
Abstract
The nickel-based catalysts supported on MgO-modified α-Al2O3, CeO2, and SBA-15 were prepared by impregnation method and investigated by N2 physisorption measurements, powder X-ray diffraction, Raman spectroscopy, H2 temperature-programmed reduction, CO2 temperature-programmed desorption, and transmission electron microscopy. Investigation of the kinetics of the dry reforming of methane (DRM) was carried out in gradientless circulating micro-flow system at atmospheric pressure and temperature range of 600–800 °C. The results showed that carriers have a prominent role in characterising the physico-chemical properties of catalysts such as specific surface area, dispersity of active metal, reducibility and basicity that greatly affect the adsorption feature and activity of NiO catalyst. However, the kinetic equation of DRM on three catalysts was found to be written by a common fractional equation, following a dual-site Langmuir–Hinshelwood Hougen Watson model. The order in the catalyst reducibility and apparent rate constant was observed as follows: NiMg/Al<Ni/Ce<Ni/SBA, while the apparent activation energy (E) is in the opposite order. The highest activity was observed on the catalyst containing 31.2 wt% Ni supported on SBA-15.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dry reforming of methane (DRM) is a promising way to convert two major greenhouse gases, CO2 and CH4, into valuable semi-product, the synthetic gas. Many metals used as active components of catalysts have been reported for reforming of CH4. Among these metals, due to the relatively high activity, low cost and availability, Nickel has been used frequently in researches. However, current restrictions on the activity and stability caused by strong coke-formation and sintering of the nickel catalysts make them inapplicable commercially. So, many ways have been proposed to improve the Ni-based catalysts for methane reforming.
The most commonly used support for DRM is Al2O3 while other supports such as MgO, TiO2, SiO2, and La2O3 are also used [1]. Both Ni/α–Al2O3 and Ni/γ–Al2O3 were unstable with time on stream (TOS) for methane reforming. While Ni/α–Al2O3 has been reported to be unstable due to carbon deposition, phase change of γ-Al2O3 at high temperatures (700 °C) was the reason for instability of NiO/γ–Al2O3 catalyst [2]. Nakamura et al. [3] found that the effect of the support in the performance of reforming catalysts decreased in following order: Al2O3>TiO2>SiO2.
To overcome the limitations of Al2O3 support, the alkali metals [4] and lanthanides [5] had been added to Al2O3. The addition of basic oxide as MgO led to increase CO2 adsorption as well as enhance the coke resistance of the catalyst [4]. Core–shell catalysts are also studied for abating the aggregation of Ni particles under high reaction temperature. The core–shell catalysts [(Ni/MgAl2O4)@SiO2] had better coke-resistance and stability than the uncoated catalysts (Ni/MgAl2O4) in DRM [6]. The addition of lanthanum in the catalysts led to the formation of LaNiO3 perovskite, which reduces the sintering of the active phase, increases the degree of dispersion of the catalyst and provides better Ni–La interaction in dry reforming of methane [7]. Xu et al. [8] presented that the addition of lanthanum in the NiO–La2O3–Al2O3 catalyst improves the absorptive capacity of CO2 by improving the reactivity of the reaction.
As a material with high thermal stability (SBA-15 [9]) and unique properties (CeO2 [10]), they recently are considered as the promising supports for DRM reaction. CeO2 has been known as a material with high oxygen storage and release by shifting between CeO2 under oxidizing conditions and Ce2O3 under reducing conditions [11]. This property originates from the relative ionic radius of Ce4+/O2− of 0.71 which results in a coordination number of Ce/O atoms of 8 and that of O/Ce atoms of 4 [12], producing oxide defects in CeO(2−x) (x ~ 0.5) [13]. High oxygen storage (OSC) at the cerium oxide surface, reaching 357 μmol O2/g [12], was proved to improve the yield of CO and H2 in gasification of cellulose [14], the activity of three-way catalysts [11, 15] as well as enhance coke-resistant of DRM catalysts [16]. Studies have shown that the affection of the support could be attributed to direct activation of CH4 or CO2 by metal oxides and differences in particle size. As a support, CeO2 can provide strong metal-support interaction, leading to the high dispersion of active metal onto CeO2 [17]. Meanwhile, the controllable and uniform pore size distribution, high surface area and large pore volume of Santa Barbara Amorphous 15 (SBA-15) facilitate improving the nickel catalyst dispersion for CO2 reforming of CH4 [18, 19].
Recently, hexagonal boron nitride (h-BN) is considered as a promising nanomaterial in the heterogeneous catalysis due to the superior chemical and thermal stability [20]. The boron nitride defect-confined Ni catalysts (Ni/d-BN) [21] and Ni nanoparticles embedded on vacancy defects of hexagonal boron nitride nanosheets (Ni/h-BNNS) [22] exhibited very high catalytic activity, excellent stability and coking-resistance for DRM. It was found that the defect sites of BN play a key role in adsorption and activation of reaction gases in the first case and the synergistic effect of abundant surface defects and nano-sized Ni species significantly improve the conversion of CH4 and CO2 in the second case. Meanwhile, a combined effect of both h-BN interface and nano-sized Ni species result in the excellent catalytic stability and coke-resistance of h-BN supported mesoSiO2-confined Ni catalysts Ni/BN@mSiO2 in DRM [23].
Ni catalysts confined between boron nitride (BN)-nanoceria (NC) interfaces have been also demonstrated as efficient and stable DRM catalysts, which exhibit high activity and high resistance towards carbon deposition [24]. The stronger interaction between nickel and BN-nanoceria interfaces led to higher concentration of Ce3+ species, which promotes adsorption and activation of CO2 that facilitates the fast formation of –OH species. The active –OH species prevent coke formation and improve the stability of Ni catalysts. In addition, an efficient and stable boron nitride interface-confined and layered double hydroxides (LDHs)-derived Ni catalysts (NiMA-BN-M-R) was also developed for DRM [25]. The confinement derived from the interface between h-BN and LDHs-derived (Ni, Mg)Al2O4-sheets were responsible for well-dispersed Ni nanoparticles and anti-sintering of Ni nanoparticles during DRM reaction were demonstrated.
To widely apply this process in simulation and industry, design reactors and find the optimal conditions for the process, the mechanism and kinetics of the process was extremely important [26]. Although kinetics and mechanism of dry reforming of CH4 have been studied for many years, it is still highly controversial [27,28,29]. Different mechanisms for DRM reactions are proposed depending on the used catalysts and reaction conditions [30, 31]. Correspondingly, the various kinetic models, including the Power Law [32], Eley–Rideal [33], and Langmuir–Hinshelwood models [34], have been published.
The simple power-law rate Eq. (1) was reported in numerous publications [35,36,37]:
Here r is reaction rate; k is the reaction rate constant; n and m are the orders of corresponding reactants.
The rate of the CO2–CH4 reforming is first-order in CH4 and zero-order in CO2 was found on Ni/MgO [35] and Rh/NaY catalysts [36]. Further, the kinetically relevant step of the C–H bond activation and fast steps of hydrogen desorption to form H2 and CO2 reactions with CH4-derived chemisorbed species to form CO was suggested [35]. Meanwhile, on Rh/Al2O3 catalyst both CH4 and CO2 limit overall rates of DRM reaction [38] and first-order rate dependencies on CO2 and CH4 were proposed [37]. Zheng et al. [39] reported that the reaction order of CH4 (n) is in the range of 0.419–0.479, lower than that of CO2 (m) ranging within 0.523–0.734 for the reaction rate of CH4, while the reaction order of CO2 in the range of 0.434–0.567, lower than that of CH4 for rate of CO2 conversion (Eq. 1) for the DRM on silica-coated LaNiO3 nanoparticles. The Power Law model was used quite universally to calculate the kinetics for dry reforming. The main advantage of this model is the simplicity of applying and estimating parameters as the reaction order. However, it cannot fully explain the various steps in the reaction mechanism that occur on the catalyst surface [40].
The Eley–Rideal model is also used to describe DRM reaction [41]. According to this mechanism, the reaction takes place between the substances adsorbed on the surface of catalyst with the other in the gas phase to produce the product. The following kinetic equations for DRM on the Ir/Al2O3 catalyst was established based on Eley-Rideal model [41]:
Eley − Rideal I (ER I):
and Eley − Rideal II (ER II):
Here \({K}_{C{H}_{4}}\) and \({K}_{C{O}_{2}}\) are the adsorption equilibrium constant of CH4 or CO2, respectively; Pi is the partial pressure of corresponding substance, and Keq is equilibrium constant of the reaction at certain temperature and pressure.
Langmuir–Hinshelwood (LH) model received much attention from scientists [42] due to the concordance between mechanisms and experimental results. For this model, both reactants were firstly adsorbed on the catalyst surface before proceeding the reaction between adsorbed species to form product. Based on kinetic research and isotopic measurements [27, 29, 43, 44], a common sequence of elementary steps for reactions of CO2–CH4 reforming on different catalysts was proposed, from which different kinetics equation was offered.
It has been shown in numerous publications that there are two main models under the Langmuir–Hinshelwood mechanism: single [34] and dual-site model [45]. For the single-site model, the following general kinetic equation is often used to express the reaction rate on different catalysts [31, 41, 46,47,48]:
Here α is surface coverage, the other symbols in Eq. 4 having the same meaning as in Eqs. 1, 2, and 3. In this equation the second factor of the right part of equation takes into account the inverse reaction.
This kinetic equation described the reaction between CH4 and CO2 both chemisorbed to the one kind of surface site. Different values of exponents create different LH models. The LH1 model of molecular adsorption of both CH4 and CO2 on the singe site with bimolecular surface reaction (n = m = 1) is reported in most of the researches [30, 31, 41, 46, 48, 49]. Meanwhile, LH2 (n = 1 and m = 0.5), the associate adsorption of CH4 and dissociative adsorption of CO2 and LH3 (n = 0.5 and m = 1), the dissociative adsorption of CH4 and associate adsorption of CO2 with bimolecular surface reaction models, were proposed for DRM on lanthania supported cobalt catalyst [46]. The LH4 model described the reaction, in which both materials is involved in reaction in dissociated adsorption state (n = m = 0.5) was considered in the studies [46, 47]. In addition, the reaction took place in the high surface coverage (α = 1) in LH1, LH2, LH4 models, while in the medium surface coverage in LH3 model (α = 0.5). On Ni/TiO2, Ni/MgO, Ni/SiO2 [50], and Ni/La2O3 catalysts [51], the reaction rate is expressed by Eqs. 5 and 6, in which reaction products (H2 and CO) exhibit reaction suppression.
Nevertheless, other study [52] proposed a dual-site Langmuir–Hinshelwood mechanism, corresponding to the following equation:
From this general equation different LH models were drawn. The dual site associate adsorption of CH4 and CO2 with bimolecular surface reaction (n = m = 1) was proposed by Pichas [53] for DRM on Ni/γ–Al2O3 and perovskite-type oxides. Dual site dissociative adsorption of CH4 and associate adsorption of CO2 (n = 0.5 and m = 1) [54] on bimetallic Co–Ni/Al2O3 catalysts and dissociative adsorption both of CH4 and CO2 (n = 0.5 and m = 0.5) with bimolecular surface reaction [47] on the catalysts based on Ni–Co/Al2O3 was considered. Other study [52] indicated that over a Ni/La/Al2O3 catalyst the CH4 consumption rate was inhibited by product CO and the kinetic equation has form:
Furthermore, considering DRM as a reversible reaction Olsbye et al. [52] has expressed the reaction rate in the form of Eq. 9:
In the references, the different opinions in intermediates and rate-determining step (RDS) for the dry methane reforming reaction have been published. Some authors [27, 55] suggest that the intermediate compound is CHxO formed via quasi-equilibrated reaction of CHx with O, which are originated from reversible dissociation of CH4 and dissociative adsorption of CO2. Further, the interaction of CHx with surface oxygen to form CHxO is fast, and the absence of CHx species on the catalyst surfaces was reported [50, 56]. Finally, CHxO dissociates to form adsorbed CO and H, which then desorb to form CO and H2. In this case, the CH4 activation and CHxO decomposition as kinetically relevant steps were suggested and the kinetics of the reaction was described by Eq. 5.
In other studies [29, 57, 58], the decomposition of CH4 to chemisorbed carbon Cads in a series of elementary H-abstraction steps was accepted. Because activation energy for the first H-abstraction step in CH4 on Ni clusters is much higher than that for CH2 formation from CH3 [59], the CHx,ads coverages are low and Cads is the most abundant carbon-containing reactive. In the next step the resulting chemisorbed carbon is removed by oxidation using CO2 dissociatively adsorbed on the catalyst surface as CO and O. Based on the dependence of the reaction rate on the partial pressure of CH4 and CO2 and the kinetic isotope effects, Junmei Wei et al. [35] asserted that the exclusive kinetic relevance of C–H bond activation and the absence of any species derived from CO2 in rate-limiting steps during DRM reaction.
However, there are several studies reporting the role of CO2 activation in the kinetics of DRM reaction. On Ni/La2O3 and Ni/SiO2 [60,61,62] as well as noble metals catalysts [55], the significant involvement of CO2 activation and of chemisorbed oxygen in kinetically relevant steps are recognized. Based on the thermodynamic isotope effects, CO2 activation or reaction of chemisorbed carbon with oxygen [60] or CO2 dissociation [63] or its reaction with adsorbed CHx species [64] was proposed to limit CO2-reforming rates. However, Junmei Wei et al. [35] confirmed the quasi-equilibrated nature of CO2 activation and H2 formation.
The single rate-determining step (RDS) in the dry reforming was accepted in some investigations. The CH4 decomposition over Ni/MgO catalyst [35], the decomposition of CHxO (x = 1–2) into CO gas and adsorbed H species on the KNi/Al2O3 [65], or the reaction between the carbon species originated from CH4 dissociation and the oxygen atoms resulting from CO2 decomposition to produce CO gas over Ni/Al2O3 [66], Ni/SiO2 [60], and KNiCa [61] catalysts were considered as single RDS in DRM.
The other opinion in DRM reaction is two RDS involving in the reforming reaction. Both CH4 dissociation and CHxO decomposition were the RDS in the reforming reaction over the supported Ni catalysts suggested in several publications [50, 56, 67]. The CH4 dissociation and the reaction of surface carbon species with La2O2CO3 were RDSs in the reforming reaction over Ni/La2O3 catalyst was also concluded [51].
As can be found from the literature review, although there are various mechanisms and kinetic equations proposed for the DRM reaction, however, CH4 dissociative adsorption via sequential elementary H-abstraction steps, and its surface chemical reaction with CO2 molecular adsorption [55] or O originated from CO2 dissociatively adsorbed on the catalyst surface [27, 29] as the rate-determining step was commonly accepted over different catalysts. By density-functional theory, Burghgraef et al. [59] had determined that the activation energy for the first H-abstraction step in CH4 molecular on Ni clusters is 142 kJ/mol and reduces to 25–40 kJ/mol for CH2 formation from CH3. So, the first H-abstraction is the kinetically relevant step. Junmei Wei and Enrique Iglesia [35] also reported that only the rate constant for the activation of the first C–H bond in CH4 appears in the rate expression.
Recently, for kinetic modelling the Langmuir Hinshelwood Hougen Watson (LHHW) theory have been used [68, 69]. For example, the kinetic modelling of methanol to olefin (MTO) over SAPO-34 nanocatalyst [68], butane catalytic cracking over La/HZSM-5 [69], biogas dry reforming over a platinum–rhodium alumina catalyst [70], and Fischer–Tropsch synthesis [71, 72] was performed using LHHW theory. According to the LHHW theory, in reactions over heterogeneous catalyst, the reactive molecules are absorbed onto the active sites of catalyst and the reactive molecules are formed. When this weak bond is broken, the reaction products leave the active sites mechanism [68, 69]. In the LHHW model the substance adsorption step or the surface reaction of the adsorbed species was accepted as the reaction determined step (RDS). For the catalytic process of biogas dry reforming, the reaction takes place in the 5 following stages: (1) CH4 adsorption; (2) CO2 adsorption; (3) the surface reaction of adsorbed species; (4) desorption of CO and (5) desorption of H2 [70]. In this mechanism system, step 3 was considered as RDS [70].
Generally, there have been many studies on kinetics and mechanism of DRM reaction, but no agreement on the kinetic model of this reaction has been released, shows that the reaction kinetics strongly depends on the catalyst used. Furthermore, we have not found previous studies on the kinetics of methane dry reforming over NiO catalyst supported on CeO2 or SBA-15. The purpose of this study is to investigate properties and the activity of Nickel catalysts supported on different supports (MgO-modified Al2O3, CeO2, and SBA-15) as well as kinetics of DRM reaction over given catalysts. On these results, the effect of various supports would be clarified.
Experimental
Preparation of the catalysts
α-Al2O3 obtained by calcination of γ-Al2O3 (≥ 99.9%, Merck) at 1200 °C for 3 h. CeO2 nanorods were obtained by following procedure outlined before [73]. SBA-15 was prepared by hydrothermal method, described in detailed in our previous paper [74]. The NiO catalysts supported on CeO2 and SBA-15 were prepared by impregnation method from Ni(NO3)2⋅6H2O (Prolabo, > 99%) precursor. The Ni content in the catalysts was 7.8 and 31.2 wt% on CeO2 nanorod and SBA-15 supports, based on the result of previous investigations [73, 74]. After drying, samples were calcined in air at 800 °C for 0.5 and 2 h. The two catalysts prepared are symbolized as Ni/Ce and Ni/SBA. Thirdly, the catalyst containing 5.2 wt% Ni; 12.0 wt% Mg on α-Al2O3 was prepared by co-impregnating Ni(NO3)2⋅6H2O and Mg(NO3)2⋅6H2O (≥ 99%, Xilong) solution on α-Al2O3 according to the procedure described in [75]. The resulted suspension was then stirred regularly in 1 h at 80 °C before overnight aging and drying in air at 80 °C, 100 °C and 120 °C within 2 h at each temperature. Finally, the sample was calcined in air at 900 °C for 3 h and denoted as NiMg/Al.
Characterization of the catalysts
The crystalline structure of prepared catalysts was investigated by X–ray diffraction using Bruker D2 Phaser powder diffractometer with Cu Kα radiation (λ = 0.15406 nm). The specific surface area of the catalysts was measured by BET isothermal adsorption of nitrogen at − 196 °C (Nova Station B, Quantachrome NovaWin Instrument). The Raman spectra were obtained at room temperature with a laser Raman spectrometer (Invia, Renishaw, UK). The reducibility of catalysts was characterized by Temperature-Programmed Reduction (H2-TPR) and the basicity of catalysts activating at 450 °C for 1 h was evaluated by Carbon dioxide Temperature-Programmed Desorption (CO2-TPD) measurements, both using a Gas Chromatograph GOW-MAC 69-350 with a Thermal Conductivity Detector (TCD). The size of metal particle dispersed on support was characterised by scanning electron microscope on FE–SEM JEOL 7401 instrument and transmission electron microscopy (TEM) using TEM-JEOL 1400 instrument. The amount of coke deposited on the catalysts working at 700 °C during 30 h was determined by temperature programmed oxidation (TPO) technique [73].
Investigation of the catalyst activity and the reaction kinetics
The activity for DRM of the prepared catalysts was tested in a micro-flow reactor under atmospheric pressure at 600–800 °C, feed flow velocity of 6 L h−1, the mol ratio of CH4:CO2 in feed of 3:3 and the loading mass of catalyst sample 0.2 g. Before conducting the reaction, catalyst was reduced in-situ in 40% H2/N2 gas mixture flow (3 L h−1) for 2 h at 800 °C. The reaction mixture was analysed on the Agilent 6890 Plus Gas Chromatograph (HP-USA) using both TCD detector (capillary column TG-BON Q) and FID detector (capillary column DB-624).
The kinetics of DRM was studied in a gradientless flow-circulating system [76] using the circulating pump with a flow rate of 150 L h−1, which is much higher than feedstock flow velocity (varying from 6 L h−1 to 36 L h−1). The kinetic investigation was conducted at atmospheric pressure and 600–800 °C. The ranges of initial partial pressures of CH4, CO2, CO, and H2 were 15–30, 15–30, 0–15, and 0–15 hPa. Under these conditions, the conversion of CH4 (\({X}_{{CH}_{4}}\)) was varied in the range of 0.25 to 0.95.
The conversion (X) of CH4 and CO2, selectivity (S) of H2 and CO are defined as follows [39]:
Here: \({h}_{i,in}\) and \({h}_{i,out}\) are the moles number of substance i at the inlet and out of the reactor, respectively.
The reaction rate in the flow system in gas phase is calculated by the Temkin’s formula [77]:
Here \({P}_{{CH}_{4}}^{o}\) is the initial partial pressure (hPa) of methane in the inlet gas mixture; \({X}_{{CH}_{4}}\) is the methane conversion; g is the catalyst loading mass (gram); and v is the total flow velocity of reaction gas mixture (L.h−1).
Results and discussion
Characterization of the catalysts
The low-angle XRD pattern for bare SBA-15 support (Fig. S1) exhibited three intensive main diffraction peaks at 2θ of 0.90°, 1.60 and 1.84°, indexed as the (100), (110) and (200) reflections, indicating the ordered hexagonal meso-structure SBA-15 was successfully synthesized [78, 79]. The low-angle diffraction pattern of the NiO/SBA-15 catalysts (Fig. S1) exhibits a broader (100) peak shifts toward a larger angle to around 1.1°, with low intensity. The (110) and (200) peaks completely disappeared. This is explained by the presence of NiO particles out and into mesoporous structure of SBA-15 channels [80], as evidenced in the TEM image (Fig. 3c).
The PXRD patterns of 5.2%Ni/Al2O3 (Ni/Al), NiMg/Al, Ni/Ce and Ni/SBA samples (Fig. 1) were compared with the reported standard JCPDS data for NiO (JCPDS cards No.71-1179). It could be seen that diffraction peaks characterizing NiO phase appear strongly on Ni/SBA sample at 2θ = 37.2°, 43.3°, 62.9°, 75.4°, and 79.6° corresponding to (101), (200), (220), (311), and (222) plans with high intensities [18]. Meanwhile, on Ni/Al, NiMg/Al and Ni/Ce catalysts, these characteristic peaks are very weak (Ni/Al, NiMg/Al) or absent (Ni/Ce). This indicates that NiO exists in the crystals of 18.3 nm on the first catalyst and in the highly dispersed or amorphous phase on the two rest catalysts. In addition, on the XRD patterns of NiMg/Al the weak peaks characterizing MgAl2O4 spinel appeared at 2θ = 19.1°; 31.5° and 65.4° (JCPDS cards No.77-1193) [81] were also observed, meanwhile, on Ni/Al samples, the very weak peaks characteristic for spinel NiAl2O4 at 2θ = 18.9° and 44.39° (JCPDS Card no. 73-0239) [82] appeared.

For supports, in contrast, α-Al2O3 and CeO2 exist in crystalline state, which are characterized by strong diffraction peaks at 2θ = 25.5°, 35.1°, 37.8°, 43.3°, 52.5°, 57.5°, 61.3°, 66.5°, 68.2°, 76.8° (JCPDS cards No.82-1399) [83] and 2θ = 28.6°, 33.1°, 47.5°, 56.4°, 59.2°, 69.4°, 76.7°, 79.2° (JCPDS cards No.34-394) [84, 85]. While the broad diffraction peak observed at about 2θ = 20–30° is attributed to amorphous SiO2 (ICDD PDF No. 00-29-0085), frameworks of SBA-15 [86, 87].
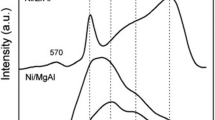
Raman spectroscopy was used to further validate the phase purity of the synthesized samples. Fig. 2 shows that on all samples the peaks appear in the range of 500–650 cm−1 were attributed to the presence of the NiO phase due to the oscillation of the Ni–O bond in line [88]. It shows that no other peaks were detected on the Ni/SBA sample, indicating the amorphous properties of SBA-15 material [18]. On Ni/Ce sample, a main peak at 465 cm−1 was observed which corresponds to the first order F2g mode of the cubic fluorite structure of CeO2 [89]. There is the additional bands around 625 and 1170 cm−1 attributed to defect-induced (D) [90], being induced by the oxygen vacancy originated from reducing Ce4+ to Ce3+ [91, 92] and longitudinal optical (2LO). The concentration of oxygen vacancies of CeO2 catalysts were estimated by the ratio of intensity of these peaks (I(625+1170)/I465) from Fig. 2 [93], to be 0.28.

Raman spectra of the catalysts
From the SEM image in Fig. S2, it can be asserted that NiMg/Al catalyst exists in the form of a solid bar, size (60–100 nm) × 300 nm, Ni/Ce is in the form of small, long rods, size (10–20 nm) × (40–100 nm), while Ni/SBA is in large cocoon shape particles, size (70–150 nm) × (150–700 nm). Compared with Ni/Al sample, the modified NiMg /Al catalyst showed smaller bar size. Further, on SBA-15 cocoons, there are 50–100 nm particles attached to the surface. The rod shape of CeO2 in Ni/Ce catalyst and the hexagonal mesostructured along channels of SBA-15 in Ni/SBA was also observed in their TEM images (Fig. 3b, c).

TEM images of the catalysts: a NiMg/Al, b Ni/Ce, and c Ni/SBA
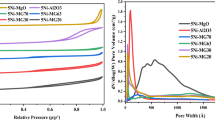
The pore diameter of the catalysts, determined from nitrogen adsorption isotherms, is approximately 2 nm for Ni/Ce, Ni/Al and NiMg/Al and 6 nm for Ni/SBA catalysts, being favourable for diffusion of CH4 and CO2 into the pores, as their kinetic diameter are 0.38 and 0.44 nm. The TEM image (Fig. 3c) indicated that Ni/SBA sample is a highly porous material. However, the mean average crystallite size for NiO was superior to the mean pore diameter for SBA-15 support (18.3 nm vs 6.08 nm), thus, a large number of aggregated NiO blocks can be seen on the outer part of the support.
The block of the parts of pores of SBA-15 and the cover of surface and pores of the supports by NiO particles as well as the agglomeration of catalyst particles, as observed from SEM and TEM images, were responsible for a sharp reduction of the specific surface area (SBET) of catalyst as compared to corresponding supports. Specifically, the value of SBET reduced from 18 to 7.9 m2 g−1 and 7.1 m2 g−1 for the Ni/Al and NiMg/Al, from 103.2 to 46.8 m2 g−1 for Ni/Ce, and from 630 to 232.6 m2 g−1 for Ni/SBA catalysts. As it follows from Table 1, the value of the specific surface area and pore volume, as well as optimal Ni content of three catalysts are in the same decreasing order as Ni/SBA≫Ni/Ce > NiMg/Al ≈ Ni/Al.
On the H2-TPR diagram of the Ni/Al catalyst (Fig. 4) three reduction peaks with the maximum reduction temperatures at 425 °C, 504 °C and 820 °C were observed. The two low-temperature reduction peaks are characteristic for the reduction of small and large NiO particle [94], while the reduction peak at 820 °C is attributed to the reduction of strong interaction NiO-support species or NiAl2O4 [95]. H2-TPR pattern of NiMg/Al catalyst showed the main reduction peak with maxima at about 875 °C, corresponded to reduction of Ni2+ in the mixed metal oxide phase (MgxNi1−xO) [96] and a very weak peak at 336 °C, assigning to the reduction of the relatively free NiO species. In H2-TPR pattern of Ni/Ce, the broad H2 consumption peak consists of three reducing peaks. The peak with maxima at 325 °C belongs to the reduction of NiO to metallic Ni [97, 98]. The main H2 consumption peaks concentring at 360 °C, which represented the reduction of Ni from the mixed Ni–Ce hydroxides [97, 98]. While the peak at 380 °C is characterized the strongly interactive NiO species with CeO2 reduction [99]. In addition, the broad weak peak at around 820 °C was attributed to the reduction of lattice oxygen in bulk CeO2 (elimination of O2 from the lattice and formation of Ce2O3) [100] was also seen in the H2-TPR pattern. Further, very weak peak at 250 °C, attributed to the reduction of oxygen adsorbed on the vacancies of the catalysts, which originated from the incorporation of Ni2+ ions into the ceria lattice [101] was also observed.

Meanwhile, Ni/SBA catalyst presented two main reduction zones: the first one appearing at temperature ranging 300–500 °C with higher intensity and the second one at Tmax ~ 630 °C. According to S.M. Sidik et al. [102], bulky NiO and Ni2O3 species tended to reduce at temperature lower than 400 °C, whereas NiO interacted with support was reduced at the temperature > 400 °C. The weak interaction NiO-support species can be reduced at temperature range of 400–500 °C, while the medium NiO-support strength species are reduced at temperature zone 500–600 °C [103], and the strong NiO-support interaction sites are reduced at temperature > 600 °C [104]. Then, the strongest peak concentred at 370 °C on H2-TPR pattern of Ni/SBA catalyst was ascribed to the reduction of bulky NiO. The reduction peak at Tmax = 455 °C was representing the reduction of the weak NiO-support strength species, while the peak at 630 °C likely corresponded to the reduction of NiO dispersed deeply in pores of support, as seen on TEM image, or reduction of strong NiO-support strength species [104]. These types of NiO should comprise portions of the precursors of the NiO species which acted as the active component during the reaction [105, 106]. In addition, the amount of hydrogen consumed in the reduction process of Ni/SBA catalyst has estimated approximately 0.814 mmol g−1, 3.0 and 6.5 times of Ni/Ce and NiMg/Al, as seen in Table 1. Relatively low reducibility of NiMg/Al sample is likely related to the fact that on this catalyst, nickel exists largely in the form of the hardly reducible mixed metal oxide phase MgxNi1−xO, which is not completely reduced even at 900 °C, as indicated on Fig. 4. Meanwhile, on Ni/Ce and Ni/SBA catalysts nickel exists in the form of easily reducible NiO particles, as observed in TEM image (Fig. 3b, c), and H2-TPR pattern (Fig. 4), having higher reducibility. The high reducibility of Ni/Ce and Ni/SBA samples is probably due to a high dispersion of small size NiO particles inside the rod/pore [107, 108], as seen from TEM images (Fig. 3). The good dispersion of NiO particles in the Ni/Ce sample is explained by the strong interaction of Ni2+ with the CeO2 support forming Ce3+ ions and oxygen vacancies that disperse NiO particles better on the catalyst surface [109]. Meanwhile, the high dispersion of inside NiO particles on Ni/SBA sample was supported by the high specific surface area and well orderly channel structure with large pores of SBA-15, facilitating diffusion and dispersion of NiO inside the pores.
CO2-TPD patterns of the three activated samples (Fig. S3) reflected that there was one main broad desorption zone stretching from 60 to 250 °C. The peak maxima at 130–150 °C corresponding to weak CO2 adsorption on OH groups [101]. On the CO2-TPD pattern of the two catalysts Ni/Ce and Ni/SBA also observed weak desorption peak at ca 250 °C attributing to moderate CO2 adsorption on metal–oxygen pairs [92]. The peak appeared at ca 520 °C in Ni/SBA catalysts was ascribed to strong CO2 adsorption on O2−anions [101]. The appearance of strong base sites in NiO/SBA-15 catalysts has also been observed in previous work [110]. The order in the desorbed CO2 amount obtained from the CO2–TPD results decreased in following order: Ni/Ce>Ni/SBA>NiMg/Al (Table 1).
The exceptionally high basicity of the Ni/Ce sample, as seen in Table 1, was explained as follows, the oxygen vacancies on the surface of Ni/Ce catalyst as demonstrated in Raman spectroscopy above, promoted the reduction of Ce4+ ions to Ce3+ ions to make the system charge neutral [111], which is expressed as reducing peak at 820 °C on H2-TPR diagrams (Fig. 4). On the reduced Ce3+ ions CO2 adsorbed to form carbonate − \(\text{{CO}}_{2}^{2-}\)species, which have a higher thermal stability than those on the Ce4+ sites [112,113,114]. It resulted in increasing of the basic sites that enhance the adsorption of acid gas CO2. In the CeO2 crystal, oxygen has good migration ability and the electron delocalization formed by the oxygen vacancy that can increase the electron density in the nano-CeO2 structure [115, 116] and the strength and the amount of the basic sites.
As is well known, the high basicity of catalysts stimulates the adsorption and dissociation of CO2, which in turn reduces the carbon deposition over the surface of the catalyst and therefore, suppressing the catalyst deactivation [117, 118]. In DRM reaction, CO2 on the one hand participates in main reaction (15), on the other hand oxidizes deposited coke in the reverse Boudouard reaction (16) that improved the coke resistance and stability of nickel catalysts.
The high basicity of Ni/SBA and Ni/Ce samples (Table 1) should create their superior coke resistance.
Catalytic performance for DRM of the catalysts
The results showed that the conversion of both CH4 and CO2 increased as reaction temperature is raised from 600 to 750 °C because DRM (15) is a strongly endothermic reaction. It is noticeable that the order in the activity, based on CH4 and CO2 conversions, decreased in the following order: Ni/SBA>NiMg/Al>Ni/Ce (Fig. S4). The conversion of CH4 and CO2 on Ni/SBA catalysts reached 88% and 76% at 650 °C. Meanwhile, on catalysts Ni/Ce and NiMg/Al, in the same condition reached the conversion of CH4 and CO2 75% and 59%, and 84% and 76%. The best activity of Ni/SBA catalyst could be explained by its higher surface area for dispersing a bigger number of active sites and good reducibility, as seen in Table 1. The results of studying the activity of three catalysts in the temperature range of 600–800 °C showed that, the CO2 conversion in all cases is lower than that of CH4. This can be explained by the occurrence of Water Gas Shift (WGS) (17) in DRM conditions, producing additional CO2 [119].
Although Ni/Ce catalyst had much higher basicity than NiMg/Al and Ni/SBA samples, CO2 conversion of Ni/Ce was lower than that of the remaining two catalysts. This is likely related to the fact that excessive basicity stimulated higher extent of the CO2 dissociation (CO2 → C + O2) to be happened and the phenomenon worsens when Boudouard reaction start to happen at high temperature due to the enriched composition of CO upon DRM activity, thus resulted in higher quantity of coke deposited on the catalyst surface and reduce CO2 conversion [120]. Indeed, the value of H2/CO ratio on Ni/Ce catalyst reached 1.33 while this value on the two others remained at a theoretical level, approximately 1. In general, excessive basicity of the catalyst is not favourable over DRM [120, 121]. Thus, it was deduced that Ni/Ce was less favourable for DRM activity due to its excessive basicity.
In DRM reaction conditions, in addition to the main reaction (15), several side-reactions may be involved in the process, including the Water Gas Shift (17) consumes part of the product CO, methane decomposition (18), the Boudouard reaction and the carbon gasification reverse reaction (19) generate or gasify coke [122].
The thermodynamic analysis showed that in the condition of DRM reaction the reverse water gas shift reaction operates in or very close to thermodynamic equilibrium [55], and its influence on the DRM can be neglected. Thermodynamic studies also show that other side reactions of DRM such as CO disproportionation, CO/H2 reduction (19), CO/H2 methanation, CO2/H2 methanation also could be neglected above 550 °C. Furthermore, except unconverted methane, in the gas chromatographic spectrum of the gas reaction mixture other hydrocarbons, originating from side-reaction (20), were not detected.
Therefore, the only by-product, which can appear in the kinetic equation of the DRM reaction, is coke generated from Eq. (18) and from the H-abstraction of CH4 [59]. However, at high temperatures, this deposited coke could be consumed in the reverse Boudouard reaction (Eq. 16), the reaction favourable at high temperatures [123]. Then, the amount of coke formed in the reaction should be small and could be ignored.
Conducting the reaction at 700 °C for 30 h, showed that all catalysts had stable activity (expressed through CH4 and CO2 conversion) (Fig. S5). The coke formation on the catalysts, determined by TPO method, was 5.25, 0.70 and 0.63 \({mg}_{C} {g}_{cat}^{-1}\) on NiMg/Al, Ni/Ce and Ni/SBA catalysts. In the same condition, the amount of coke deposited on the Ni/Al catalyst was determined to be 37.52 mgC g−1. Despite high coke depodition, the Ni/Al catalyst remained stable for 30 h TOS. The amount of carbon (C) involved in coke formation, calculated based on the above results, accounts for 0.035; 0.005; 0.001 and 0.001% and the amount of coke was 3.5; 0.5; 0.1 and 0.1% of catalyst weight to Ni/Al, NiMg/Al, Ni/Ce and Ni/SBA catalysts. Obviously, on NiMg/Al, Ni/Ce and Ni/SBA catalysts, the coke deposited (in %) is very small compared to the catalyst weight, and the activity of the catalysts is stable during the reaction process. So, the obtained results indicated that the amount of coke formed on NiMg/Al, Ni/Ce, and Ni/SBA catalysts is negligible and does not affect reaction kinetics.
Adding MgO to Ni/Al catalyst results in a sevenfold reduction in the coke deposition. This result is due to the fact that MgO is a typical base oxide, its addition generates the base sites, enhances CO2 adsorption [2], that promoted the reverse Boudouard reaction consumed coke [122]. Another reason contributing to the reduction of coke on the MgO-promoted catalyst is the presence of MgO-NiO layers on the catalyst surface [124], as seen in TEM image.
The NiO supported on CeO2 and SBA-15 (Ni/Ce and Ni/SBA) catalysts have lower coke deposition after 30 h TOS as compared to Ni/Al. This may be explained by the fact that CeO2 acts as a storage and suply of oxygen for coke oxidation. The interaction of Ni2+ with the CeO2 support forming Ce3+ ions in the form of Ce2O3 and oxygen vacancies. On Ce2O3, CO2 adsorbs and dissociates to form CO and CeO2, the produced CeO2 reacts with the coke to regenerate Ce2O3 and CO2 [109, 125]. In addition, small-sized NiO crystals also distributed to lower coke deposition of Ni/Ce catalyst. CeOx doping induces strong metal-support interactions, that stabilize Ni single atoms towards sintering, and favour selective activation of only the first C–H bond in methane, resulting in a high activity and stability with negligible carbon deposition was found in Ni/hydroxyapatite (HAP) catalyst [16]. As seen from the XRD analysis results (Fig. 1), NiO in the Ni/Ce catalyst exists in highly dispersed or amorphous form, that significantly reduced in coke deposition. The existence of numerous NiO particles of less than 6 nm insize in the channels of SBA-15, as observed in the TEM image, also contributed to the reduction of coke formation on the Ni/SBA catalyst. It has been shown in several studies that on Ni nanoparticle size below 2 nm [126] or 7–10 nm [62] the carbon deposition significantly decreases. Another reason for high coke resistance of two NiO catalysts carried on CeO2 and SBA-15 is their high basicity (Table 1), that enhanced CO2 adsorption, providing O adatoms for the oxidation of the resulting carbon [57, 58, 127]. However, as analyzed above, the excessive basicity of Ni/Ce to some extent is unfavorable to its coke resistance. Therefore, despite its high coke oxidation capacity in nature, the amount of coke deposited on Ni/Ce is not lower than on Ni/SBA.
To ensure the catalytic activity was the same in all experiments, the standard reaction was repeated after each experiment. Once the activity has changed, the catalyst was regenerated by burning the coke at 600 °C with a stream of air to remove the formed coke before in-situ reduction. Thus, all the experimental data can be used in kinetic calculations.
The kinetics of methane dry reforming
The influence of internal diffusion was examined by comparing the reaction rate of DRM on Ni/SBA catalyst beads of different sizes, d = 0.01–0.25; 0.25–0.50; 0.50–0.75; 0.75–1.00 mm at a constant reaction condition. The results exhibited that the reaction rate remained almost unchanged when the catalyst bead size was varied from 0.01–0.25 to 0.50–0.75 mm. Thus, there is no effect of internal diffusion when the bead size of the catalyst is less than 0.75 mm.
The influence of external diffusion was examined by correlating CH4 conversion on Ni/SBA catalyst with different total gas flows, v = 6, 9, 12, 18, 30 and 36 L h−1 at a constant reaction condition. The results showed that the CH4 conversion decreased with increasing gas flow velocity, corresponding to the reduction in dwelling time. This means external diffusion does not affect the reaction when total gas flow was changed from 6 to 36 L h−1. In the experiment, the catalyst particles of 0.25–0.5 mm and total gas flow ranging from 6 to 36 L h−1 were used to avoid the effect of diffusions.
To determine the dependence of the reaction rate on the partial pressure of feedstocks, from the kinetic data set select the reaction rate at the different partial pressure of CH4 (or CO2) at constant temperature and partial pressure of the remaining substances.
The curves expressing the dependence of the reaction rate on the partial pressure of CH4 and CO2 on three catalysts (Fig. 5) is a convex form, describing nonlinear increase of r with concentration of both feedstocks. This suggests that the partial pressure of CH4 and CO2 enter both the numerator and the denominator of the kinetic equation.

The reaction rate (r) versus the partial pressure of CH4 (\({P}_{{CH}_{4}}\)) (above) and CO2 (\({P}_{{CO}_{2}}\)) (below) at 700 °C on the catalysts
Fig. 6 showed that at a constant composition of the reaction mixture the reaction rate (r) decreased with increasing of the partial pressure of reaction products, H2 and CO. This demonstrates, the two products inhibiting the reaction. The dependence of reversed values of reaction rate (1/r) vs partial pressure of hydrogen (\({P}_{{H}_{2}}\)) and carbon monoxide (\({P}_{CO}\)) is nearly linear, indicating the quantities \({\mathrm{P}}_{{\mathrm{H}}_{2}}\) and PCO must appear in the denominator of the kinetic equation in power may be unit.

Variation of reaction rate (r) with partial pressure of H2 (\({P}_{{H}_{2}}\)) (above) and partial pressure of CO (\({\mathrm{P}}_{\mathrm{CO}}\)) (below) on the catalysts at 700 °C
From the research results, it is possible to draw the following feature of kinetics of the DRM reaction:
-
1.
The partial pressure of the feed reactants enters both the numerator and the denominator of the kinetic equation;
-
2.
The reaction is inhibited by the products of reaction and the partial pressure of hydrogen and carbon monoxide (PH2 and PCO) present only in the numerator with exponent may be 1.
The above characteristics of the kinetics of the methane DRM reaction are similar to the 5-steps LHHW mechanism was proposed for DRM of biogas [70]. In the LHHW model the adsorption of reactants or the surface reaction between the adsorbing particles can be rate-limiting steps [128]. Since DRM reaction takes place at high temperatures range:
This stage is considered to be the rate determining step (RDS). Then, the rate of reaction CO2–CH4 reforming is proposed to be written in the following kinetic equation [70]:
Here CCH4, CCO2, CCO, and CH2 are the gas phase concentration of CH4, CO2, CO, and H2, k is the rate constant for the forward direction of reaction (Eq. 21), θυ is the fraction of vacant sites, given by
The mean of CCH4, CCO2, CCO, and CH2 in Eqs. 22 and 23 are equivalent to θCH4, θCO2, θCO, and θH2—the fraction of adsorption of corresponding surfactants to the surface. Therefore, Eq. 22 can be written as
Here r is the reaction rate; k is the rate constant, \({\theta }_{{CH}_{4}}\) and \({\theta }_{{CO}_{2}}\) are the fraction of methane and carbon dioxide adsorbed to the catalyst surface, respectively, which were determined by the Langmuir formulas:
Here Ki represents the Langmuir adsorption constant of the corresponding species to the surface of the catalyst, J is the intermediates; and ξi is the dissociation coefficient of i adsorbed species. γ is the coefficient, taking into account the inverse reaction, given by (15) [52].
Here KEq is the equilibrium constant of the reversible reaction (15).
Replacing quantities \({\theta }_{C{H}_{4}}\) and \({\theta }_{C{O}_{2}}\) in Eq. (24) with expressions (25) and (26) and divide both the numerator and the denominator of the equation by \({\left({K}_{{CH}_{4}}\right)}^{{n}_{1}}{\left({K}_{{CO}_{2}}\right)}^{{n}_{2}}\) obtain the general kinetic equation for DRM:
Here \((-{r}_{{CH}_{4}}\)) is the reaction rate of methane consumption; \({\mathrm{P}}_{\mathrm{i}}\) are the partial pressures of corresponding substances; n1, n2, m1, m2, m3, m4 are reaction orders of the corresponding reactants (\({n}_{i}=\) 1/\({\xi }_{i}\,\mathrm{and}\,{m}_{i}=\) 1/\({\xi }_{i}\)); 2α is surface coverage; and k, ki, and ki’ are kinetic constants.
Here
The reaction order and the values of kinetic constants in Eq. (28) were determined by using the least-squares optimization and the solver tool in MS excel to calculate the experimental data with the conditions: n1, n2 = 0–2 (step 0.25), mi = 0–2 (step 0.25), α = 0–1 (step 0.25). The calculated results best fit the experimental data when: n1 = n2 = 1; m1 = m2 = m3 = m4 = 1, α = 0.5, A = 1 and A′ = 1.
The values of the kinetic constants have been indicated in Table 2. From the calculation results, it follows that, the rate of DRM on studied catalysts is described by the following equations:
For NiMg/Al and Ni/Ce catalysts:
For Ni/SBA catalyst:
The form of Eqs. 28, 29, and 30 proves that the dry reforming of methane follows the dual-site Langmuir–Hinshelwood-Hougen Watson model, where the surface reaction was rate-controlling step of CH4 dry reforming process and all other steps were considered at equilibrium.
The value of the root-mean-square deviations (∆) ranges from 17.1% to 18.7% on different catalysts. Furthermore, the comparison of the calculated and experimental consumption rate of CH4 for Eqs. 29 and 30 is expressed in Fig. S6 showing R2 value that reflects the amount of variance is reported as [72]:
Here
The R2 value of this model was obtained approximately 0.97–0.98 (Table 2). The obtained value of the deviations (∆) and R2 shows that the well fit of experimental data was achieved using a dual-site LHHW model.
Assuming the single-site Langmuir–Hinshelwood model for dry reforming of methane, reaction kinetics can be descried by Eq. (33).
Calculate experimental results according to Eq. 33 show that the root-mean-square deviations (∆) of calculated reaction rates from experimental values are very large (> 100%). It means this model is not suitable. Furthermore, the experimental results show that when adding 0.3% V2O5, a selective oxidation additive, the CH4 conversion on NiMg/Al catalyst at 650 °C increased 11% (from 84 to 95%), while CO2 conversion decreased from 76 to 70%. This proves that CH4 and CO2 were activated by different types of sites.
The two factors in the denominator of kinetic Eqs. (29) and (30) showed that the reaction takes place on two active sites. In many publications, adsorption and activation of CH4 and CO2 on two different sites are accepted. Specifically, CH4 was bound on the metallic phase while the CO2 was bound on the oxide phase of the catalyst [45, 51, 129]. The appearance of PCO term in the first factor in the denominator of Eq. (30) explained by CO formed and adsorbed on the metal-oxide interface before desorbing, the same as was reported in publication [45].
The value of α = 0.5 in Eq. 28, was determined from calculation, meaning that the reaction takes place in the medium coverage region. The constant k5 and k5′ in Eq. 28 on all catalysts is zero, indicating no intermediates inhibited the reaction.
From Eqs. 29 and 30, it is shown that CH4 and CO2 both participate in the reaction in the form of molecular adsorption. The presence of \({\mathrm{P}}_{{\mathrm{CH}}_{4}}\) and \({\mathrm{P}}_{{\mathrm{CO}}_{2}}\) terms in both the numerator and the denominator of the kinetic equation indicates that the adsorption of the feeds simultaneously promotes and inhibits the reaction. As is known, for heterogeneous catalytic reaction, the adsorption of raw materials on catalyst surface is a prerequisite for the reaction to take place. However, their strong adsorption may inhibit the reaction. Meanwhile, the two resulting products only inhibited the reaction, as the \({\mathrm{P}}_{{\mathrm{H}}_{2}}\) and \({\mathrm{P}}_{\mathrm{CO}}\) terms are present in the denominator of the kinetic equation, although the restraint is weak.
Supports created specificity in the physicochemical properties of nickel catalysts that greatly affect the adsorption feature and activity of NiO catalyst. The result in Table 2 indicated that the adsorption coefficients of CH4 and CO2 on the catalysts depend on the carriers used. The highest CO2 adsorption coefficient obtained on Ni/Ce catalyst is related to its highest basicity (\({m}_{{CO}_{2}}\)), while the highest adsorption constant of CH4 and H2 was observed on Ni/SBA sample, the sample with the highest number of reduced Nio (\({m}_{{Ni}^{o}}\)), as seen in Table 1. The order in the ratio of adsorption constant of CH4 and CO2 (k1/k2′) (Table 2) coincides with the order in the ratio of the quantities characterizing for catalyst reducibility and basicity (\({m}_{{Ni}^{o}}/{m}_{{CO}_{2}}\)) (Table 1), increased in the following order: Ni/Ce<NiMg/Al<Ni/SBA, the same as the order in activity in DRM (Fig. S4). This shows an intimate relationship between the adsorption affinity of the reactants and the properties of the catalysts, which directly depend on the nature of the carrier.
It has been found from Table 2 that the order in the apparent rate constant (k) of CH4 reforming coincides with the order in the reducibility (\({m}_{{Ni}^{o}}\)) of the studied catalyst: NiMg/Al<Ni/Ce<Ni/SBA and the value of the activation energy (E) is in the opposite order. The well order mesoporous structure of SBA-15 creates the outstanding physicochemical characteristics of the nickel catalyst including the high specific surface area and pore volume, leading to finely disperse large amounts of highly reducible NiO, together with the reasonable basicity of catalyst, that lowering activation energy, increasing the catalyst activity as well as reducing coke deposition. The advantage of CeO2 carrier is included in high dispersion and reducibility of NiO that reduces activation energy, increase the reaction rate constant and coke resistance as well as stability of NiO catalyst. However, the low optimal Ni content and excessive basicity, leading to overwhelming adsorption of CO2 vs CH4, that reduce catalyst activity.
Conclusion
The results show that the kinetics of the DRM reaction on Ni-based catalysts supported on different carriers is described by a common kinetic equation, following the a dual-site Langmuir Hinshelwood Hougen Watson mechanism with the surface reaction between adsorbed CH4 and CO2 was rate-controlling step. In which both CH4 and CO2 participate the reaction in form of molecules adsorbed on two active sites and the reaction is inhibited by the resulting products. However, the nature of the supports shows a strong effect on the adsorption affinity as well as activity of catalysts in the DRM reaction. On the two Ni/Ce and Ni/SBA catalysts, nickel exists in the form of highly dispersed NiO particles, leading to greatly improve the reducibility of catalyst and lower reaction activation energy, and their high basicity markedly reduces the coke deposition as compared to NiMg/Al sample. The order in the apparent activation energy (E) of reaction on the studied catalysts was observed as follows: NiMg/Al>Ni/Ce>Ni/SBA, while the the apparent rate constant (k) and the catalyst reducibility were in the opposite order. Additionally, the increasing order in catalytic activity coincides with the order in the ratio of adsorption constant of CH4 and CO2 (k1/k2′) and \({m}_{{Ni}^{o}}/{m}_{{CO}_{2}}\) ratio: Ni/Ce<NiMg/Al<Ni/SBA. The highest activity was observed on the catalyst 31.2wt%Ni/SBA-15 thank to its high surface area, high reducibility and high adsorption affinity to methane.
References
Guo J, Lou H, Zhao H, Chai D, Zheng X (2004) Dry reforming of methane over nickel catalysts supported on magnesium aluminate spinels. Appl Catal A 273(1–2):75–82
Roh H-S, Jun K-W (2008) Carbon dioxide reforming of methane over Ni catalysts supported on Al2O3 modified with La2O3, MgO, and CaO. Catal Surv Asia 12(4):239–252
Nakamura J, Aikawa K, Sato K, Uchijima T (1994) Role of support in reforming of CH4 with CO2 over Rh catalysts. Catal Lett 25(3–4):265–270
Koo KY, Roh H-S, Seo YT, Seo DJ, Yoon WL, Park SB (2008) A highly effective and stable nano-sized Ni/MgO–Al2O3 catalyst for gas to liquids (GTL) process. Int J Hydrogen Energy 33(8):2036–2043
Mazumder J, de Lasa H (2014) Fluidizable Ni/La2O3–γAl2O3 catalyst for steam gasification of a cellulosic biomass surrogate. Appl Catal B 160:67–79
Wang Y, Fang Q, Shen W, Zhu Z, Fang Y (2018) (Ni/MgAl2O4)@ SiO2 core–shell catalyst with high coke-resistance for the dry reforming of methane. React Kinet Mech Catal 125(1):127–139
Silva CK, Baston EP, Melgar LZ, Bellido JD (2019) Ni/Al2O3–La2O3 catalysts synthesized by a one-step polymerization method applied to the dry reforming of methane: effect of precursor structures of nickel, perovskite and spinel. React Kinet Mech Catal 128(1):251–269
Xu Z, Li Y, Zhang J, Chang L, Zhou R, Duan Z (2001) Ultrafine NiO–La2O3–Al2O3 aerogel: a promising catalyst for CH4/CO2 reforming. Appl Catal A 213(1):65–71
Nematollahi B, Rezaei M, Khajenoori M (2011) Combined dry reforming and partial oxidation of methane to synthesis gas on noble metal catalysts. Int J Hydrogen Energy 36(4):2969–2978
Charisiou ND, Siakavelas G, Papageridis KN, Baklavaridis A, Tzounis L, Avraam DG, Goula MA (2016) Syngas production via the biogas dry reforming reaction over nickel supported on modified with CeO2 and/or La2O3 alumina catalysts. J Nat Gas Sci Eng 31:164–183. https://doi.org/10.1016/j.jngse.2016.02.021
Fornasiero P, Dimonte R, Rao GR, Kaspar J, Meriani S, Trovarelli A, Graziani M (1995) Rh-loaded CeO2–ZrO2 solid-solutions as highly efficient oxygen exchangers: dependence of the reduction behavior and the oxygen storage capacity on the structural-properties. J Catal 151(1):168–177. https://doi.org/10.1006/jcat.1995.1019
Zhang J, Kumagai H, Yamamura K, Ohara S, Takami S, Morikawa A, Shinjoh H, Kaneko K, Adschiri T, Suda A (2011) Extra-low-temperature oxygen storage capacity of CeO2 nanocrystals with cubic facets. Nano Lett 11(2):361–364
Wu L, Wiesmann HJ, Moodenbaugh AR, Klie RF, Zhu Y, Welch DO, Suenaga M (2004) Oxidation state and lattice expansion of CeO2−x nanoparticles as a function of particle size. Phys Rev B 69(12):125415. https://doi.org/10.1103/PhysRevB.69.125415
Tomishige K, Asadullah M, Kunimori K (2003) Novel catalysts for gasification of biomass with high conversion efficiency. Catal Surv Asia 7(4):219–233
Nunan JG, Robota HJ, Cohn MJ, Bradley SA (1992) Physicochemical properties of Ce-containing three-way catalysts and the effect of Ce on catalyst activity. J Catal 133(2):309–324
Akri M, Zhao S, Li X, Zang K, Lee AF, Isaacs MA, Xi W, Gangarajula Y, Luo J, Ren Y (2019) Atomically dispersed nickel as coke-resistant active sites for methane dry reforming. Nat Commun 10(1):1–10
Ramli A, Mohamad MF, Yusup S, Hin TYY (2016) Hydrogen production from gasification of palm kernel shell in the presence of Fe/CeO2 catalysts. Malays J Anal Sci 20(2):303–308
Omoregbe O, Danh HT, Nguyen-Huy C, Setiabudi H, Abidin S, Truong QD, Vo D-VN (2017) Syngas production from methane dry reforming over Ni/SBA-15 catalyst: effect of operating parameters. Int J Hydrogen Energy 42(16):11283–11294
Zhang Q, Wang M, Zhang T, Wang Y, Tang X, Ning P (2015) A stable Ni/SBA-15 catalyst prepared by the ammonia evaporation method for dry reforming of methane. RSC Adv 5(114):94016–94024
Harley-Trochimczyk A, Pham T, Chang J, Chen E, Worsley MA, Zettl A, Mickelson W, Maboudian R (2016) Platinum nanoparticle loading of boron nitride aerogel and its use as a novel material for low-power catalytic gas sensing. Adv Func Mater 26(3):433–439
Bu K, Deng J, Zhang X, Kuboon S, Yan T, Li H, Shi L, Zhang D (2020) Promotional effects of B-terminated defective edges of Ni/boron nitride catalysts for coking-and sintering-resistant dry reforming of methane. Appl Catal B 267:118692
Cao Y, Maitarad P, Gao M, Taketsugu T, Li H, Yan T, Shi L, Zhang D (2018) Defect-induced efficient dry reforming of methane over two-dimensional Ni/h-boron nitride nanosheet catalysts. Appl Catal B 238:51–60
Cao Y, Lu M, Fang J, Shi L, Zhang D (2017) Hexagonal boron nitride supported mesoSiO2-confined Ni catalysts for dry reforming of methane. Chem Commun 53(54):7549–7552
Lu M, Zhang X, Deng J, Kuboon S, Faungnawakij K, Xiao S, Zhang D (2020) Coking-resistant dry reforming of methane over BN-nanoceria interface-confined Ni catalysts. Catal Sci Technol 10:4237–4244
Bu K, Kuboon S, Deng J, Li H, Yan T, Chen G, Shi L, Zhang D (2019) Methane dry reforming over boron nitride interface-confined and LDHs-derived Ni catalysts. Appl Catal B 252:86–97
Al-Fatesh AS, Fakeeha AH, Abasaeed AE (2011) Effects of promoters on methane dry reforming over Ni catalyst on a mixed (a-Al2O3+TiO2-P25) support. Int J Phys Sci 6(36):8083–8092
Bradford MC, Vannice MA (1999) CO2 reforming of CH4 over supported Ru catalysts. J Catal 183(1):69–75
Iyer MV, Norcio LP, Kugler EL, Dadyburjor DB (2003) Kinetic modeling for methane reforming with carbon dioxide over a mixed-metal carbide catalyst. Ind Eng Chem Res 42(12):2712–2721
Wei J, Iglesia E (2004) Structural requirements and reaction pathways in methane activation and chemical conversion catalyzed by rhodium. J Catal 225(1):116–127
Solh TE, Jarosch K, de Lasa H (2003) Catalytic dry reforming of methane in a CREC riser simulator kinetic modeling and model discrimination. Ind Eng Chem Res 42(12):2507–2515
Gokon N, Yamawaki Y, Nakazawa D, Kodama T (2011) Kinetics of methane reforming over Ru/γ-Al2O3-catalyzed metallic foam at 650–900 °C for solar receiver-absorbers. Int J Hydrogen Energy 36(1):203–215
Daza CE, Kiennemann A, Moreno S, Molina R (2009) Dry reforming of methane using Ni–Ce catalysts supported on a modified mineral clay. Appl Catal A 364(1–2):65–74
Akpan E, Sun Y, Kumar P, Ibrahim H, Aboudheir A, Idem R (2007) Kinetics, experimental and reactor modeling studies of the carbon dioxide reforming of methane (CDRM) over a new Ni/CeO2–ZrO2 catalyst in a packed bed tubular reactor. Chem Eng Sci 62(15):4012–4024
Gokon N, Osawa Y, Nakazawa D, Hatamachi T, Kodama T (2008) Kinetics of CO2 reforming of methane by catalytically activated metallic foam absorber for solar receiver-reactors. ASME 2008 2nd International Conference on Energy Sustainability collocated with the Heat Transfer, Fluids Engineering, and 3rd Energy Nanotechnology Conferences. American Society of Mechanical Engineers, Florida, pp 371–383
Wei J, Iglesia E (2004) Isotopic and kinetic assessment of the mechanism of reactions of CH4 with CO2 or H2O to form synthesis gas and carbon on nickel catalysts. J Catal 224(2):370–383
Bhat R, Sachtler W (1997) Potential of zeolite supported rhodium catalysts for the CO2 reforming of CH4. Appl Catal A 150(2):279–296
Mark MF, Maier WF (1994) Active surface carbon—a reactive Intermediate in the production of synthesis gas from methane and carbon dioxide. Angew Chem, Int Ed Engl 33(15–16):1657–1660
Rostrupnielsen J, Hansen JB (1993) CO2-reforming of methane over transition metals. J Catal 144(1):38–49
Zheng X, Tan S, Dong L, Li S, Chen H (2015) Silica-coated LaNiO3 nanoparticles for non-thermal plasma assisted dry reforming of methane: experimental and kinetic studies. Chem Eng J 265:147–156
Erdohelyi A, Cserényi J, Solymosi F (1993) Activation of CH4 and its reaction with CO2 over supported Rh catalysts. J Catal 141(1):287–299
Mark MF, Maier WF, Mark F (1997) Reaction kinetics of the CO2 reforming of methane. Chem Eng Technol Ind Chem Plant Equip Process Eng Biotechnol 20(6):361–370
Li Y, Wang Y, Zhang X, Mi Z (2008) Thermodynamic analysis of autothermal steam and CO2 reforming of methane. Int J Hydrogen Energy 33(10):2507–2514
Wei J, Iglesia E (2004) Mechanism and site requirements for activation and chemical conversion of methane on supported Pt clusters and turnover rate comparisons among noble metals. J Phys Chem B 108(13):4094–4103
Wei J, Iglesia E (2004) Reaction pathways and site requirements for the activation and chemical conversion of methane on Ru-based catalysts. J Phys Chem B 108(22):7253–7262
Verykios XE (2003) Catalytic dry reforming of natural gas for the production of chemicals and hydrogen. Int J Hydrogen Energy 28(10):1045–1063
Ginsburg JM, Piña J, El Solh T, De Lasa HI (2005) Coke formation over a nickel catalyst under methane dry reforming conditions: thermodynamic and kinetic models. Ind Eng Chem Res 44(14):4846–4854
Foo SY, Cheng CK, Nguyen T-H, Adesina AA (2011) Kinetic study of methane CO2 reforming on Co–Ni/Al2O3 and Ce–Co–Ni/Al2O3 catalysts. Catal Today 164(1):221–226
Ayodele BV, Khan MR, Lam SS, Cheng CK (2016) Production of CO-rich hydrogen from methane dry reforming over lanthania-supported cobalt catalyst: kinetic and mechanistic studies. Int J Hydrogen Energy 41(8):4603–4615
Benguerba Y, Virginie M, Dumas C, Ernst B (2017) Methane dry reforming over Ni–Co/Al2O3: kinetic modelling in a catalytic fixed-bed reactor. Int J Chem Reactor Eng 15(6):20160170
Bradford MC, Vannice MA (1996) Catalytic reforming of methane with carbon dioxide over nickel catalysts II. Reaction kinetics. Appl Catal A 142(1):97–122
Tsipouriari VA, Verykios XE (2001) Kinetic study of the catalytic reforming of methane with carbon dioxide to synthesis gas over Ni/La2O3 catalyst. Catal Today 64(1–2):83–90
Olsbye U, Wurzel T, Mleczko L (1997) Kinetic and reaction engineering studies of dry reforming of methane over a Ni/La/Al2O3 catalyst. Ind Eng Chem Res 36(12):5180–5188
Pichas C, Pomonis P, Petrakis D, Ladavos A (2010) Kinetic study of the catalytic dry reforming of CH4 with CO2 over La2−xSrxNiO4 perovskite-type oxides. Appl Catal A 386(1–2):116–123
Cheng CK, Foo SY, Adesina AA (2010) Glycerol steam reforming over bimetallic Co-Ni/Al2O3. Ind Eng Chem Res 49(21):10804–10817
Barroso Quiroga MM, Castro Luna AE (2007) Kinetic analysis of rate data for dry reforming of methane. Ind Eng Chem Res 46(16):5265–5270
Luo J, Yu Z, Ng C, Au C (2000) CO2/CH4 reforming over Ni–La2O3/5A: an investigation on carbon deposition and reaction steps. J Catal 194(2):198–210
Foppa L, Margossian T, Kim SM, Müller C, Copéret C, Larmier K, Comas-Vives A (2017) Contrasting the role of Ni/Al2O3 interfaces in water–gas shift and dry reforming of methane. J Am Chem Soc 139(47):17128–17139
Liu Z, Grinter DC, Lustemberg PG, Nguyen-Phan TD, Zhou Y, Luo S, Waluyo I, Crumlin EJ, Stacchiola DJ, Zhou J (2016) Dry reforming of methane on a highly-active Ni–CeO2 catalyst: Effects of metal-support interactions on C−H bond breaking. Angew Chem Int Ed 55(26):7455–7459
Burghgraef H, Jansen A, Van Santen R (1995) Methane activation and dehydrogenation on nickel and cobalt: a computational study. Surf Sci 324(2–3):345–356
Kroll V, Swaan H, Lacombe S, Mirodatos C (1996) Methane reforming reaction with carbon dioxide over Ni/SiO2 catalyst: II. A mechanistic study. J Catal 164(2):387–398
Chang J-S, Park S-E, Yoo JW, Park J-N (2000) Catalytic behavior of supported KNiCa catalyst and mechanistic consideration for carbon dioxide reforming of methane. J Catal 195(1):1–11
Tang S, Ji L, Lin J, Zeng H, Tan K, Li K (2000) CO2 reforming of methane to synthesis gas over sol–gel-made Ni/γ–Al2O3 catalysts from organometallic precursors. J Catal 194(2):424–430
Gamman JJ, Millar JG, Rose G, Drennan J (1998) Characterisation of SiO2-supported nickel catalysts for carbon dioxide reforming of methane. J Chem Soc Faraday Trans 94(5):701–710. https://doi.org/10.1039/A706730E
Osaki T, Horiuchi T, Suzuki K, Mori T (1997) CH4/CD4 isotope effect on the reaction of adsorbed hydrocarbon species in CO2-reforming over Ni/Al2O3 catalyst. Catal Lett 44(1–2):19–21
Osaki T, Mori T (2001) Role of potassium in carbon-free CO2 reforming of methane on K-promoted Ni/Al2O3 catalysts. J Catal 204(1):89–97
Schuurman Y, Marquez-Alvarez C, Kroll VCH, Mirodatos C (1998) Unraveling mechanistic features for the methane reforming by carbon dioxide over different metals and supports by TAP experiments. Catal Today 46:185–192
Nandini A, Pant K, Dhingra S (2006) Kinetic study of the catalytic carbon dioxide reforming of methane to synthesis gas over Ni–K/CeO2–Al2O3 catalyst. Appl Catal A 308:119–127
Azarhoosh MJ, Halladj R, Askari S (2017) Presenting a new kinetic model for methanol to light olefins reactions over a hierarchical SAPO-34 catalyst using the Langmuir–Hinshelwood–Hougen–Watson mechanism. J Phys: Condens Matter 29(42):425202
Rahimi N, Karimzadeh R (2015) Kinetic modeling of catalytic cracking of C4 alkanes over La/HZSM-5 catalysts in light olefin production. J Anal Appl Pyrol 115:242–254
Sawatmongkhon B, Theinnoi K, Wongchang T, Haoharn C, Tsolakis A (2017) Combination of Langmuir-Hinshelwood-Hougen-Watson and microkinetic approaches for simulation of biogas dry reforming over a platinum-rhodium alumina catalyst. Int J Hydrogen Energy 42(39):24697–24712
Arsalanfar M, Mirzaei A, Atashi H, Bozorgzadeh H, Vahid S, Zare A (2012) An investigation of the kinetics and mechanism of Fischer-Tropsch synthesis on Fe–Co–Mn supported catalyst. Fuel Process Technol 96:150–159
Vahid S, Mirzaei A (2014) An investigation of the kinetics and mechanism of Fischer-Tropsch synthesis on Fe–Co–Ni supported catalyst. J Ind Eng Chem 20(4):2166–2173
Loc LC, Phuong PH, Putthea D, Tri N, Van NTT, Cuong HT (2018) Effect of CeO2 morphology on performance of NiO/CeO2 catalyst in combined steam and CO2 reforming of CH4. Int J Nanotechnol 15(11–12):968–982
Phuong PH, Loc LC, Cuong HT, Tri N (2018) Effect of NiO loading and thermal treatment duration on performance of Ni/SBA-15 catalyst in combined steam and CO2 reforming of CH4. Mater Trans 59(12):1898–1902
Loc LC, Phuong PH, Thao NHP, Tri N, Van NTT, Cuong HT, Anh HC (2017) Influence of preparation method on the activity of NiO+MgO/Al2O3 catalyst in dry reforming of methane. Vietnam J Chem 55(3E):1–7
Kiperman SL (1978) Kinetic Models in Heterogeneous Catalysis. Russ Chem Rev 47(1):1
Temkin MI (1976) Relaxation speed of two-stage catalytic reaction. Kinet Catal 17(5):1095–1099
Zhao D, Feng J, Huo Q, Melosh N, Fredrickson GH, Chmelka BF, Stucky GD (1998) Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 279(5350):548–552
Bukhari S, Chin C, Setiabudi H, Vo D-VN (2017) Tailoring the properties and catalytic activities of Ni/SBA-15 via different TEOS/P123 mass ratios for CO2 reforming of CH4. J Environ Chem Eng 5(4):3122–3128
Rodriguez-Gomez A, Pereñiguez R, Caballero A (2018) Nickel particles selectively confined in the mesoporous channels of SBA-15 yielding a very stable catalyst for DRM reaction. J Phys Chem B 122(2):500–510
Aghamohammadi S, Haghighi M, Karimipour S (2013) A comparative synthesis and physicochemical characterizations of Ni/Al2O3–MgO nanocatalyst via sequential impregnation and sol–gel methods used for CO2 reforming of methane. J Nanosci Nanotechnol 13(7):4872–4882
Sun Y, Jiang E, Xu X, Wang J, Li Z (2018) Supplied oxygen properties of NiO/NiAl2O4 in chemical looping re-forming of biomass pyrolysis gas: the influence of synthesis method. ACS Sustain Chem Eng 6(11):14660–14668
Liu D, Quek XY, Cheo WNE, Lau R, Borgna A, Yang Y (2009) MCM-41 supported nickel-based bimetallic catalysts with superior stability during carbon dioxide reforming of methane: effect of strong metal–support interaction. J Catal 266(2):380–390
Walker DM, Pettit SL, Wolan JT, Kuhn JN (2012) Synthesis gas production to desired hydrogen to carbon monoxide ratios by tri-reforming of methane using Ni–MgO–(Ce, Zr)O2 catalysts. Appl Catal A 445:61–68
Ay H, Üner D (2015) Dry reforming of methane over CeO2 supported Ni, Co and Ni–Co catalysts. Appl Catal B 179:128–138
Li B, Zhang S (2013) Methane reforming with CO2 using nickel catalysts supported on yttria-doped SBA-15 mesoporous materials via sol–gel process. Int J Hydrogen Energy 38(33):14250–14260
Liu H, Wang H, Shen J, Sun Y, Liu Z (2008) Promotion effect of cerium and lanthanum oxides on Ni/SBA-15 catalyst for ammonia decomposition. Catal Today 131(1–4):444–449
Luo C, Li D, Wu W, Zhang Y, Pan C (2014) Preparation of porous micro–nano-structure NiO/ZnO heterojunction and its photocatalytic property. RSC Adv 4(6):3090–3095
Agarwal S, Zhu X, Hensen E, Mojet B, Lefferts L (2015) Surface-dependence of defect chemistry of nanostructured ceria. J Phys Chem C 119(22):12423–12433
Gamarra D, Munuera G, Hungría A, Fernández-García M, Conesa J, Midgley P, Wang X, Hanson J, Rodríguez J, Martínez-Arias A (2007) Structure−activity relationship in nanostructured copper−ceria-based preferential CO oxidation catalysts. J Phys Chem C 111(29):11026–11038
Sudarsanam P, Hillary B, Mallesham B, Rao BG, Amin MH, Nafady A, Alsalme AM, Reddy BM, Bhargava SK (2016) Designing CuOx nanoparticle-decorated CeO2 nanocubes for catalytic soot oxidation: role of the nanointerface in the catalytic performance of heterostructured nanomaterials. Langmuir 32(9):2208–2215
Zheng X, Li Y, Zhang L, Shen L, Xiao Y, Zhang Y, Au C, Jiang L (2019) Insight into the effect of morphology on catalytic performance of porous CeO2 nanocrystals for H2S selective oxidation. Appl Catal B 252:98–110
Li Y, Wei Z, Gao F, Kovarik L, Baylon RA, Peden CH, Wang Y (2015) Effect of oxygen defects on the catalytic performance of VOx/CeO2 catalysts for oxidative dehydrogenation of methanol. ACS Catal 5(5):3006–3012
Ocsachoque M, Pompeo F, Gonzalez G (2011) Rh–Ni/CeO2–Al2O3 catalysts for methane dry reforming. Catal Today 172(1):226–231
Djaidja A, Libs S, Kiennemann A, Barama A (2006) Characterization and activity in dry reforming of methane on NiMg/Al and Ni/MgO catalysts. Catal Today 113(3–4):194–200
Schulze K, Makowski W, Chyży R, Dziembaj R, Geismar G (2001) Nickel doped hydrotalcites as catalyst precursors for the partial oxidation of light paraffins. Appl Clay Sci 18(1–2):59–69
Roh H-S, Jun K-W, Dong W-S, Chang J-S, Park S-E, Joe Y-I (2002) Highly active and stable Ni/Ce–ZrO2 catalyst for H2 production from methane. J Mol Catal A: Chem 181(1–2):137–142
Li Y, Xie X, Liu J, Cai M, Rogers J, Shen W (2008) Synthesis of α-Ni(OH)2 with hydrotalcite-like structure: precursor for the formation of NiO and Ni nanomaterials with fibrous shapes. Chem Eng J 136(2–3):398–408
Du X, Zhang D, Shi L, Gao R, Zhang J (2012) Morphology dependence of catalytic properties of Ni/CeO2 nanostructures for carbon dioxide reforming of methane. J Phys Chem C 116(18):10009–10016
Jacobs G, Das TK, Zhang Y, Li J, Racoillet G, Davis BH (2002) Fischer-Tropsch synthesis: support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl Catal A 233(1–2):263–281
He L, Liang B, Li L, Yang X, Huang Y, Wang A, Wang X, Zhang T (2015) Cerium-oxide-modified nickel as a non-noble metal catalyst for selective decomposition of hydrous hydrazine to hydrogen. ACS Catal 5(3):1623–1628
Sidik S, Triwahyono S, Jalil A, Majid Z, Salamun N, Talib N, Abdullah T (2016) CO2 reforming of CH4 over Ni–Co/MSN for syngas production: role of Co as a binder and optimization using RSM. Chem Eng J 295:1–10
Das S, Ashok J, Bian Z, Dewangan N, Wai M, Du Y, Borgna A, Hidajat K, Kawi S (2018) Silica-Ceria sandwiched Ni core–shell catalyst for low temperature dry reforming of biogas: coke resistance and mechanistic insights. Appl Catal B 230:220–236
Bian Z, Li Z, Ashok J, Kawi S (2015) A highly active and stable Ni–Mg phyllosilicate nanotubular catalyst for ultrahigh temperature water-gas shift reaction. Chem Commun 51(91):16324–16326
Xu D, Li W, Ge Q, Xu H (2005) A novel process for converting coalmine-drained methane gas to syngas over nickel–magnesia solid solution catalysts. Fuel Process Technol 86(9):995–1006
Yoshida T, Tanaka T, Yoshida H, Funabiki T, Yoshida S (1996) Study on the dispersion of nickel ions in the NiO− MgO system by X-ray absorption fine structure. J Phys Chem 100(6):2302–2309
Zeng Y, Ma H, Zhang H, Ying W, Fang D (2014) Highly efficient NiAl2O4-free Ni/γ-Al2O3 catalysts prepared by solution combustion method for CO methanation. Fuel 137:155–163
Wang X, Zhu L, Liu Y, Wang S (2018) CO2 methanation on the catalyst of Ni/MCM-41 promoted with CeO2. Sci Total Environ 625:686–695
Xu B, Zhang Q, Yuan S, Zhang M, Ohno T (2015) Morphology control and characterization of broom-like porous CeO2. Chem Eng J 260:126–132
Chong CC, Teh LP, Setiabudi HD (2019) Syngas production via CO2 reforming of CH4 over Ni-based SBA-15: promotional effect of promoters (Ce, Mg, and Zr). Mater Today Energy 12:408–417
Dholabhai PP, Adams JB, Crozier P, Sharma R (2010) Oxygen vacancy migration in ceria and Pr-doped ceria: A DFT+U study. J Chem Phys 132(9):094104
Binet C, Badri A, Boutonnet-Kizling M, Lavalley J-C (1994) FTIR study of carbon monoxide adsorption on ceria: CO2–2 carbonite dianion adsorbed species. J Chem Soc Faraday Trans 90(7):1023–1028
Binet C, Daturi M, Lavalley J-C (1999) IR study of polycrystalline ceria properties in oxidised and reduced states. Catal Today 50(2):207–225
Song Z, Liu W, Nishiguchi H (2007) Quantitative analyses of oxygen release/storage and CO2 adsorption on ceria and Pt–Rh/ceria. Catal Commun 8(4):725–730
Hojo H, Mizoguchi T, Ohta H, Findlay SD, Shibata N, Yamamoto T, Ikuhara Y (2010) Atomic structure of a CeO2 grain boundary: the role of oxygen vacancies. Nano Lett 10(11):4668–4672
Ganduglia-Pirovano MV, Da Silva JL, Sauer J (2009) Density-functional calculations of the structure of near-surface oxygen vacancies and electron localization on CeO2 (111). Phys Rev Lett 102(2):026101
Naeem MA, Al-Fatesh AS, Abasaeed AE, Fakeeha AH (2014) Activities of Ni-based nano catalysts for CO2–CH4 reforming prepared by polyol process. Fuel Process Technol 122:141–152
Alipour Z, Rezaei M, Meshkani F (2014) Effect of Ni loadings on the activity and coke formation of MgO-modified Ni/Al2O3 nanocatalyst in dry reforming of methane. J Energy Chem 23(5):633–638
Wang S, Lu G, Millar GJ (1996) Carbon dioxide reforming of methane to produce synthesis gas over metal-supported catalysts: state of the art. Energy Fuels 10(4):896–904
Das S, Sengupta M, Patel J, Bordoloi A (2017) A study of the synergy between support surface properties and catalyst deactivation for CO2 reforming over supported Ni nanoparticles. Appl Catal A 545:113–126
Li Z, Das S, Hongmanorom P, Dewangan N, Wai MH, Kawi S (2018) Silica-based micro-and mesoporous catalysts for dry reforming of methane. Catal Sci Technol 8(11):2763–2778
Zambrano D, Soler J, Herguido J, Menéndez M (2019) Kinetic study of dry reforming of methane over Ni–Ce/Al2O3 catalyst with deactivation. Top Catal 62:456–466
Cj L, Ye J, Jiang J, Pan Y (2011) Progresses in the preparation of coke resistant Ni-based catalyst for steam and CO2 reforming of methane. ChemCatChem 3(3):529–541
Danilova M, Fedorova Z, Kuzmin V, Zaikovskii V, Porsin A, Krieger T (2015) Combined steam and carbon dioxide reforming of methane over porous nickel based catalysts. Catal Sci Technol 5(5):2761–2768
Li KZ, Wang H, Wei YG, Yan DX (2009) Selective oxidation of carbon using iron-modified cerium oxide. J Phys Chem C 113(34):15288–15297
Lercher J, Bitter J, Hally W, Niessen W, Seshan K (1996) Design of stable catalysts for methane-carbon dioxide reforming. Stud Surf Sci Catal 101:463–472
Bradford MC, Vannice MA (1999) The role of metal–support interactions in CO2 reforming of CH4. Catal Today 50(1):87–96
Kapil A, Wilson K, Lee AF, Sadhukhan J (2011) Kinetic modeling studies of heterogeneously catalyzed biodiesel synthesis reactions. Ind Eng Chem Res 50(9):4818–4830
Ayodele BV, Hossain SS, Lam SS, Osazuwa OU, Khan MR, Cheng CK (2016) Syngas production from CO2 reforming of methane over neodymium sesquioxide supported cobalt catalyst. J Nat Gas Sci Eng 34:873–885
Acknowledgements
This work was supported by the program code VAST03.01/2019-2020 from the Materials Science Council, Vietnam Academy of Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nguyen, T., Luu, C.L., Phan, H.P. et al. Methane dry reforming over nickel-based catalysts: insight into the support effect and reaction kinetics. Reac Kinet Mech Cat 131, 707–735 (2020). https://doi.org/10.1007/s11144-020-01876-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-020-01876-8