
Abstract
Red arils of Pithecellobium dulce fruit, commonly known as guamuchil, show high antioxidant (AOx) and α-glucosidase inhibitory (IαG) activities, which have been mainly associated with the content of unknown anthocyanins. In this study, the AOx (i.e., DPPH and ABTS as Trolox equivalents, μmol TE/g) and IαG (as half-maximal inhibitory concentration, IC50, mg/mL) activities of the anthocyanin-rich fraction (ARF) obtained from red arils were contrasted with those of the methanol extract (ME), and the main ARF anthocyanins were characterized by HPLC-DAD-ESI-MS, GC-MS and 1H-NMR. The AOx and IαG values of the ARF (DPPH = 597.8; ABTS = 884.01; IαG = 0.06) were better than those of the ME (DPPH = 41.5; ABTS = 142.3; IαG = 17.5); remarkably, the ARF IαG value was about 42 times lower than that of acarbose. The main anthocyanins in ARF were pelargonidin 3-O-glucoside and cyanidin 3-O-glucoside. Thus, the consumption of red P. dulce arils could provide health benefits for prevention/treatment of chronic degenerative diseases such as diabetes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pithecellobium dulce (Roxb.) Benth. (Fabaceae) is a tree native to America from where it was introduced into Asia and East of Africa. In Mexico, P. dulce shows a wide distribution and is commonly known as guamuchil [1]. This tree is used as wood and for leather tanning, whereas its fruits are used as food and feed [1], and in traditional medicine to treat different symptoms/illnesses (e.g., dysentery, inflammation) [2]. Scientific studies of P. dulce have identified several chemical compounds (e.g., steroids, saponins, lipids, tannins, phenolics) and demonstrated several biological activities that play important roles in disease prevention (e.g., antioxidant, antidiabetic, anti-inflammatory, inhibitory of digestive enzymes) [3].
Pithecellobium dulce fruits are pods (10–15 cm long) containing white or red sweet arils [1] that differ in chemical composition and biological activities. Several red aril properties are better than those of white arils: DPPH antioxidant activity (AOx) (223.4 and 170.9 mg of vitamin C equivalents, respectively), inhibitory activity of α-glucosidase (IαG) (IC50 = 2.9 and 16.7 mg/mL, respectively), and content of total phenolics (517.8 and 392.2 mg gallic acid equivalents per 100 g f.w., respectively) [4]. The differences in biological activities of the two P. dulce arils have not been explained, but they could be associated to the unidentified anthocyanins in red arils [4, 5] as suggested by the activities previously reported for these compounds (e.g., antimutagenic, anti-inflammatory, antioxidant, inhibitory of digestive enzymes) [6]. The most abundant anthocyanidins in nature are cyanidin (30%) > delphinidin (22%) ≈ pelargonidin (18%) > peonidin, malvinidin and petunidin (20%); and they are commonly found as glycosides (anthocyanins) of glucose, galactose and rhamnose [6].
The aim of this study was to identify the main pigments of an anthocyanin-rich fraction of P. dulce fruit that showed high antioxidant and α-glucosidase inhibitory activities; this information of P. dulce fruit will contribute to the consumer appreciation and to be considered as functional food.
Materials and Methods
Reagents
The reagents were of analytical grade from Sigma-Aldrich (St. Louis, MO, US) and the solvents were of HPLC grade from Baker Inc. (Phillipsburg, NJ, US).
Plant Material
The guamuchil red aril fruits (Pithecellobium dulce Roxb. Benth.) were hand-harvested at commercial ripeness in the municipality of Culiacan, Sinaloa, Mexico, during May and June 2015. The species was corroborated by PhD Rito Vega-Aviña, School of Agronomy of the Autonomous University of Sinaloa. Arils (moisture 76.6%) were recovered from the pods, freeze-dried, milled, and passed through a 0.44 mm mesh. The obtained flour was stored at −20 °C/darkness.
Methanol Extract Preparation
One gram of flour was mixed with methanol (1:20 w/v), sonicated (FS 30 h, Fisher Sci., US) for 15 min, centrifuged (10,000 rpm/ 20 min/ 4 °C) (RC5C, Sorvall® Instruments DuPont, US), and the supernatant was recovered. The resulting pellet was extracted again as described, the supernatants were mixed, and the solvent was evaporated under vacuum at 38 °C (R-124, BUCHI, US) to obtain the methanol extract (ME).
Preparation of the Anthocyanin-Rich Fraction (ARF)
Guamuchil flour (10 g) was extracted with acidified methanol (0.01% HCl) as described in the previous section to obtain the acidified methanol extract (aME). The ARF was obtained by solid phase extraction as previously reported [7]. A 300 mg/mL aME solution was prepared with acidified water (0.01% HCl). A Sep-pack C-18 cartridge (CHROMAFIX, Macherey-Nagel, US) was conditioned with 2 mL of acidified water and then 1 mL of the aME solution was passed through it. Undesirable compounds (e.g., sugars, organic acids, lipids) were washed away the cartridge with 2 mL of acidified water and 2 mL of ethyl acetate. Then, the ARF was eluted with 2 mL of acidified methanol (0.01% HCl) or the required volume until the eluting solvent was colorless. The obtained ARF was concentrated under vacuum at 38 °C and stored until use at −20 °C in darkness under N2(g) atmosphere.
Purification of the ARF Anthocyanins
Anthocyanins were purified by thin layer chromatography (TLC) (cellulose on TLC-PET foils, 20 × 20 cm, Sigma, US). The ARF was dissolved in methanol:H2O (2:1, v/v) (75 mg/mL), charged onto the TLC plate, and resolved with CH3CO2C2H5:HCO2H:CH3CO2H:H2O (100:11:11:26 v/v) [8]. The anthocyanins in the chromatographic bands were recovered with acidified methanol (0.2% TFA) and the solvent was removed at 40 °C under vacuum.
For sugar identification of the anthocyanin, 1 mg of each recovered anthocyanin was hydrolyzed with 2 mL of 2 N HCl at 90 °C for 2 h [9]. Each compound was separated through a Sep-pack C-18 cartridge (CHROMAFIX, Macherey-Nagel, US) previously conditioned with 2 mL of acidified water; then, sugars were eluted with acidified water (0.01% HCl) [10]. The recovered sugar fraction was freeze-dried and the residue was mixed with 100 μL of pyridine and 100 μL of BSTFA +1% TMCS (N,O-bis(trimethylsilyl)trifluoroacetamide in trimethylchlorosilane); the mixture was incubated under N2(g) atmosphere at 70 °C for 1 h to obtain the derivatized sugars [11], and the solvent was eliminated. The obtained sample was dissolved in hexane, passed through a PVDF filter (0.45 μm, Titan, US), and analyzed by gas chromatography-mass spectrometry (GC-MS). Standards of glucose and galactose were derivatized and analyzed as described, and used for sugar identification.
HPLC-DAD-ESI-MS Analysis of the ARF
The anthocyanins in the ARF were identified by HPLC-DAD-ESI-MS [12]. Five microliters of the ARF (1 mg/mL) was injected into the HPLC-DAD ACCELA coupled with the ESI and the LTQ-XL mass spectrometer (Thermo Sci., US). Separation was carried out with a FORTIS C18 column (50 × 2.1 mm, 3 μm) (Fortis Technologies, US). The mobile phase contained 1% phosphoric acid (A) and acetonitrile (B), and the following gradient was used: 0–2 min, 100% A; 2–20 min, 80% A; 20–30 min 60% A; and 30–35 min 100% B; flow rate 0.3 mL/min.
For compound identification, the mass spectra of the eluted peak were acquired in positive mode over the range m/z 115–1000, resolution of 30,000. The MS worked at 275 °C and 35 V in the capillary tube, source voltage at 5 kV, and tube lens voltage at −200 V. The sheet gas flow was 8 units, whereas the flow rate of both the auxiliary and sweep gases were 0 units. For tandem mass spectrometry analysis (MSn), ultrahigh-purity helium was used for the collision-induced dissociation (CID) and the energy was adjusted between 15 and 25%. Data were acquired and processed using the Xcalibur 2.2 software. Peaks were identified by their fragmentation patterns and by comparison with MS data published in the literature.
Carbohydrate Identification by GC-MS
The derivatized sugars were analyzed in a GC Agilent (HP 6890) coupled to a mass selective detector (5973 Network) (Agilent Technologies, US). The separation was performed using a QUADREX 007 CARBOWAX 20 M column (30 m × 0.25 mm i.d., film thickness 0.25 μm) (Quadrex Corporation, US). Helium was used as the carrier gas at 0.9 mL/min. The injector temperature was 250 °C and the oven temperature varied as follows: initial 60 °C, lineal gradient 5 °C/ min up to 200 °C and 10 °C/ min up to 275 °C, and 275 °C until the end of the chromatographic run (60 min). The temperatures in the detector and quadrupole were 245 °C and 150 °C, respectively. The mass spectrometer was operated in mode of electron-impact ionization at 70 eV. Spectra were collected in the range 50–800 u at 2 scans/s [11]. The compounds were identified by comparing the retention times and mass spectra with those of derivatized commercial standards.
1H-NMR Analysis
The 1H-NMR spectra were recorded at 400 MHz with a Bruker Avance III spectrometer (Bruker, Germany) at 30 °C. 1H NMR chemical shifts were reported in ppm referenced to residual solvent resonances (1H NMR, 4.78 and 3.31 ppm for CHD2OH in methanol-d4). Coupling constants (J) were given in Hertz (Hz).
Inhibitory Activity of α-Glucosidase (IαG)
The IαG was measured as previously published [13], using the Saccharomyces cerevisiae α-glucosidase (21 U/mg, Sigma-Aldrich, US). Sample and acarbose (positive control) were dissolved in DMSO and dilutions were prepared. In a 96 microwell plate, 50 μL of sample or acarbose at different concentrations were mixed with 100 μL of α-glucosidase (1 U/mL in 0.1 M PBS, pH 6.9). The final concentration of DMSO per well was ≤10%. The microplate was incubated at 37 °C for 10 min (Stat Fax-2200, Awareness Technology, US); then, 50 μL of 5 mM p-nitrophenyl-α-D-glucopyranoside was added and incubated again under the same conditions. Finally, the absorbance at 405 nm was measured (Multiskan Bichromatic, Fisher Scientific, US). Solutions without inhibitor and without substrate were used as controls of reaction and color, respectively.
The percentage of IαG (% IαG) was calculated with the equation % IαG = ((Ac-As)/Ac) × 100; where Ac and As are the absorbances of the control (without inhibitor) and the sample or acarbose, respectively. The results were expressed as the half-maximal inhibitory concentration of the enzyme activity (IC50).
Antioxidant Activity (AOx) by DPPH
The DPPH AOx was measured as previously published [14]. A 0.2 mL aliquot of ME (5 mg/mL) or ARF (0.3 mg/mL) was mixed with 1.8 mL of 150 μM DPPH (1,1-diphenyl-2-picrylhydrazyl) radical in methanol, the mixture was incubated at 27 °C/darkness for 30 min, and the absorbance was measured at 517 nm. The results were expressed as micromoles of Trolox equivalents per gram of sample (μmol TE/g).
Antioxidant Activity by ABTS
The ABTS AOx was measured as previously published [15]. The ABTS (2,2-azino-bis-3-ethylbenzothiazoline-6-sulfonic acid) radical was generated by mixing 5 mL of 7 mM ABTS with 88 μL of 140 μM potassium persulfate, and the mixture was allowed to stand for 12 h at 27 °C/darkness. The ABTS radical was diluted with methanol to reach an absorbance of 0.70 (± 0.02) at 734 nm. For the assay, 50 μL of sample and 1.95 mL of diluted ABTS were mixed, the mixture was allowed to stand for 10 min and its absorbance was measured at 734 nm. The results were expressed as μmol TE/g.
Statistical Analysis
Measurements were made at least by triplicate and registered as the mean ± the standard deviation. Data was analyzed by one-way ANOVA and the means were contrasted by the Fisher test (α = 0.05), using the STATGRAPHICS v. 5.1 software (Statistical Graphics Corporation, US). For the IαG assay, the IC50 was calculated with the GraphPad Prism v. 6.1 software (GraphPad Prism ® software, Inc., US).
Results and Discussion
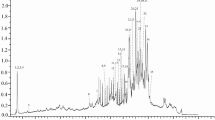
The HPLC-DAD analysis of the ARF (yield = 0.192% d.w.) showed two main peaks with relative areas of 25.32% (1) and 65.22% (2); their UV-vis spectra were characteristic of glycosylated anthocyanins at position 3 (λmax 505–520 nm, shoulder 430 nm) (Fig. 1a) [16, 17]. The ESI-MS spectrum of peak 1 (22.29 min) showed a protonated molecule [M + H]+ at m/z 449 whose MS2 yielded a main fragment ion at m/z 287 by the loss of an hexose residue (162 Da) of the cyanidin aglycone [18, 19] (Fig. 1b). GC-MS analysis of the derivatized sugar obtained from the hydrolyzed anthocyanin showed the same retention time (TR: glucose = 22.32 min, galactose = 22.68 min) and fragmentation pattern (abundance ratio 191/21: glucose >1, galactose <1) than that of the glucose standard, this fragmentation pattern is reported for the first time (Online Resources 1 and 2 ). Based on this information, peak 1 was identified as cyanidin 3-O-glucoside (C3G). The peak 2 (24.89 min) showed the protonated molecule [M + H]+ at m/z 433 and its MS2 main fragment ion at m/z 271, corresponding to the loss of a hexose of the pelargonidin aglycone (Fig. 1c) [18, 19], and the hexose was also characterized as glucose (Online Resources 1 and 2 ); thus peak 2 was identified as pelargonidin 3-O-glucoside (P3G).

Spectroscopic and spectrometric data of the anthocyanin-rich fraction: HPLC chromatogram (λ = 520 nm) and UV spectra of the main peaks 1 and 2 (a); and mass spectra of cyanidin 3-O-glucoside 1 (b) and pelargonidin 3-O-glucoside 2 (c)
The identity of the TLC purified anthocyanins 1 and 2 was corroborated by 1H-NMR (methanol-d4). The spectrum of 1 (yield = 13.7 mg) showed the proton signals for cyanidin (δ, ppm) (Online Resource 3 ): 9.02 (H4, s), 8.26 (H6′, dd, J1 = 2.4, J2 = 8.8 Hz), 8.05 (H2′, d, J = 2.4 Hz), 7.03 (H5′, d, J = 8.8 Hz), 6.89 (H8, d, J = 2 Hz), and 6.65 (H6, d, J = 2 Hz). The presence of the glucose fraction was confirmed by the signal for the anomeric proton at 5.29 ppm (H1″, d, J = 7.6 Hz) and the value of the coupling constant showed a β-configuration [20]. The proton signals of 2 (yield = 24.3 mg) corresponded to pelargonidin (δ, ppm) (Online Resource 4 ): 9.08 (H4, s), 8.60 (H2′, H6′, d, J = 9.2 Hz), 7.05 (H3′, H5′, d, J = 9.2 Hz), 6.92 (H8, d, J = 2 Hz), 6.67 (H6, d, J = 2 Hz), and 5.27 (H1″, d, J = 7.6 Hz); and the signal for the anomeric proton of the β-glucosidic bond was observed at 5.27 (H1″, d, J = 7.6 Hz). These spectroscopic and spectrometric data corresponded with those previously reported for the cyanidin 3-O-β-glucoside and pelargonidin 3-O-β-glucoside [9, 10, 18]. Cyanidin and pelargonidin are common in food [6]; the main anthocyanin component in purple corn and black soybean is C3G [21], whereas in strawberry and red merlot bean is P3G [19] as in the P. dulce arils. Compared to the P. dulce anthocyanins, the ripe pods of Prosopis spp. (Fabaceae) showed anthocyanins derived of cyanidin, malvinidin, peonidin, and petunidin, being cyanidin 3-O-hexoside the main component [22].
The consumption of foods enriched in anthocyanins has been associated with a lower risk of diabetes and hypertension; these compounds inhibit the digestive enzymes α-amylase and α-glucosidase and decrease the postprandial hyperglycemia [23]. The ARF showed the best IαG value, being up to 42 times lower than that of acarbose (Table 1). In this regard, the IαG of an ARF from black rice (90% of C3G) was 70 times lower than that of acarbose [24]; in the present study the ARF of P. dulce arils contained C3G but in lower proportion. Other studies have demonstrated the IαG of cyanidin and derivative compounds and their synergism with acarbose [25]. Cyanidin 3-galactoside and C3G were better enzyme inhibitors of the intestinal sucrase than acarbose [26]. In addition, it was demonstrated that C3G inhibits the α-glucosidase activity by competitive and non-competitive mechanisms [27]. Matsui et al. [28] showed that diacylated anthocyanins inhibited the intestinal maltase following the order pelargonidin > peonidin ≈ cyanidin; they demonstrated that the substitution at 4′ of ring B is important for the IαG. In this regard, it has been suggested that certain structural characteristics of anthocyanins are essential for their inhibitory effect on digestive enzymes, e.g., C2-C3 double bond on ring C, methoxyl groups at positions 4 and 7, number of hydroxyls in ring B, and glycosylation [23, 26, 28]. The chemical structures of C3G and P3G are similar and both anthocyanins could be involved in the IαG of the ARF.
Most anthocyanins have shown AOx activity, which has been associated with disease prevention/treatment (e.g., cardiovascular diseases, diabetes, cancer) [29]. This study showed that the AOx of the ARF was about 6 and 14 times higher than that of the ME by ABTS and DPPH, respectively (Table 1). The highest difference in AOx observed with the DPPH assay may be because this method is better to measure the activity of hydrophilic compounds such as anthocyanins, corresponding with the good correlation between DPPH AOx and anthocyanin content reported by other authors [30]. Pío-León et al. [4] showed higher AOx values (mg EVC) in the ME of P. dulce red arils (DPPH = 223.4, ABTS = 224.8) than those of ME from white arils (DPPH = 170.9, ABTS = 155.9), which was associated with a higher content of phenolics and anthocyanins in red than white arils; assumption that was supported by a bioguided ME fractionation (unpublished data). Ponmozhi et al. [3] reported higher AOx values in the aME of P. dulce arils (color was not indicated) (DPPH = 66.16%, metal chelating activity = 92.95%) than those obtained in the ME (DPPH = 40.33%, metal chelating activity = 11.08%), which could be explained by a more efficient extraction of anthocyanins and phenolics with acidified methanol.
The association between the AOx activity and the anthocyanin content of the P. dulce red arils is similar to that found in previous reports. Cyanidin and its glycosides show a wide distribution in plants [29] and are the best antioxidants in berries [31]. Jakobek et al. [30] showed high correlation between the anthocyanin content and DPPH AOx of red fruit juices; the highest AOx values were obtained in chokeberry (Aronia melanocarpa) and elderberry (Sambucus nigra), both enriched in C3G, whereas a lower AOx value was observed in strawberry (Fragaria ananassa) that contains mainly P3G. Another study contrasted the AOx of different cyanidin 3-O-glycosides isolated from Ribes biebersteinii Berl. and found that C3G showed the highest value [32]. In general, the AOx of anthocyanins depends on their structure and the method used in the evaluation, being DPPH and ABTS some of the most commonly used assays [29]. In this regard, Kähkönen and Heinonen [33] measured the DPPH AOx of anthocyanidins and anthocyanins and the order of activity for the aglycones was delphinidin > cyanidin ≈ peonidin > pelargonidin > malvidin > petunidin, finding also that aglycones > monoglycosides (glucose > galactose) > diglycosides. In addition, several reports have shown that the AOx increases with the number of hydroxyls on ring B, being the 4′ position determinant for the activity, but a methoxyl group at the 5′ position decreases the AOx [29, 33]. Consequently, although the content of C3G in P. dulce red arils is lower than that of P3G, its contribution to the registered AOx activity could be high. Nevertheless, the biological activity of these compounds depends on their bioavailability; in this regard, intact anthocyanins have been found in the blood after intestinal absorption [34], whereas other reports suggested they are metabolized by the intestinal microflora and transformed in metabolites that also showed remarkable biological activities [34, 35].
The anthocyanin-rich fraction of P. dulce red arils (ARF), characterized by its high antioxidant and α-glucosidase inhibitory activities, contains pelargonidin 3-O-glucoside and cyanidin 3-O-glucoside as the main anthocyanins; thus, the consumption of this fruit or its ARF could provide health benefits considering that anthocyanin-containing foods help to prevent or treat chronic degenerative diseases [6, 29].
Abbreviations
- AOx:
-
Antioxidant activity
- IαG:
-
α-glucosidase inhibitory
- ME:
-
Methanol extract
- aME:
-
Acidified methanol extract
- ARF:
-
Anthocyanin-rich fraction
- C3G:
-
Cyanidin 3-O-glucoside
- P3G:
-
Pelargonidin 3-O-glucoside
References
Parrotta J (1991) Pithecellobium dulce (Roxb.) Benth. Guamúchil, Madras thorn., vol SO-ITS-SM-40. Forest Service, North Carolina
UNAM (2009) Biblioteca Digital de la Medicina Tradicional Mexicana. Universidad Nacional Autónoma de México. http://www.medicinatradicionalmexicana.unam.mx/termino.php?l=1&t=diarrea&letra=D&opcion=D&id=2968. Accessed 23 Feb 2018
Ponmozhi P, Geethá M, Saravana Kumar M et al (2011) Extraction of anthocyanin and analysing its antioxidant properties from Pithecellobium dulce fruit pericarp. Asian J Pharm Clin Res 4(Suppl. 1):41–45 https://innovareacademics.in/journal/ajpcr/Vol44Suppl41/377.pdf
Pío-León JF, Díaz-Camacho S, Montes-Avila J, López-Angulo G, Delgado-Vargas F (2013) Nutritional and nutraceutical characteristics of white and red Pithecellobium dulce (Roxb.) Benth fruits. Fruits 68(5):397–408. https://doi.org/10.1051/fruits/2013084
Wall-Medrano A, Gonzalez-Aguilar GA, Loarca-Pina GF et al (2016) Ripening of Pithecellobium dulce (Roxb.) Benth. [guamuchil] fruit: physicochemical, chemical and antioxidant changes. Plant Foods Hum Nutr 71(4):396–401
Andersen ØM, Jordheim M (2006) The anthocyanins. In: Andersen ØM, Markham KR (eds) Flavonoids. Chemistry, Biochemistry and Applications, 1st edn. Taylor and Francis Group, New York, pp 471–551. https://doi.org/10.1201/9781420039443.ch10
Rodriguez-Saona LE, Wrolstad RE (2001) Extraction, isolation, and purification of anthocyanins. Curr Protoc Food Analyt Chem F1.1.1–F1.1.11. https://doi.org/10.1002/0471142913.faf0101s00
Hema A, Palé E, Duez P et al (2012) Two diglucosylated anthocyanins from Combretum paniculatum flowers. Nat Sci 4(3):166–169. https://doi.org/10.4236/ns.2012.43024
Tatsuzawa F, Saito N, Yukawa T, Honda T, Shinoda K, Kato K, Miyoshi K (2014) Acylated cyanidin 3,7-diglucosides in the red-purple flowers of Sophronitis wittigiana (Orchidaceae). J Jpn Soc Hortic Sci 83(1):64–71. https://doi.org/10.2503/jjshs2501.CH-2084
Qin CG, Li Y, Niu WN, Ding Y, Shang XY, Xu CL (2011) Composition analysis and structural identification of anthocyanins in fruit of waxberry. Czech J Food Sci 29(2):171–180. https://doi.org/10.17221/17177/12010-CJFS
López-Angulo G, Montes-Avila J, Díaz-Camacho SP, Vega-Aviña R, Báez-Flores ME, Delgado-Vargas F (2016) Bioactive components and antimutagenic and antioxidant activities of two Echeveria DC. species. Ind Crop Prod 85:38–48. https://doi.org/10.1016/j.indcrop.2016.1002.1044
Durst RW, Wrolstad RE (2001) Separation and characterization of anthocyanins by HPLC. Curr Protoc Food Analyt Chem F1.3.1–F1.3.13. https://doi.org/10.1002/0471142913.faf0103s00
da Silva Pinto M, Kwon Y-I, Apostolidis E, Lajolo FM, Genovese MÍ, Shetty K (2008) Functionality of bioactive compounds in Brazilian strawberry (Fragaria x ananassa Duch.) cultivars: evaluation of hyperglycemia and hypertension potential using in vitro models. J Agric Food Chem 56(12):4386–4392
Brand-Williams W, Cuvelier ME, Berset C (1995) Use of a free radical method to evaluate antioxidant activity. LWT-Food Sci Technol 28(1):25–30. https://doi.org/10.1016/S0023-6438(1095)80008-80005
Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C (1999) Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med 26(9–10):1231–1237
Aguilar O, Hernandez-Brenes C (2015) Use of modified phenolic thyme extracts (Thymus vulgaris) with reduced polyphenol oxidase substrates as anthocyanin color and stability enhancing agents. Molecules 20(12):22422–22434
Jansom C, Bhamarapravati S, Itharat A (2008) Major anthocyanin from ripe berries of Cleistocalyx nervosum var. paniala. Thammasat Med J 8(3):394–370 https://www.researchgate.net/publication/242190711
Cui C, Zhang S, You L, Ren J, Luo W, Chen W, Zhao M (2013) Antioxidant capacity of anthocyanins from Rhodomyrtus tomentosa (Ait.) and identification of the major anthocyanins. Food Chem 139(1–4):1–8
Kajdžanoska M, Gjamovski V, Stefova M (2010) HPLC-DAD-ESI-MSn identification of phenolic compounds in cultivated strawberries from Macedonia. Maced J Chem Chem Eng 29(2):181–194 http://www.mjcce.org.mk/index.php/MJCCE/article/view/165
Berardini N, Schieber A, Klaiber I, Beifuss U, Carle R, Conrad J (2005) 7-O-Methylcyanidin 3-O-β-D-galactopyranoside, a novel anthocyanin from mango (Mangifera indica L.) cv. ‘Tommy Atkins’ peels. Z Naturforsch B 60:801–804. https://doi.org/10.1515/znb-2005-0718
Lee JH, Kang NS, Shin S-O, Shin SH, Lim SG, Suh DY, Baek IY, Park KY, Ha TJ (2009) Characterisation of anthocyanins in the black soybean (Glycine max L.) by HPLC-DAD-ESI/MS analysis. Food Chem 112(1):226–231. https://doi.org/10.1016/j.foodchem.2008.1005.1056
Pérez MJ, Cuello AS, Zampini IC, Ordoñez RM, Alberto MR, Quispe C, Schmeda-Hirschmann G, Isla MI (2014) Polyphenolic compounds and anthocyanin content of Prosopis nigra and Prosopis alba pods flour and their antioxidant and anti-inflammatory capacities. Food Res Int 64:762–771
Rasouli H, Hosseini-Ghazvini SM, Adibi H et al (2017) Differential alpha-amylase/alpha-glucosidase inhibitory activities of plant-derived phenolic compounds: a virtual screening perspective for the treatment of obesity and diabetes. Food Funct 8(5):1942–1954
Bae IY, An JS, Oh IK, Lee HG (2017) Optimized preparation of anthocyanin-rich extract from black rice and its effects on in vitro digestibility. Food Sci Biotechnol 26(5):1415–1422. https://doi.org/10.1007/s10068-10017-10188-x
Adisakwattana S, Charoenlertkul P, Yibchok-Anun S (2009) Alpha-glucosidase inhibitory activity of cyanidin-3-galactoside and synergistic effect with acarbose. J Enzyme Inhib Med Chem 24(1):65–69
Akkarachiyasit S, Charoenlertkul P, Yibchok-Anun S et al (2010) Inhibitory activities of cyanidin and its glycosides and synergistic effect with acarbose against intestinal alpha-glucosidase and pancreatic alpha-amylase. Int J Mol Sci 11(9):3387–3396
He H, Lu YH (2013) Comparison of inhibitory activities and mechanisms of five mulberry plant bioactive components against alpha-glucosidase. J Agric Food Chem 61(34):8110–8119
Matsui T, Ueda T, Oki T, Sugita K, Terahara N, Matsumoto K (2001) Alpha-glucosidase inhibitory action of natural acylated anthocyanins. 1. Survey of natural pigments with potent inhibitory activity. J Agric Food Chem 49(4):1948–1951
Martin Bueno J, Ramos-Escudero F, Sáez-Plaza P et al (2012) Analysis and antioxidant capacity of anthocyanin pigments. Part I: general considerations concerning polyphenols and flavonoids. Crit Rev Anal Chem 42(2):102–125. https://doi.org/10.1080/10408347.10402011.10632312
Jakobek L, Šeruga M, Medvidović-Kosanović M et al (2007) Anthocyanin content and antioxidant activity of various red fruit juices. Deut Lebensm-Rundsch 103:58–64 https://bib.irb.hr/datoteka/210741.Jakobek_et_al_DLR_10322007.10322058-10322064.PDF
Lee SG, Vance TM, Nam TG, Kim DO, Koo SI, Chun OK (2015) Contribution of anthocyanin composition to total antioxidant capacity of berries. Plant Foods Hum Nutr 70(4):427–432
Delazar A, Khodaie L, Afshar J, Nahar L, Sarker S (2010) Isolation and free-radical-scavenging properties of cyanidin 3-O-glycosides from the fruits of Ribes biebersteinii Berl. Acta Pharma 60(1):1–11
Kähkönen MP, Heinonen M (2003) Antioxidant activity of anthocyanins and their aglycons. J Agric Food Chem 51(3):628–633
Kay CD, Pereira-Caro G, Ludwig IA, Clifford MN, Crozier A (2017) Anthocyanins and flavanones are more bioavailable than previously perceived: a review of recent evidence. Annu Rev Food Sci Technol 8:155–180
Gowd V, Bao T, Wang L, Huang Y, Chen S, Zheng X, Cui S, Chen W (2018) Antioxidant and antidiabetic activity of blackberry after gastrointestinal digestion and human gut microbiota fermentation. Food Chem 269:618–627
Acknowledgements
Authors acknowledge this research was partially funded by CONACYT-Mexico and PROFAPI-Universidad Autonoma de Sinaloa.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Authors declare they have no conflict of interest.
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
López-Angulo, G., Montes-Avila, J., Sánchez-Ximello, L. et al. Anthocyanins of Pithecellobium dulce (Roxb.) Benth. Fruit Associated with High Antioxidant and α-Glucosidase Inhibitory Activities. Plant Foods Hum Nutr 73, 308–313 (2018). https://doi.org/10.1007/s11130-018-0693-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11130-018-0693-y