilibrium phase diagram (by Brown and Odell and Fan Fu-Kanu and Keler) [13]. The component masses (97 mol% ZrO2/3 mol% Y2O3) in units of g per 100 g of mixture were 94.62/5.38.
Samples of powder mixtures of Al2O3 and SiO2, TiC and c-BN, and c-ZrO2 were prepared by the published method [12] at compression load 70 MPa.
Method for determining properties of powders and sintered samples
The phase composition of the synthesized and sintered powders, microstructure, relative density ρrel, open porosity φ, linear shrinkage ∆l, elasticity modulus E, Vickers hardness HV, and surface area S of each sample (Table 1) were calculated as before [12]. The theoretical densities (g/cm3) of the powder components were 3.17, mullite; 4.93, TiC; 3.49, c-BN; and 6.27, ZrO2.
Toughness of the sintered samples was determined by a Vickers microcompression method using an AVK-A Hardness Testing Machine (Akashi Co., Japan) and was calculated using the formula
where KIc is the critical strain coefficient or toughness (MPa·m1/2); P, load applied to test sample surface (kg/cm2); c, half-length of microcracks formed around indenter impression corners (mm).
Results and Discussion
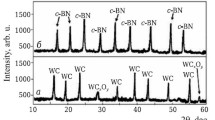
Figure 1 illustrates the phase compositions of TiC and c-BN powders synthesized by a plasma-chemical method. It shows mainly strong diffraction peaks for TiC and c-BN with a small amount of TiCxOy. This phase was nonstoichiometric TiC containing unreacted TiO2 and C.

Phase composition of TiC (a) and c-BN powders (b ) synthesized by a plasma-chemical method at 1600°C: TiCxOy is titanium oxycarbide.
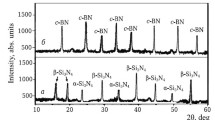
Figure 2 illustrates the phase composition of sintered c-ZrO2, which was characterized by distinct diffraction peaks for this phase. This was explained by strain and rearrangement of the ZrO2 structure from tetragonal to cubic under the compression load and diffusion of Y3+ into the c-ZrO2 structure, according to the ZrO2–Y2O3 equilibrium phase diagram (by Brown and Odell and Fan Fu-Kanu and Keler) [13].

Phase composition of c-ZrO2 spark-plasma sintered at 1400°C.
Figure 3 shows the phase compositions of mixtures of starting components spark-plasma sintered at 1200 – 1600°C.

Phase composition of M92TiC3BN5ZrO2 (a) and M92TiC5BN3ZrO2 samples (b ) sintered at 1200 – 1600°C: M, mullite (3Al2O3·2SiO2); h-BN, hexagonal boron nitride; B4C, boron carbide; TiN, titanium nitride.
Samples of all compositions typically underwent extensive mullitization at 1200 – 1600°C due to structuring and stoichiometric mullite formation. In a similar manner, TiC resulted from a transition into a viscoelastic (plastic) state that promoted diffusion and structuring of TiC under these sintering conditions. Strong ingrowth of c-BN and less of c-ZrO2 were observed with increasing c-BN/c-ZrO2 ratio at 1200 – 1600°C. However, c-BN and c-ZrO2 grew less vigorously than mullite and TiC. This was explained by the denser structures of these components with covalent bonds in c-BN. This limited diffusion and structuring of c-BN and c-ZrO2 in the solid phase. Also, the ingrowth of c-ZrO2 in the sample with 5 mol% c-ZrO2 was slightly greater than that of c-BN in the sample with 5 mol% c-BN at 1200 – 1600°C. This was explained by better diffusion into c-ZrO2 than into c-BN. In addition, h-BN was detectable in the sample with 5 mol% c-BN but not in that with 3 mol% c-BN because of a partial phase transformation of c-BN into h-BN in the solid in a sintered powder mixture with 5 mol% c-BN. These samples had different quantitative ratios of diffraction maxima for c-BN and c-ZrO2 at 1200 – 1600°C (Fig. 3) because of different amounts of crystalline phases. The x-ray pattern of the sample with 5 mol% c-BN showed weak diffraction peaks for B4C and TiN that were missing for the sample with 3 mol% c-BN at 1500 – 1600°C. These side phases formed from the reaction of TiC and h-BN in the sintered powder mixture. Mullite and c-ZrO2 did not react simultaneously with TiC and c-BN because decomposition products of mullite and oxidation products of TiC and c-BN did not form at 1200 – 1600°C (Fig. 3).
Figure 4 shows microstructures of samples spark-plasma sintered at 1500°C. The microstructure of sintered composition M92TiC3BN5ZrO2 (Fig. 4a ) was more uniformly and densely sintered, crystalline, and fine-grained with fewer pores and weakly sintered sections than that of composition M92TiC5BN3ZrO2 (Fig. 4b ). This was explained by the different c-BN/c-ZrO2 ratios in the sintered compositions (Table 1). Sintering was promoted more by diffusion of the oxide component in the solid phase at high c-ZrO2 and c-BN ratios. Viscous flow of mullite and TiC melts also helped to form microstructures with different uniformities and densities. This was explained by the different sintering behaviors of c-BN and c-ZrO2 particles in the presence of these melts.

Microstructure of M92TiC3BN5ZrO2 (a) and M92TiC5BN3ZrO3 samples (b ) sintered at 1500°C.
Figures 5 – 8 show measured ρrel, φ, ∆l, E, KIc, HV, and a photograph of impressions and microstructures of samples with different c-BN/c-ZrO4 ratios at 1200 – 1600°C and 1500°C.

Parameters ρrel, φ, and ∆l of samples with different c-BN/c-ZrO2 ratios that were sintered at 1200 – 1600°C: M92TiC3BN5ZrO2 (1 ) and M92TiC5BN3ZrO2 (2 ).
Composition N92TiC3BN5ZrO2 had a decreased φ and the maximum degree of sintering (91.2%) at 1600°C. This was due to the more even filling of pores because of viscous flow of mullite and TiC melts and initiation of c-ZrO2 diffusion at 1200 – 1300°C. Diffusion of c-ZrO2 became greater than that of c-BN in the solid phase at 1300 – 1500°C (Fig. 3a ). Sintering slowed at 1500 – 1600°C with φ > 8% at 1600°C. This was explained by less extensive and incomplete pore filling during solid-state sintering of c-BN and c-ZrO2 particles. The sintering results correlated with the sample microstructure at 1500°C (Fig. 4a ).
The rises of ρrel and ∆l and fall of φ for composition M92TiC5BN3ZrO2 at 1200 – 1600°C were irregular. Extensive sintering up to 1400°C was explained by pore filling resulting from viscous flow of mullite and TiC melts (Fig. 3b ). Slow sintering at 1400 – 1500°C was due to weaker diffusion of c-ZrO2 (at 3 mol%) and c-BN because of the increasing partial phase transformation c-BN → h-BN in the solid phase (Fig. 3b ) with the latter formed as a layer on the sintered particles that hindered diffusion of matter between particles and pore filling. This was confirmed by studying microstructures of boundary regions of sample crystalline phases (Fig. 6b1 – b4). The sintering was reduced somewhat because of the formation of a certain amount of h-BN, similar crystalline phases B4C and TiN (Fig. 3b ), and a slight porosity increase at 1500 – 1600°C. This correlated with the microstructure of the sample sintered at 1500°C (Fig. 4b ).

Microstructure of boundary regions of mullite, c-ZrO2, TiC, c-BN, and h-BN in compositions M92TiC3BN5ZrO2 (a, a1) and M92TiC5BN3ZrO2 (b, b1 – b4) sintered at 1500°C.
The crystalline, more uniform, and denser sintered fine-grained microstructure of the sample with 3 mol% c-BN (Fig. 4a ) helped significantly to improve the elastic properties and, as a result, to increase the resistance of the sample to external applied loads with high KIc and HV values at 1500°C (Fig. 7). On the other hand, the high parameters for these properties were due to insignificant mullite–TiC (Fig. 6a ), mullite–c-ZrO2–c-BN, and c-ZrO2–c-BN boundary regions (Fig. 6a1). The sample had short microcracks along straight-line trajectories (Fig. 8a ). The impression showed slight damage as a chip (Fig. 8a ), indicating little accumulation of internal stresses and minimally brittle boundary regions of these crystalline phases (Fig. 6a, a1). This corresponded to extensive solid-state sintering of c-ZrO2 and c-BN particles at 1400 – 1600°C (Fig. 5).

Parameters E, KIc, and HV of samples with different c-BN/c-ZrO2 ratios that were sintered at 1200 – 1600°C: M92TiC3BN5ZrO2 (1 ) and M92TiC5BN3ZrO2 (2 ).

Impressions from measuring HV on samples M92TiC3BN5ZrO2 (a) and M92TiC5BN3ZrO2 (b ) sintered at 1500°C.
The sample with 5 mol% c-BN had less drastic changes of elastic properties (KIc and HV) up to 1400°C. The increase of physicomechanical properties slowed at 1400 – 1500°C and decreased slightly at 1500 – 1600°C (Fig. 7). This was due mainly to h-BN formation at boundary sections of sample crystalline phases (Fig. 6b, b1 – b4) because of partial phase transformation of c-BN into h-BN at 1500 – 1600°C (Fig. 3b ). The resulting h-BN embrittled the structure with noticeable boundary regions of crystalline phases (Fig. 6b ) that were less extensive at mullite–c-BN and c-ZrO2–c-BN boundary regions (Fig. 6b1, b3) and more extensive at TiC–c-BN and c-BN–h-BN boundary regions (Fig. 6b1 – b4). These properties decreased because the microstructure was incompletely sintered and the sample had many pores (Fig. 4b ). As a result, the crack resistance of the sample to formation of many microcracks propagating along tortuous trajectories of more weakly sintered regions (Fig. 6b2, b4) and chips (Fig. 8b ) was diminished. This was caused by the high brittleness of the sample at the TiC–c-BN and c-BN–h-BN boundary regions (Fig. 6b2, b4).
Figure 9 shows the linear correlation of E and KIc. A comparison of the R2 parameters of the approximations with 3 and 5 mol% c-BN showed that the difference was small at 0.01 and was greater for the sample with 3 mol% BN, which agreed with the greater increase of physicomechanical properties (Fig. 7). This was due to the formation of a uniformly and densely sintered fine-grained microstructure with few pores (Fig. 4a ) and strengthening of the structure at mullite – TiC (Fig. 6a ), mullite – c-ZrO2 – c-BN, and c-ZrO2 – c-BN boundary regions at 1500°C (Fig. 6, a1). As a result, the E and KIc values of the sample correlated well with the straight line at 1200 – 1600°C and slightly less at 1400°C. This was related to initiation of solid-state sintering because of extensive diffusion of c-ZrO2 and less of c-BN at this temperature (Fig. 5). The high correlation of the sample properties with the straight line was indicative of uniform viscous-flow sintering at 1200 – 1300°C and the corresponding solid-state sintering at 1300 – 1600°C.

Linear correlation of E and KIc for samples sintered at 1200 – 1600°C.
The lower R2 values for the sample with 5 mol% c-BN were explained by irregularly and incompletely sintered microstructure with many pores (Fig. 4b ) and a weaker structure with formation of thicker h-BN in the TiC – c-BN and c-BN – h-BN boundary regions at 1500°C (Fig. 6, b2, b4). As a result, the E and KIc values deviated more from the straight line at 1600°C than those at 1200, 1300, 1400, and 1500°C, which did not deviate. This indicated that the effects of these processes were greatest and caused nonuniform solid-state sintering of the composition at 1600°C. The different effects of the correlated properties on the R2 values in both samples were explained by the different sintering mechanisms and, as a result, the different degrees of sintering of the corresponding compositions at 1200 – 1600°C (Fig. 5). The E and KIc points (values) correlated best with the straight line and, as a result, the R2 values were greater for the sample with 3 mol% c-BN at 1200 – 1600°C because of the complete and uniform solid-phase sintering.
Conclusion
Different c-BN/c-ZrO2 ratios had different effects during spark-plasma sintering of compositions at compression load 70 MPa and 1200 – 1600°C on the phase composition, microstructure, ρrel, φ, ∆l, physicomechanical properties, and linear correlation of E and KIc of mullite–TiC–c-BN–c-ZrO2 samples. The synthesized TiC and c-BN powders and c-ZrO2 spark-plasma sintered at 1400°C were extensively crystallized.
Sintered samples with different c-BN/c-ZrO2 ratios showed extensive mullite and TiC ingrowth. Increasing the c-BN/c-ZrO2 ratio promoted c-BN ingrowth more than c-ZrO2 at 1200 – 1600°C and caused a less uniformly and densely sintered crystalline microstructure with many pores to form at 1500°C. As a result, the sample had lower ρrel, ∆l, and physicomechanical properties at 1200 – 1600°C, less crack resistance at 1500°C, and a poorer linear correlation of E and KIc at 1200 – 1600°C.
References
M. Hotta and T. Goto, “Densification and microstructure of Al2O3–cBN composites prepared spark plasma sintering,” J. Ceram. Soc. Jpn., 116(6), 744 – 748 (2008).
S. Chuan, L. Yunkai, W. Yunfei, and Z. Lingbo, “Effect of alumina addition on the densification of boron carbide ceramics prepared by spark plasma sintering technique,” Ceram. Int., 40(8), 12723 – 12728 (2014).
A. V. Khmelev, “Preparation of mullite–TiC–ZrC ceramic materials by a plasma-arc method and their properties,” Refract. Ind. Ceram., 57(6), 645 – 650 (2017).
P. Klimczyk, M. E. Cura, and A. M. Vlaicu, “Al2O3–cBN composites sintered by SPS and HPHT methods,” J. Eur. Ceram. Soc., 36(7), 1783 – 1789 (2016).
O. Fabrichnaya, G. Savinykh, T. Zienert, and G. Schreiber, “Phase relations in the ZrO2–Sm2O3–Y2O3–Al2O3 system: Experimental investigation and thermodynamic modelling,” Int. J. Mater. Res., 103(12), 1469 – 1487 (2012).
S. Guo and Y. Kagawa, “High-strength zirconium diboride-based ceramic composites consolidated by low temperature hot pressing,” Sci. Technol. Adv. Mater., 13(4), 1 – 6 (2012).
X. Zhang, X. Li, J. Han, and W. Han, “Effect of Y2O3 on microstructure and mechanical properties of ZrB2–SiC,” J. Alloys Compd., 465(1/2), 506 – 511 (2008).
W. Li, Z. Xinghong, H. Changquing, and H. Jiecai, “Hotpressed ZrB2–SiC–YSZ composites with various yttria content: Microstructure and mechanical properties,” Mater. Sci. Eng., A, 494(1/2), 147 – 152 (2008).
A.-K. Wolfrum, B. Matthey, A. Michaelis, and M. Herrmann, “On the stability of c-BN reinforcing particles in ceramic matrix materials,” Materials, 255(11), 1 – 17 (2018).
D. Chakravarty and G. Sundararajan, “Microstructure, mechanical properties and machining performance of spark plasma sintered Al2O3–ZrO2–TiCN nanocomposites,” J. Eur. Ceram. Soc., 33(13/14), 2597 – 2607 (2013).
X. Zhang, W. Li, C. Hong, and W. Han, “Microstructure and mechanical properties of ZrB2-based composites reinforced and toughened by zirconia,” Int. J. Appl. Ceram. Technol., 58(5), 499 – 504 (2008).
A. V. Khmelev, “Preparation of mullite–TiC–TiN materials by plasma spark method and their properties,” Refract. Ind. Ceram., 58(4), 418 – 425 (2017).
A. Hmelov, “Synthesis of ceramic powders in the Al2O3–SiO2–ZrO2 (Y2O3) system and production of materials,” Doctoral Thesis, Riga Technical University, 2011, pp. 1 – 27.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Novye Ogneupory, No. 2, pp. 23 – 29, February, 2019.
Rights and permissions
About this article

Cite this article
Hmelov, A.V. Mullite–TiC–c-BN–c-ZrO2 materials Produced by Spark-Plasma Sintering and Their Properties. Refract Ind Ceram 60, 86–91 (2019). https://doi.org/10.1007/s11148-019-00314-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11148-019-00314-0