
Abstract
10%NiO–La0.3Sr0.7Co0.7Fe0.3O3−δ (10%NiO-LSCF3773) was synthesized using the EDTAcitrate complexing method. Non-catalytic and catalytic nitrous oxide decomposition and methane partial oxidation using 10%NiO–LSCF3773 was experimentally studied, assuming that the reactions occurred separately in a membrane reactor at feed side and permeate side. The experimental results are in good agreement with the chemical equilibrium composition calculated using Aspen Plus, and the changes of standard Gibbs free energy of each relevant elementary reactions. The mechanism of the reactions was proposed to follow Eley–Rideal surface reaction. The optimal temperature was 800 °C, under atmospheric pressure, where (1) NO2 formation was not detected (2) no production of C2 + and C3 + (3) complete conversion of N2O, CH4 and O2 were achieved (4) high purity syngas was obtained with no significant amount of undesired products and (5) readily utilizable syngas at the ratio of two was achieved.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nitrous oxide (N2O) is one of the greenhouse gases that causes global warming. It has 300 times of the heat-trapping ability in the atmosphere more than carbon dioxide (CO2) compared to the same amount within 100 years [1,2,3]. N2O comes from both man-made and natural sources. Examples of the former are petrochemical industries (adipic acid, nitric acid plant, agriculture (use of nitrogen fertilizer), combustion processes of fossil fuels and biomass; while the latter’s are volcano emission and animal wastes. [1, 4,5,6,7]. The current industrial nitrous oxide abatement in Thailand is nitrous oxide decomposition [8]. The nitrous oxide is converted to nitrogen (N2) and oxygen (O2) at 600–800 °C, where the non-toxic gaseous product could be released to the environment. A part from being environmentally friendly, this method is costly because it requires a lot of external energy and does not give any valuable products.
Nitrous oxide can be utilized as oxygenated feedstock in chemical and energy production (i.e. fuel cells, production of syngas, carbonyl compounds, methanol, dimethyl ether, formaldehyde and so on) [9,10,11,12,13]. This work focuses on syngas production, as it is not only an energy source but also an importance feedstock in which it further conversion can give other valuable chemicals such as synthetic fuel, methanol, dimethyl ether, acetic acid etc. [9,10,11,12,13,14]. The nitrous oxide will be used as an oxidant providing a synergy of N2O utilization and syngas production simultaneously. Transition metals (Co, Fe, and Ni) [15,16,17,18,19,20], precious metals (Pd, Pt, Re, Rh) [11,12,13], and metal oxide compound alumina (Al2O3), lanthanum cobaltite (LaCoO3), lanthanum cobalt iron oxide (LaCoFeO3), lanthanum ferrite (LaFeO3), lanthanum manganite (LaMnO3), ruthenium oxide (RuO2), yttria-stabilized zirconia (YSZ) [15, 16, 18, 21,22,23,24,25,26,27].
The methane partial oxidation by nitrous oxide was studied widely in a packed-bed reactor [11]. Since both methane partial oxidation and nitrous oxide decomposition are exothermic reaction, therefore, a large amount of heat could be created causing temperature runaway. The conventional packed-bed reactor also requires a separation unit leading to a high investment cost. Membrane reactors have attracted significant attention due to its ability to function as a reactor and separator at the same time. However, the methane partial oxidation using nitrous oxide has not yet been researched in a membrane reactor. The membrane reactor could eliminate the complexity of the separation process and to ease complication of the reaction, narrow down the product distribution, increase selectivity, reduce energy consumption [12, 28,29,30,31,32,33]. This work studied methane partial oxidation and nitrous oxide decomposition which supposed to occur at the feed side and permeate side of the membrane reactor, respectively. Thermodynamics is useful for process assessment and optimal condition prediction, thus in this work, chemical equilibrium of the reactions were estimated using Aspen Plus program. The reactions were experimented using 10%NiO–La0.3Sr0.7Co0.7Fe0.3O3−δ (10%NiO–LSCF) to investigate actual reactions and their surface mechanisms. Ni and LSCF3773 was chosen according to its economically acceptable yield and its oxygen storage capacity, respectively [34,35,36,37].
Methodology
Possible reactions
The possible reactions in the membrane reactor are tabulated in Table 1. Table 2 represents relevant mathematical formulas for the changes of the standard Gibbs free energy calculation.
At the feed side, nitrous oxide was decomposed on the surface of the membrane giving gaseous oxygen and nitrogen (1). Nitrous oxide could react with oxygen giving nitrogen dioxide as the undesired product (2). Oxygen was assumed to flow through the membrane to the permeable side and partially oxidize methane giving synthesis gas (4) and other by-products via associated reactions (5–11) [38,39,40]. The chemical equilibrium properties were calculated using Aspen Plus program. Changes of the standard Gibbs free energy were calculated based on heat capacity of each reactions [41, 42] using mathematical formulas presented in Fig. 5, where i denotes species of substance, γi is the stoichiometric coefficient of species i, ∆Cp is heat capacity. ∆rH, ∆rS, and ∆rG are the different of enthalpy, entropy, and Gibbs free energy of reaction, respectively. T is temperature. A, B, C, D, and E are characteristic constants [43,44,45].
Catalyst preparation and characterization
The aqueous solution of lanthanum (III) nitrate hexahydrate (La(NO3)3·H2O), strontium nitrate (Sr(NO3)2), cobalt Nitrate (Co(NO3)2·6H2O), and iron(III) nitrate (Fe(NO3)3·9H2O) were added stoichiometrically and stirred until homogeneous. EDTA-NH4OH, citric acid and ethylene glycol was added drop-wise while stirred continuously. The pH of the solution was kept at 6 to 7 using NH4OH. The resulting solution was evaporated at 80–90 °C. The residual muddy dark red gel was left in an oven overnight at 200 °C. The sample was calcined in moving air at 1100 °C for 12 h. giving LSCF3773 powder. The powder was mixed into Ni(NO3)2·6H2O solution. The same procedure of drying, evaporation and calcination were applied. The crystallinity of the catalysts were characterized using X-ray diffraction technique (XRD, Rigaku TTRAX III using Cu Kα radiation, λ = 1.5418 Å in a range of 30° < 2θ < 80° with a scanning step of 0.01°). The reducibility of the catalyst was studied by H2-TPR technique (H2 temperature-programmed reduction). Interaction between CH4 and catalyst surface was investigated by CH4-TPSR technique (CH4 temperature-programmed surface reaction). The catalysts was packed in a lab-scaled tubular reactor. 100 ml/min of 5%H2/Ar (for H2-TPR) or 5%CH4/Ar (for CH4-TPSR) was introduced into the system while temperature was increased at 5 °C/min from 25 to 950 °C. The consumption of the reactant and the formation of the product gases were measured by real-time quadrupole mass spectrometer gas analyzer, based on molecular weights for gases CH4 (16), CO2 (44), CO (28), H2 (2), and Ar (40).
Experimental set-up
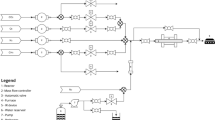
The schematic diagram of the rig was presented in Fig. 1. 2 g of 10%NiO–LSCF3773 was packed into a quartz tubular reactor with an inside diameter of 10 mm. The reactor was placed in the middle of an electrical furnace (Chavachote, Thailand) with 30 cm heating zone. The catalyst was reduced in 40%H2 in Ar at 500 °C for 2 h before the reaction for all experiments. For nitrous oxide decomposition, total flow rate of 100 ml/min of N2O (99.95%, company) balanced in Ar (99.995%, BIG) at 1:1 ratio was fed into the system using mass flow controllers (Brooks instrument flow, 0–220 ml/min). A mixture of 50 ml/min of CH4 (40%CH4/Ar, company), 25 ml/min of O2 (40%O2/Ar, company), and 25 ml/min of Ar was introduced into the reactor for methane partial oxidation at 2:1 molar ratio of CH4 to O2. All the experiments were isothermally operated under atmospheric pressure at temperature range of 500–1000 °C using type K thermocouple and PID temperature controller. Gas compositions were analyzed using gas chromatography method (Shimadzu GC-2014ATP).

Rig’s diagram for nitrous oxide decomposition and methane partial oxidation
Conversions percentages were estimated using the following equations;
Here i denotes to N2O, CH4, and O2 and ni is mole or mole flow rate of species i.
Result and discussion
Characterization techniques
X-ray diffraction
Fig. 2 shows diffractograms of (a) NiO, (b) LSCF3773, and (c) 10%NiO–LSCF3773. The XRD pattern of 10%NiO–LSCF3773 has perovskite structure and NiO as major and minor crystalline phase, respectively. The Ni-based LSCF catalyst shown the mixed XRD diffraction peaks at 2θ = 23.20, 33.04, 37.32, 40.74, 43.36, 47.42, 53.94, 58.96, 62.92, 69.22, 75.46, 78.84 and 80.16, the diffractograms also confirmed the presence of NiO (ICDD card No.03-065-5745; 37.32, 43.36, 62.92, 75.46, and 80.16 corresponding to (111), (200), (220), (311), and (222) planes) with a cubic crystal structure. However, there was no peak shift in XRD pattern when more doped Ni content on LSCF, which noticed that the introduction of Ni loading did not alter the crystal structure of prepared-catalyst.

The XRD patterns; (a) NiO, (b) LSCF, and (c) 10%NiO–LSCF
H2 Temperature-programmed reduction (H2-TPR)
Reduction property of the synthesized NiO-based LSCF catalyst were evaluated using H2-TPR. H2 consumption profiles of the catalysts were compared and shown in Fig. 3. The TPR profile of pure NiO showed two main reduction peaks centered at 390 and 500 °C, indicating reduction of Ni3+ ions to Ni2+ ions and reduction of Ni2+ ions to Ni0 ions, respectively [46,47,48,49,50]. For LSCF, the lowest temperature of reduction at 270 °C was attributed to surface oxygen reduction [51], while bulk oxygen reduction took placed during higher temperatures at 505, 625, and 735 °C. The bulk oxygen reduction indicate three stages of reduction (1) reduction of Co3+/Fe4+, Fe5+ ions to Co2+/Fe3+ ions at about 505 °C, (2) reduction of Co2+/Fe3+ ions to Co0/Fe2+ ions at 625 °C, and (3) reduction of Co0/Fe2+ ions to Co0/Fe0 ions at 735 °C [51,52,53]. The reduction temperatures of 10%NiO–LSCF showed more similarity to that of LSCF rather than of NiO. This is because the NiO level was not high enough to dominate the crystal structure [46, 54,55,56]. The H2 consumption could be interpreted as oxygen vacancy/oxygen lattice/oxygen storage capacity/oxygen deficiency, which was 11,188 µmol/gsolid, 5754 µmol/gsolid, and 6442 µmol/gsolid, for NiO, LSCF3773 and 10%NiO–LSCF3773, respectively. The 10%NiO was in situ reduced before each run at 500 °C, achieving Ni0 ions which acts as an active site for the partial oxidation of methane.

H2-TP–R profiles of synthesized catalysts; (a) NiO, (b) LSCF, and (c) 10%NiO–LSCF
CH4 Temperature-programmed surface reaction (CH4-TPR)
CH4 dissociation on the surface of 10%NiO-LSCF was characterized by CH4-TPR technique, shown in Fig. 4. Four regions of temperature range can be considered according to its mechanisms.

CH4-TPSR profiles of 10%NiO–LSCF catalyst
No CH4 consumption was detected at temperature lower than 450 °C, in this region I.
During temperature from 450 to 600 °C, referred to as region II in Fig. 4, CH4 dissociation and combustion were presumably occurred on Ni active site (Eq. 23), and adsorbed oxygen on the surface \(\left( {O_{\left( {ads} \right)}^*} \right)\) (Eq. 24) via Eley–Rideal mechanism,.
A slight CH4 consumption and rapid H2 formation were correspondingly observed at the beginning of this region. Meanwhile, adsorbed oxygen on LSCF’s surface was reduced by CH4, giving H2O and CO2 as gaseous products. During the process, the oxygen vacancies was achieved (Eq. 24). These oxygen vacancies are to be re-oxidized by gaseous N2O during the N2O decomposition.
During temperature from 600 to 900 °C, shown as region III in Fig. 4, CH4 was significantly consumed while all the gaseous products, H2, CO, CO2 and H2O were found. Two main redox mechanisms were proposed as below:
-
(1)
CH4 partial oxidation/combustion occurred on the oxygen surface of LSCF, giving H2, H2O, CO, CO2 and oxygen vacancies as products, shown below as Eq. 25.
$$C{{H}_{4,(g)}}+O_{(ads)}^{*}\to \left[ \begin{matrix} {{H}_{2,(g)}} \\ {{H}_{2}}{{O}_{(g)}} \\\end{matrix} \right]+\left[ \begin{matrix} C{{O}_{(g)}} \\ C{{O}_{2,(g)}} \\\end{matrix} \right]+{{*}_{(s)}}$$(25) -
(2)
Langmuir–Hinshelwood surface reaction between the coked Ni site \(\left( {{{(C \times N{i^*})}_{\left( {ads} \right)}}} \right)\) (obtained from Eq. 23) and surface oxygen \(\left( {O_{\left( {ads} \right)}^*} \right)\) was occurred, producing gaseous products of CO and CO2. The oxygen vacancy \(\left( {_{\left( s \right)}^*} \right)\) was obtained as the result of coke combustion, between the adsorbed coke \(\left( {{{(C \cdot N{i^*})}_{\left( {ads} \right)}}} \right)\) and the lattice oxygen \(\left( {O_{\left( {ads} \right)}^*} \right)\), represented by Eq. 26, where the coked-Ni site \(\left( {{{(C \times N{i^*})}_{\left( {ads} \right)}}} \right)\) was regenerated back to its original form of active Ni \(\;\left( {Ni_{\left( s \right)}^*} \right)\). This regenerated Ni \(\;\left( {Ni_{\left( s \right)}^*} \right)\) could react cyclically with CH4 giving more H2 and coked-Ni \(\left( {{{(C \times N{i^*})}_{\left( {ads} \right)}}} \right)\), according to Eq. 23. The coked-Ni \(\left( {{{(C \times N{i^*})}_{\left( {ads} \right)}}} \right)\) would consequently reduce the LSCF surface by reacting with lattice oxygen \(\left( {O_{\left( {ads} \right)}^*} \right)\). The oxygen vacancy \(\left( {_{\left( s \right)}^*} \right)\) was then achieved via Eq. 26.
$$C\times Ni_{(ads)}^{*}+O_{(ads)}^{*}\to \left[ \begin{matrix} C{{O}_{(g)}} \\ C{{O}_{2,(g)}} \\\end{matrix} \right]+Ni_{(s)}^{*}+{{*}_{(s)}}$$(26) -
(3)
In the 4th region where the temperature was higher than 900 °C, CH4 concentration returned back to its initial level due to the absence of the adsorbed oxygen on the surface of LSCF before started to slightly decrease again by CH4 cracking over the coked-Ni active site. This resulted in an increase in H2 formation, where the CO, CO2 and H2O concentration, more or less, remain unchanged. However, H2O, CO, and CO2 re-adsorption could be suggested.
The results conclude that CH4 combustion was dominant when the temperature was lower than 600 °C. The desired CH4 partial oxidation occurred at temperature between 600 and 900 °C, while the CH4 cracking took part at 900 °C and beyond.
Chemical equilibrium estimation and effect of operating temperature
Changes of the standard Gibbs free energy \(\left( {{\Delta_r}{G^0}} \right)\;\) of the possible reactions listed in Table 1 were calculated using formulas in Table 2 shown below in Fig. 5. The selected temperature for the study was ranged from 500 to 1000 °C.

Changes of the standard Gibbs free energy of each reactions
The result showed that the NO2 formation (reaction 2) was theoretically not possible for all temperature range while CH4 cracking could occur at temperature higher than 700 °C, approximately. From 500 to 600 °C, combustion of methane would be evidenced (reactions 4, 10, 11). Methane partial oxidation (reaction 4) would occur during temperature from 700 to 1000 °C. Coke formation could be deposited on the surface of the catalyst at temperature higher than 700 °C.
Figs. 6 and 7 present conversions of reactants and products of N2O decomposition and CH4 partial oxidation, which supposed to occurred at the feed side and permeate side of the membrane reactor, respectively.

a Conversion of the reactant (N2O) as a function of temperature. Mole fraction product of N2O decomposition using b no catalyst (homogeneous) and c 10%NiO–LSCF catalyst compared with equilibrium mole fraction using Aspen Plus program (line) run WHSV = 50 ml/g/min at 1 atm from 500 to 1000 °C

a The temperature dependence of methane conversion. Mole fraction product at the permeate side of the membrane process using b no catalyst (homogeneous) and c 10%NiO–LSCF catalyst compared with equilibrium mole fraction using Aspen Plus program (line) run is WHSV = 50 ml/g min at 1 atm from 500 to 1000 °C
Fig. 6a shows the highest N2O conversion of 68.71 was in the non-catalytic reaction and complete N2O conversion (99.13%) when using 10%NiO–LSCF as a catalyst. The conversion was close to the theoretical when using the 10%NiO–LSCF catalyst.
Fig. 6b represents non-catalytic nitrous oxide decomposition. The highest N2O conversion was obtained at 1000 °C giving N2 and O2 of 56.39 and 17.93 mol%, respectively, although both products began to be detectable at 800 °C. The productivities were increased with increasing temperature. The ratio of N2 to O2 was around 2.3–2.4 to 1 which disagreed with theoretical ratio from stoichiometry (2 to 1). There was no NO2 detected for all temperature range. In addition, NO was not measured in the experiment, although NO formation as an intermediate could be suggested via N2O oxidation/decomposition.
Fig. 6c shows that complete N2O conversion started to be achievable at 600 °C, giving 66.59 mol% of N2 and 32.78mol% of O2. Ratio of N2 to O2 was 2:1 during temperature from 600 to 800 °C, agreeing with its stoichiometry theoretically. At temperature higher than 800 C, the ratio was found to increase by the influence of oxygen inhibition, explained in the later part. The following reaction pathway could be suggested.
N2O was adsorbed and dissociated on the oxygen vacancy, achieved by CH4 partial oxidation via reaction (23)–(26), giving N2 and surface oxygen \(\left( {O_{\left( {ads} \right)}^*} \right)\), represented by Eq. 28. The resulting surface oxygen \(\left( {O_{\left( {ads} \right)}^*} \right)\), was reacted further with gaseous N2O via Eley–Rideal surface mechanism via Eq. (29), where more N2 and adsorbed oxygen molecule \(\left( {O_{2\;\left( {ads} \right)}^*} \right)\) were obtained. Afterwards, the adsorbed oxygen molecule \(\left( {O_{2\;\left( {ads} \right)}^*} \right)\) could be; (i) desorbed giving gaseous O2 while leaving the oxygen lattice vacant again \(\left( {_{\left( s \right)}^*} \right)\), demonstrated in Eq. 30, (ii) react with oxygen vacancy \(\left( {_{\left( s \right)}^*} \right)\) via Langmuir–Hinshelwood surface reaction, providing more surface oxygen \(\left( {O_{\left( {ads} \right)}^*} \right)\), shown in Eq. 31.
However, the gaseous oxygen produced via Eq. 30 could be re-adsorbed on the oxygen vacancy \(\left( {_{\left( s \right)}^*} \right)\), represented by Eq. 32. The surface oxygen was obtained.
Oxygen inhibition, where full surface coverage was achieved by oxygen re-adsorption, could occur if k1, k4 and/or k5 is larger than k2 and/or k3. This would be evident by N2 to O2 ratio higher than 2. However, as the ratio of N2 to O2 was approximately 2 to, this implies that k1, k4 and/or k5 is less than k2 and/or k3, meaning that reactions 28, 31 and/or 32 is the rate limiting step. At temperature from 900 to 1000 °C, the unwanted NO2 formation was utilized the surface oxygen, allowing Eq. 32 to go more forward. This leads to the decrease of O2, resulted in the higher ratio of N2 to O2. Mechanism of NO2 formation was proposed below, where the N2O adsorption/dissociation (reaction 28) occurred, then followed by the below Eley–Rideal surface reactions.
Fig. 6 (line) shows the equilibrium molar fraction of N2 and O2 at 0.67 and 0.33, calculated using Aspen Plus program, for all temperatures. NO2 was not found. The results agreed with the calculation reported in Fig. 5, where reaction 1 was spontaneous and reaction 2 was thermodynamically limited. The catalytic conversion of N2O and its productivities (Fig. 6c) were found to approach the equilibrium approximation as shown in (Fig. 6 (line)) especially during temperature from 500 to 900 °C.
Fig. 7 shows CH4 partial oxidation using no catalyst (b), in the presence of 10%NiO-LSCF (c), and (line) equilibrium concentration calculated using Aspen Plus program. From Fig. 7a, it can be seen that maximum CH4 conversion in non-catalyst, achieved at 1000 °C, was 53.51. CH4 consumption began to be observable at 800 °C, giving conversions of 2.87. In using 10%NiO–LSCF catalyst, complete conversion of CH4 was obtained at 1000 °C, which gives conversion that are close to the theoretical.
From Fig. 7b, less than 3mol% fraction of H2, CO2, H2O, C2H4, and C2H6 were produced at this temperature. Reverse water gas shift reaction (RWGS), which is the combination of reaction (10) and 11, was evident as H2O production was noticed. The Boudouard reaction, the combination of reactions 4, 9, and 10, was not observed, as there was no CO2 formation, therefore, coke formation did not occurred at this temperature range. On the other hand, CO2 was detected at higher temperature ranging from 900 to 1000 °C, suggesting the possibility of the Boudouard reaction, therefore, coke formation was also predicted. In addition, H2, CO, CO2, and H2O molar fraction were increased with increasing temperature. This indicated that complete combustion, reverse water gas shift and the Boudouard reaction were presumably taken place. However, C2 + was increased at temperature from 800 to 900 °C, then decreased again at 900 to 1000 °C. This implied that reactions 5 and 7 were driven backward at temperature lower than 900 °C, and moved forward at temperature higher than 900 °C.
From Fig. 7c, H2O and CO2 were detected at temperature lower than 700 °C and found to decrease with increasing temperature. Small amount (less than 1mol%) of H2O and CO2 were found at temperature higher than 700 °C. On the other hand, H2 and CO were increased with increasing temperature. The reaction temperatures of the CH4 partial oxidation, CH4 combustion and CH4 cracking shown in Fig. 7c were correspondingly agreed with those shown in Fig. 4. The catalytic experimental result was found to approach the equilibrium composition, shown in Fig. 7c, and corresponded with calculation of Gibbs free energy changes tabulated in Fig. 5. The negative value of ∆Go of reaction 4 became larger with increasing temperature, while that of reactions 10 and 11 became smaller. This means that reaction 4 was more spontaneous at higher temperature leading to higher yield of H2 and CO, while reaction 10 and 11 were more thermodynamically limited resulting in less amount of CO2 and H2O production. At temperature higher than 700 °C, the possible mechanism was proposed as follows;
The surface oxygen or lattice oxygen \(\left( {O_{\left( {ads} \right)}^*} \right)\), produced from reaction (28), assumed to diffuse through the membrane from the feed side and reacted with CH4, giving adsorbed CO \(\left( {CO_{\left( {ads} \right)}^*} \right)\) and gaseous H2 as shown in Eq. 35. The adsorbed CO \(\left( {CO_{\left( {ads} \right)}^*} \right)\) was afterwards desorbed to gaseous CO and left the active site \(\left( {_{\left( s \right)}^*} \right)\) free again, represented by Eq. 36:
CO2 and H2O splitting were occurred on the oxygen vacancy \(\left( {_{\left( s \right)}^*} \right)\) as shown in Eq. (37) and (38), producing CO and H2 as gaseous products. On the other hand, the oxygen vacancy \(\left( {_{\left( s \right)}^*} \right)\) was simultaneously re-oxidized by CO2 and H2O becoming the adsorbed oxygen \(\left( {O_{\left( {ads} \right)}^*} \right)\);
From Fig. 7c, no significant amount of C2 + and C3 + were found for all temperature range, indicating the occurrence of reaction (5), (6), (7), and (8) where C2 + and C3 + were reacted with oxygen giving H2 and CO. The result agreed with the standard Gibbs free energy changes, calculated and shown in Fig. 5. From Fig. 5, the standard Gibbs free energy changes of reaction (9) was positive at temperature from 500 to 600 °C, and became negative at temperature higher than 600 °C. This means that coke formation via reaction (9) was not occurred at 500 to 600 °C, but instead was likely to occur at temperature higher than 600 °C.
Figure 8 compared syngas productivities and its ratio between obtained from catalytic process using 10%NiO–LSCF, and from the chemical equilibrium calculation. The experimental result was found to be in good agreement with the calculation. Syngas ratio was approaching 2 at temperature from 700 to 1000 °C.

Experimental catalytic performance using 10%NiO–LSCF compared with equilibrium composition
Conclusion
10%NiO–LSCF was a promising catalyst for high purity syngas production via methane partial oxidation using nitrous oxide as an oxidant in a membrane reactor. Surface mechanisms of the reactions, at both feed side and permeate side were proposed, based on the experimental results. The catalytic experimental result was in good agreement with the chemical equilibrium properties, calculated using Aspen Plus. The optimal operating temperature for the reaction carried out in a membrane reactor is 800 °C under atmosphere pressure. At this temperature, NO2 formation was not occurred, and full conversion of N2O decomposition at the feed side, and methane partial oxidation at the permeate side were achieved. Heavier hydrocarbons, i.e. C2 + and C3 + , should not be found. High purity syngas could be obtained at the ratio of H2 to CO equal to 2.
References
Li C, Shen Y, Zhu S, Shen S (2014) Supported Ni–La–Ox for catalytic decomposition of N2O I: component optimization and synergy. RSC Adv 4(55):29107
Xue L, Zhang C, He H, Teraoka Y (2007) Catalytic decomposition of N2O over CeO2 promoted Co3O4 spinel catalyst. Appl Catal B 75(3–4):167–174
Liu Z, Zhou Z, He F, Chen B, Zhao Y, Xu Q (2017) Catalytic decomposition of N2O over NiO–CeO2 mixed oxide catalyst. Catal Today 293–294:56–60
Vitousek PM, Aber JD, Howarth RW, Likens GE, Matson PA, Schindler DW, Schlesinger WH, Tilmen DG (1997) Human alteration of the global nitrogen cycle: sources and consequences. Ecol Appl 7:737–750
van den Brink RW, Booneveld S, Verhaak MJFM, De Bruijn FA (2002) Selective catalytic reduction of N2O and NOx in a single reactor in the nitric acid industry. Catal Today 75:227–232
Ravishankara AR, Daniel JS, Portmann RW (2009) Nitrous oxide (N2O): the dominant ozone-depleting substance emitted in the 21st century. Science 326(5949):123–125
Li L, Xu J, Hu J, Han J (2014) Reducing nitrous oxide emissions to mitigate climate change and protect the ozone layer. Environ Sci Technol 48(9):5290–5297
Pedros PB, Askari O, Metghalchi H (2016) Reduction of nitrous oxide emissions from biological nutrient removal processes by thermal decomposition. Water Res 106:304–311
Abu-Zied BM, Schwieger W, Unger A (2008) Nitrous oxide decomposition over transition metal exchanged ZSM-5 zeolites prepared by the solid-state ion-exchange method. Appl Catal B 84(1–2):277–288
Rivallan M, Ricchiardi G, Bordiga S, Zecchina A (2009) Adsorption and reactivity of nitrogen oxides (NO2, NO, N2O) on Fe-zeolites. J Catal 264(2):104–116
Khan NA, Kennedy EM, Dlugogorski BZ, Adesina AA, Stockenhuber M (2014) Partial oxidation of methane with nitrous oxide forms synthesis gas over cobalt exchanged ZSM-5. Catal Commun 53:42–46
Ruiz-Trejo E, Boldrin P, Medley-Hallam JL, Darr J, Atkinson A, Brandon NP (2015) Partial oxidation of methane using silver/gadolinia-doped ceria composite membranes. Chem Eng Sci 127:269–275
Indarto A (2016) Partial oxidation of methane to methanol with nitrogen dioxide in dielectric barrier discharge plasma: experimental and molecular modeling. Plasma Sources Sci Technol 25(2):025002
Lee S-J, Ryu I-S, Kim B-M, Moon S-H (2011) A review of the current application of N2O emission reduction in CDM projects. Int J Greenhouse Gas Control 5(1):167–176
Zeng HC, Pang XY (1997) Catalytic decomposition of nitrous oxide on alumina-supported ruthenium catalysts Ru/Al2O3. Appl Catal B 13:113–122
Slagterna Å, Swaan HM, Olsbyea U, Dahla IM, Mirodatosb C (1998) Catalytic partial oxidation of methane over Ni-, Co- and Fe-Based catalysts. Catal Today 46:107–115
Yuzaki K, Yarimizu T, Aoyagi K, Ito S, Kunimori K (1998) Catalytic decomposition of N2O over supported Rh catalysts: effects of supports and Rh dispersion. Catal Today 45:129–134
Marnasidou KG, Voutetakis SS, Tjatjopoulos GJ, Vasalos IA (1999) Catalytic partial oxidation of methane to synthesis gas in a pilot-plant-scale spouted-bed reactor. Chem Eng Sci 54:3691–3699
Ohno Y, Kimura K, Bi M, Matsushima T (1999) The translational energy of desorbing products in NO and N2O decomposition on Pd (110). J Chem Phys 110(17):8221–8224
Huang C, Zhu Y, Wang X, Liu X, Wang J, Zhang T (2017) Sn promoted BaFeO3-δ catalysts for N2O decomposition: optimization of Fe active centers. J Catal 347:9–20
Li K, Wang XF, Zeng HC (1997) Kinetics of N2O Decomposition on a RuO2/Al2O3 Catalyst. Chem Eng Res Des 75(8):807–812
Zhu J, Vanommen J, Lefferts L (2006) Effect of surface OH groups on catalytic performance of yittrium-stabilized ZrO2 in partial oxidation of CH4 to syngas. Catal Today 117(1–3):163–167
Fleys M, Shan W, Simon Y, Marquaire P (2007) Detailed kinetic study of the partial oxidation of methane over La2O3 catalyst. Part 1: experimental results. Ind Eng Chem Res 46:1063–1068
Fleys M, Simon Y, Marquaire P (2007) Detailed kinetic study of the partial oxidation of methane over La2O3 catalyst. Part 2: mechanism. Ind Eng Chem Res 46:1069–1078
Dacquin JP, Lancelot C, Dujardin C, Da Costa P, Djega-Mariadassou G, Beaunier P, Kaliaguine S, Vaudreuil S, Royer S, Granger P (2009) Influence of preparation methods of LaCoO3 on the catalytic performances in the decomposition of N2O. Appl Catal B 91(3–4):596–604
Wu Y, Cordier C, Berrier E, Nuns N, Dujardin C, Granger P (2013) Surface reconstructions of LaCo1-xFexO3 at high temperature during N2O decomposition in realistic exhaust gas composition: impact on the catalytic properties. Appl Catal B 140–141:151–163
Ivanov DV, Pinaeva LG, Sadovskaya EM, Isupova LA (2016) Isotopic transient kinetic study of N2O decomposition on LaMnO3+δ. J Mol Catal A 412:34–38
Yang Z, Zhang Y, Ding W (2014) Investigation on the reforming reactions of coke-oven-gas to H2 and CO in oxygen-permeable membrane reactor. J Membr Sci 470:197–204
Cheng H, Lu X, Hu D, Zhang Y, Ding W, Zhao H (2011) Hydrogen production by catalytic partial oxidation of coke oven gas in BaCo0.7Fe0.2Nb0.1O3-δ membranes with surface modification. Int J Hydrogen Energy 36(1):528–538
Zhu X, Li Q, He Y, Cong Y, Yang W (2010) Oxygen permeation and partial oxidation of methane in dual-phase membrane reactors. J Membr Sci 360(1–2):454–460
Song S, Zhang P, Han M, Singhal SC (2012) Oxygen permeation and partial oxidation of methane reaction in Ba0.9Co0.7Fe0.2Nb0.1O3-δ oxygen permeation membrane. J Membr Sci 415–416:654–662
Jin W, Li S, Huang P, Xu N, Shi J, Lin YS (2000) Tubular lanthanum cobaltite perovskite-type membrane reactors for partial oxidation of methane to syngas. J Membr Sci 166:13–22
Li S, Jin W, Huang P, Xu N, Shi J, Lin YS (2000) Tubular lanthanum cobaltite perovskite type membrane for oxygen permeation. J Membr Sci 166:51–61
Figen HE, Baykara SZ (2018) Effect of ruthenium addition on molybdenum catalysts for syngas production via catalytic partial oxidation of methane in a monolithic reactor. Int J Hydrogen Energy 43(2):1129–1138
Swamy CS, Christopher J (1992) Decomposition of N2O on perovskite-related oxides. Catal Rev 34(4):409–425
Babiniec SM, Coker EN, Miller JE, Ambrosini A (2015) Investigation of LaxSr1-xCoyM1-yO3-δ (M = Mn, Fe) perovskite materials as thermochemical energy storage media. Sol Energy 118:451–459
Koh A, Chen L, Keeleong W, Johnson B, Khimyak T, Lin J (2007) Hydrogen or synthesis gas production via the partial oxidation of methane over supported nickel-cobalt catalysts. Int J Hydrogen Energy 32(6):725–730
Kangsadan T, Srisurat T, Kim P, Laosiripojana N, Jindasuwan S, Hartley UW (2015) Hydrogen production from palmitic acid through autothermal reforming: thermodynamic analysis. Eng J 19(4):153–165
Zhang J, Haribal V, Li F (2017) Perovskite nanocomposites as effective CO2-splitting agents in a cyclic redox scheme. Sci Adv 3(8):e1701184
Kapteijn F, RodriguezMirasol J, Moulijn JA (1996) Heterogeneous catalytic decomposition of nitrous oxide. Appl Catal B 9:25–64
Smith JM, Van Ness HC, Abbott MM (2005) Introduction to chemical engineering thermodynamics, 7th edn. McGraw Hill, New York
Felder RM, Rousseau RW (2005) Elementary principles of chemical processes, 3rd edn. Wiley, New York
Yaws CL (1996) Handbook of thermodynamics diagram. Gulf Publishing Company, Houston
Standard thermodynamic properties of chemical substances.
Speight JG (2017) Rules of thumb for petroleum engineers, 1st edn. Wiley, Hoboken
Pavlova S, Kapokova L, Bunina R, Alikina G, Sazonova N, Krieger T, Ishchenko A, Rogov V, Gulyaev R, Sadykovab V, Mirodatosc C (2012) Syngas production by CO2 reforming of methane using LnFeNi(Ru)O3 perovskites as precursors of robust catalysts. Catal Sci Technol 2:2099–2108
Xu X, Li L, Yu F, Peng H, Fang X, Wang X (2017) Mesoporous high surface area NiO synthesized with soft templates: remarkable for catalytic CH4 deep oxidation. Mol Catal 441:81–91
Bai G, Dai H, Deng J, Liu Y, Qiu W, Zhao Z, Li X, Yang H (2013) The microemulsion preparation and high catalytic performance of mesoporous NiO nanorods and nanocubes for toluene combustion. Chem Eng J 219:200–208
Araujo GCd, Lima SMd, Assaf JM, Peña MA, Fierro JLG, do Carmo Rangel M (2008) Catalytic evaluation of perovskite-type oxide LaNi1-xRuxO3 in methane dry reforming. Catal Today 133–135:129–135
Shan W (2003) Reduction property and catalytic activity of Ce1-XNiXO2 mixed oxide catalysts for CH4 oxidation. Appl Catal A General 246(1):1–9
Cihlar J, Vrba R, Castkova K, Cihlar J (2017) Effect of transition metal on stability and activity of La-Ca-M-(Al)-O (M = Co, Cr, Fe and Mn) perovskite oxides during partial oxidation of methane. Int J Hydrogen Energy 42(31):19920–19934
Zhao K, Zheng A, Li H, He F, Huang Z, Wei G, Shen Y, Zhao Z (2017) Exploration of the mechanism of chemical looping steam methane reforming using double perovskite-type oxides La1.6Sr0.4FeCoO6. Appl Catal B 219:672–682
Li R, Yu C, Shen S (2002) Partial oxidation of methane to syngas using lattice oxygen of La1-xSrxFeO3 perovskite oxide catalysts instead of molecular oxygen. J Nat Gas Chem 117:137–144
Singha RK, Shukla A, Yadav A, Sivakumar Konathala LN, Bal R (2017) Effect of metal-support interaction on activity and stability of Ni-CeO2 catalyst for partial oxidation of methane. Appl Catal B 202:473–488
Zheng Y, Li K, Wang H, Tian D, Wang Y, Zhu X, Wei Y, Zheng M, Luo Y (2017) Designed oxygen carriers from macroporous LaFeO3 supported CeO2 for chemical-looping reforming of methane. Appl Catal B 202:51–63
Pal P, Singha RK, Saha A, Bal R, Panda AB (2015) Defect-induced efficient partial oxidation of methane over nonstoichiometric Ni/CeO2 nanocrystals. J Phys Chem C 119(24):13610–13618
Acknowledgements
Grants from Thailand Research Fund (MSD5910030) and National Research Council of Thailand (KMUTNB-GOV-58-54) and (KMUTNB-GOV-59-43) are acknowledged for the support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Khajonvittayakul, C., Tongnan, V., Kangsadan, T. et al. Thermodynamic and mechanism study of syngas production via integration of nitrous oxide decomposition and methane partial oxidation in the presence of 10%NiO–La0.3Sr0.7Co0.7Fe0.3O3−δ. Reac Kinet Mech Cat 127, 839–855 (2019). https://doi.org/10.1007/s11144-019-01600-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-019-01600-1