
Abstract
In oxygenic photosynthetic systems, the cytochrome b6f (Cytb6f) complex (plastoquinol:plastocyanin oxidoreductase) is a heart of the hub that provides connectivity between photosystems (PS) II and I. In this review, the structure and function of the Cytb6f complex are briefly outlined, being focused on the mechanisms of a bifurcated (two-electron) oxidation of plastoquinol (PQH2). In plant chloroplasts, under a wide range of experimental conditions (pH and temperature), a diffusion of PQH2 from PSII to the Cytb6f does not limit the intersystem electron transport. The overall rate of PQH2 turnover is determined mainly by the first step of the bifurcated oxidation of PQH2 at the catalytic site Qo, i.e., the reaction of electron transfer from PQH2 to the Fe2S2 cluster of the high-potential Rieske iron–sulfur protein (ISP). This point has been supported by the quantum chemical analysis of PQH2 oxidation within the framework of a model system including the Fe2S2 cluster of the ISP and surrounding amino acids, the low-potential heme b6L, Glu78 and 2,3,5-trimethylbenzoquinol (the tail-less analog of PQH2). Other structure–function relationships and mechanisms of electron transport regulation of oxygenic photosynthesis associated with the Cytb6f complex are briefly outlined: pH-dependent control of the intersystem electron transport and the regulatory balance between the operation of linear and cyclic electron transfer chains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
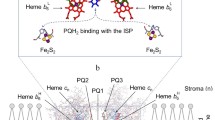
Oxygenic photosynthetic organisms (plants, algae, and cyanobacteria) assimilate carbon dioxide and produce molecular oxygen due to the energy of light quanta absorbed by the light-harvesting pigments of photosynthetic apparatus. The light-induced excitation of Photosystems I and II (PSI and PSII) initiate electron transfer along the chloroplast electron transport chain (ETC) from the water-oxidizing complex (WOC) of PSII to NADP+, a terminal physiological acceptor of PSI (Nelson and Yocum 2006; Mamedov et al. 2015). Two photosystems, PSII and PSI, are interconnected via the membrane-bound cytochrome b6f complex (Cytb6f), and mobile electron carriers, plastoquinone (PQ) and plastocyanin (Pc). The Cytb6f complex belongs to the Cytbc family of Cyt complexes (Berry et al. 2000; Crofts 2004a; Cramer et al. 2006; Tikhonov 2014, 2018; Malone et al. 2019, 2021; Sarewicz et al. 2021, 2023). This complex stands between PSII and PSI and operates as plastoquinol:plastocyanin oxidoreductase, oxidizing PQH2 and reducing Pc (Fig. 1). Two electrons, extracted from H2O by WOC (Bhowmick et al. 2023), are used to reduce PQ to plastoquinol (PQH2, the double-reduced form of PQ): H2O + PQ + 2 \({\text{H}}_{{{\text{out}}}}^{ + }\) → ½O2 + PQH2 + \({\text{2H}}_{{\text{in}}}^{ + }\). Here, \({\text{H}}_{{{\text{out}}}}^{ + }\) and \({\text{H}}_{{\text{in}}}^{ + }\) symbolize the protons taken up from the chloroplast stroma and released into the thylakoid lumen. The PQH2/PQ pool is a hub that mediates the intersystem electron transfer and serves as one of the crucial factors regulating oxygenic photosynthesis. PQH2 donates two electrons to the Cytb6f, which reduces cytochrome f (Cytf). The operation of the Cytb6f complex as the PQH2 oxidase is associated with the following events: (1) the formation of PQH2 in PSII, (2) the PQH2 diffusion in the thylakoid membrane toward the Cytb6f, (3) the penetration of PQH2 into the catalytic site of the Cytb6f, and (4) PQH2 oxidation followed by electron transfer to Cyt f. Reduced Cyt f is oxidized by Pc; reduced Pc donates an electron to P700+ (the oxidized primary electron donor in PSI) (Haehnel et al. 1980; Gross 1993). Thus, two electrons extracted from H2O by WOC of PSII are transferred sequentially via PSI to NADP+ (through ferredoxin, Fd, and ferredoxin-NADP-oxidoreductase, FNR), reducing NADP+ to NADPH. The Cytb6f complex plays a crucial role in regulation of oxygenic electron transport.

A schematic presentation of the linear and cyclic electron transport pathways in chloroplasts. a Depicts the arrangement of the membrane-bound protein complexes (Photosystem I, Photosystem II, Cytb6f, NDH-1, PTOX); FNR, ferredoxin-NADP-reductase; FQR, illusive ferredoxin-plastoquinone-reductase; PTOX, photosynthetic terminal oxidase; and the diffusion routes of mobile electron carriers: plastoquinone (PQ), plastoquinol (PQH2), plastocyanin (Pc), and ferredoxin (Fd). Reduced molecules NADPH are consumed in the Calvin-Benson cycle (CBC). Electron transport processes are shown by red arrows, they are accompanied by translocation of hydrogen ions into the thylakoid lumen. b Presents the enlarged side view of the monomer of the dimeric Cytb6f complex from Chlamydomonas reinhardtii (PDB entry 1Q90, Stroebel et al. 2003). This view is perpendicular to the membrane plane. Symbols Qo and Qi depict the catalytic centers involved into the reactions of PQH2 oxidation and PQ reduction, respectively. Plastoquinone binding site Qo is positioned between heme b6L and the Fe2S2 cluster of the iron–sulfur protein (ISP). Plastoquinone binding site Qi is placed between hemes b6H and cn. Color code of main polypeptides: cyan, Cyt f; grey, Cyt b6; purple, the iron–sulfur (Fe2S2) protein; blue, subunit IV. Cofactors: red, hemes b6L, b6H, and cn, as indicated; orange, heme f; Fe atoms are shown as dark red spheres; green, Chl a. For true colors of subunits and cofactors see the online version of this article. Figures were produced using Accelerys DV visualizer software package (http://www.accelrys.com)
The oxidation of PQH2 by the Cytb6f is the step that virtually determines the rate of the intersystem electron transport (Haehnel 1984; Berry et al. 2000; Crofts 2004a; De Vitry et al. 2004; Tikhonov 2013, 2014; Höhner et al. 2020; Malone et al. 2021; Johnson and Berry 2021; Sarewicz et al. 2021). The Cytb6f complex works as a proton pump: two protons, taken up from the stroma upon the reduction of PQ (PQ + 2e− + 2H+out → PQH2), are released into the lumen (the internal volume of thylakoids) as a result of PQH2 oxidation at the Qo site (PQH2 → PQ + 2e− + 2H+in). Thus, the Cytb6f contributes to generation of \(\Delta {\tilde \mu _{{{\rm{H}}^ + }}}\), the trans-thylakoid difference in electrochemical potentials of hydrogen ions, which serves as the driving force for ATP formation from ADP and inorganic phosphate Pi (Mitchell 1969, 2011; Williams 1988; Boyer 1997; Walker 2013; Romanovsky and Tikhonov 2010; Junge and Nelson 2015). According to the Q-cycle model first suggested by Peter Mitchell (Mitchell 1975, 1976), the overall number of protons translocated through the Cytb6f complex is two times higher than a number of electrons delivered from PQH2 to PSI (H+/e− = 2; for review, see Sacksteder et al. 2000). ATP and NADPH (the macroergic products of photosynthesis) are used mainly in biosynthetic reactions of the Calvin-Benson cycle (CBC) (Edwards and Walker 1983).
Despite the progress in understanding the molecular structure of the Cytb6f complex, its interaction with PQH2 is far from being satisfactorily understood (for comprehensive reviews, see (Malone et al. 2021; Sarewicz et al. 2021). In this paper, I briefly overview the molecular architecture of the Cytb6f complex in chloroplasts and mechanisms of PQH2 oxidation. The analysis of a bifurcated (two-electron) oxidation of PQH2 suggests that the first step of PQH2 oxidation (electron transfer from PQH2 to the iron–sulfur protein, ISP) is the basic factor that determines the rate of electron transfer between PSII and PSI. The second step of the bifurcated oxidation of PQH2 would be prompted by the plastosemiquinone radical (PSQ·) movement toward the low-potential heme of Cyt b6L (an electron acceptor) and the carboxy group of the Glu78 residue (a proton acceptor). Finally, the participation of the Cytb6f complex in regulation of oxygenic photosynthesis is briefly outlined.
Photosynthetic electron transport in chloroplasts
Lateral heterogeneity of thylakoids and the routes of photosynthetic electron transfer
Lateral heterogeneity of thylakoid membranes is characteristic of chloroplasts (Anderson and Anderson 1980; Anderson et al. 1988, 2012; Albertsson 2001; Staehelin 2003; Dekker and Boekema 2005; Pribil et al. 2014; Ruban and Johnson 2015). Figure 2 shows schematic representation of electron transport complexes locations in granal and stromal domains of thylakoids. The piles of granal thylakoids are comprised of appressed circular disk-like vesicles (typically, an average size of ~ 0.4–0.6 μm). Grana has been found to be plastic structure, with variable diameter and a number of layers, depending on the intensity and spectral quality of light (Rozak et al. 2002; Wood et al. 2018, 2019; Gu et al. 2022). Most of PSII complexes are positioned in appressed thylakoids of grana, while PSI and ATP synthase complexes are localized mainly in stroma-exposed domains of thylakoids and grana margins. Cytb6f complexes are distributed between stacked (granal) and unstacked (stromal) regions of chloroplasts. The populations of Cytb6f complexes localized in granal and stromal domains of thylakoid membranes can be involved into different routes of electron flow, linear and cyclic pathways. Due to a high mobility of PQH2 in thylakoid membranes and a rapid lateral diffusion of Pc within the lumen (Höhner et al. 2020), granal Cytb6f complexes ensure the long-distance linear electron flow (LEF) from PSII to PSI. The stroma-exposed Cytb6f complexes can participate in cyclic electron flow (CEF) around PSI, supporting the trans-thylakoid proton transfer and ATP synthesis, but without the reduction of NADP+. In the course of CEF, electrons return from PSI to the intersystem ETC via the illusive ferredoxin-plastoquinone reductase (FQR) (Bendall and Manasse 1995; Joliot and Joliot 2002; Munekage et al. 2002, 2004, 2008; Puthiyaveetil et al. 2016); this route of electron transfer may be termed as a ‘short’ cycle (CEF1). There are good reasons to believe, based on biochemical experiments, that the illusive FQR can be identified with the electron transfer mediators PGR5 and PGRL-1 bound to PSI (DalCorso et al. 2008). The operation of the CEF1 pathway is likely to proceed through a supercomplex formed by the PSI and Cytb6f complexes (Buchert et al. 2020; Yadav et al. 2017). Within this supercomplex, PSI and Cytb6f are able to exchange electrons, mediating the electron flow around PSI (Iwai et al. 2010). Since CEF1 returns electrons from Fd to the PQ/PQH2 pool through the Cytb6f complex, one can say that the Cytb6f virtually plays the role of the FQR.

A schematic representation of the location of electron transport complexes in the granal and stromal domains of thylakoid membranes. The alternative pathways of electron transport, LEF (linear electron flow), CEF1 and CEF2 denote the “short” and “long’ routes of cyclic electron transfer around PSI. Electron transport processes are shown by red arrows. Abbreviations: CBC, the Calvin-Benson cycle; PSI, PSII, photosystems I and II; b6f designates the Cytb6f complex; CF0-CF1 denote the ATP synthase complex; FNR, ferredoxin-NADP-reductase; FQR, illusive protein ferredoxin-plastoquinone reductase; NDH-1, NADH-dehydrogenase-like complex type-1; PQ and PQH2 denote plastoquinone and plastoquinol; PQA and PQB are the PQ molecules bound to PSII. LEF, CEF1 and CEF2 symbolize alternative pathways of electron transport (for other details, see explanations in the main body of the text)
Another route of cyclic electron transfer around PSI (nominated as CEF2, Fig. 1) is realized through the photosynthetic NADH dehydrogenase-like complex type-1 (NDH-1), which accepts an electron from reduced Fd (not NADPH) and then donates it to the PQ/PQH2 pool (Shikanai 2007, 2016; Strand et al. 2016, 2017; Laughlin et al. 2019, 2020; Schuller et al. 2019). Similar to mitochondrial Complex I, the NDH-1 complex operates as a Fd-dependent proton-pumping oxidoreductase, which pumps two protons into the lumen per electron transferred to PQ (for review, see Strand et al. 2017; Laughlin et al. 2020). A proper partitioning of electron fluxes between the LET, CEF-1 and CEF-2 pathways would support an optimal balance between ATP and NADPH, which is necessary for functioning the Calvin-Benson cycle (ATP/NADPH = 3/2; Edwards and Walker 1983).
The rate-limiting steps in the intersystem electron transfer
It is generally recognized that the rate of the intersystem electron transfer is determined mainly by the rate of PQH2 oxidation (Rumberg and Siggel 1969; Haehnel 1984; Tikhonov et al. 1984; Ryzhikov and Tikhonov 1988; Hope et al. 1994; Kramer et al. 1999; Takizawa et al. 2007; Foyer et al. 2012; Hasan and Cramer 2012; Malone et al. 2021). The rate of PQH2 turnover is determined by the following events: (i) the PQH2 diffusion from PSII to the Cytb6f, (ii) the penetration of PQH2 into the quinol-oxidizing catalytic site Qo of the enzyme and the formation of the PQH2-ISP complex, and (iii) the oxidation of PQH2 by the ISP. In this section, we start with the diffusion-controlled steps of the intersystem electron transfer. Then, after a brief consideration of the Cytb6f architecture, we will focus on the mechanism of PQH2 oxidation in the catalytic center of the Cytb6f.
Plastoquinone diffusion
Exchange of PQH2 and PQ molecules at the catalytic sites of the enzyme needs the percolation of quinol/quinone molecules through the lipid membrane and their diffusion inside the protein moiety of the Cytb6f. The PQH2 diffusion in the thylakoid membrane over-crowded with protein obstacles may retard electron transfer from PSII to the Cytb6f (Kirchhoff et al. 2000, 2002, 2011). In chloroplasts, however, under a wide range of physiological conditions (pH, temperature), the overall rate of the intersystem electron transport is not limited by PQH2 diffusion, being determined predominantly by the processes proceeding after the PQH2 binding to the catalytic site Qo of the Cytb6f complex (Haehnel 1976; Tikhonov et al. 1984). Note that the PSII and Cytb6f complexes may be localized in distant domains of thylakoid lamellas; however, significant amounts of these complexes are close to each other (e.g., the complexes localized in and nearby the grana). This provides shortening a way for obstructed diffusion of PQH2 from PSII to the nearest Cytb6f. In plants, grana diameter varies within a small range (about 360–600 nm; for references, see Rozak et al. 2002; Staehelin 2003; Ruban and Johnson 2015). According to (Höhner et al. 2020), the restriction of the grana diameter might exert a strong evolutionary pressure, providing a fast communication between PSII and the Cytb6f complexes.
In chloroplasts, the formation of PQH2 in PSII and its diffusion toward the Cytb6f complex occur within ∆τ ≤ 2–4 ms (at room temperatures; for references, see Haehnel 1976; Tikhonov et al. 1984, 2014; Höhner et al. 2020). This time is shorter than the half-time of electron transfer from PQH2 to \({\text{P}}_{700}^{ + }\) (via the Cytb6f and Pc, t1/2 ≥ 5–20 ms), demonstrating that the overall rate of PQH2 turnover is determined mainly by direct interaction of PQH2 with the Cytb6f, but not the PQH2 diffusion from PSII to the Cytb6f complexes. This statement has been first proved by measuring the flash-induced redox transients of P700 in chloroplasts with the PSI electron acceptor methylviologen used to support efficient electron efflux from PSII to PSI and further to O2 (Haehnel 1976; Tikhonov et al. 1984). In order to illustrate this point, I reproduce below the results of our earlier study of bean chloroplasts (Fig. 3). In this set of experiments, before kinetic measurements, chloroplasts were pre-conditioned either by the far-red (FR) pre-illumination by light exciting predominantly PSI (λmax ≈ 707 nm) or by continuous white light (WL) exciting both PSI and PSII. In the first case, the plastoquinone pool and P700 centers were kept oxidized; in response to a short pulse of WL, oxidized centers P700+ reduced with the half-time τ1/2 ~ 15–20 ms. The reduction of P700+ started, however, only after a lag-phase Δτ (Fig. 3a). The length of the lag-phase involves the times of PQH2 formation in PSII and its traffic toward the Cytb6f. The release of PQH2 from PSII after a short light flash takes about 0.6 ms (at room temperatures; Haehnel 1984); thus, the lag-phase mainly reflect the migration of PSII from PSII to the Cytb6f complex. Otherwise, parameter τ1/2 is determined by the events that occur after the PQH2 molecules reached the Cytb6f complex: the oxidation of PQH2 by the ISP and further electron transfer to P700+ (PQH2 → b6f → Pc → PSI).

Effects of chloroplast pre-conditioning on the time-course of the amplitude the EPR signal of P700+ in bean chloroplasts with 20 μM methylviologen, pH 7.5. Redox changes of P700 were induced either by the pulse (t1/2 = 750 μs) of white light given simultaneously with a background far-red (λmax = 707 nm) light (a), or recorded after switching off a continuous white light, illumination time 12 s, in the presence of 10 mM NH4Cl (b). Kinetic curves represent the modified time-courses of P700+ borrowed from the original publication (Tikhonov et al. 1984)
After the pre-illumination of chloroplasts by WL exciting both photosystems, the PQ/PQH2 pool becomes reduced. In this case, the post-illumination reduction of P700+ with the half-time τ1/2 ≥ 15–20 ms (depending on temperature and pH) starts immediately after switching the WL off, without the lag-phase Δτ (Fig. 3b). The loss of the lag-phase after the WL pre-illumination can be explained: reduced PQH2 molecules already reached the Cytb6f complexes, being able to deliver electrons to the Cytb6f without the PQH2 diffusion delay. The relationship Δτ < τ1/2 has been observed under a wide range of experimental conditions (variations of temperature in the range from 5 to 35 °C, and pH variations between 5.0 and 8.5 (Tikhonov et al. 1984)). This gives clear evidence that the light-induced formation of PQH2 in PSII and its diffusion toward the Cytb6f complex proceed more rapidly than the intrinsic events of PQH2 oxidation after the PQH2 binding to the catalytic center Qo.
Recently, in order to elucidate the influence of the thylakoid architecture on the diffusion-dependent electron transport mediated by PQH2 and Pc, Höhner et al. (2020) assayed the Arabidopsis mutants with different grana diameters (varied in the range from ≈370 to 1,600 nm).
They examined electron transport in chloroplasts of three genotypes of Arabidopsis, in which thylakoid architecture was modified by inducing membrane curvature. The curt1abcd mutant was characterized by significantly extended grana diameter (up to ~ 1,600 nm), in the overexpressor mutant CURT1A-oe grana diameter shrinked to ~ 350 nm (Armbruster et al. 2013; Pribil et al. 2018). It has been found that the time of the PQH2 diffusion from PSII to the Cytb6f, estimated from the length of the lag phase, is not affected (Δτ ~ 3.2–3.6 ms) with significant variations of grana diameter (from ~ 370 to 1,600 nm). This is in agreement with the conclusion that the rate of PQH2 oxidation is determined predominantly by the intrinsic events of PQH2 oxidation inside the Cytb6f complex, rather than the PQH2 diffusion in the lipid domains of the thylakoid membrane.
The notion of a relatively fast diffusion of PQH2 in thylakoid membranes is consistent with theoretical evaluations of the plastoquinone diffusion coefficient based on the random work modelling of plastoquinone motions in two-dimensional lipid systems. According to (Tremmel et al. 2003), the apparent coefficient of the PQ diffusion in the thylakoid membrane approaches to DPQ ~ 2 × 10−8cm2 s−1. This implies that PQH2 could travel farther than 400 nm in 20 ms, suggesting that PQH2 migration from PSII to the Cytb6f complexes located near the grana margins should not limit linear electron transport between PSII and PSI, i.e., PQ-diffusion limitations would be mitigated by close localization of the Cytb6f and PSII complexes.
Note that the literature data on the rates of partial reactions of electron transfer in the Cytb6f complex are often scattered, depending on the plant species and plant pre-illumination history. Relatively short times of Cyt f and Cyt b reduction (t½ ≈ 3–6 ms) are typical of intact C. reinhardtii cells (Soriano et al. 1996; Ponamarev and Cramer 1998) and the cyanobacterium Synechococcus sp. PCC 7002 (Yan and Cramer 2003). One of the reasons for the dispersion of kinetic data might be related to differences between the species and variability of stoichiometry between the PSII, Cytb6f and PSI complexes (Schöttler et al. 2015). Different capacities of donor and acceptor species interacting with the Cytb6f may also influence the apparent rates of kinetic processes, exaggerating or underestimating rapid and slow phases post-illumination reduction of P700+. At any rate, however, we can safely state that the oxidation of PQH2 inside the Cytb6f is one of basic factors that determine the rate-limiting step in the chain of electron transport between PSII and PSI.
Plastocyanin diffusion
Obstructed diffusion of Pc within a narrow gap of the lumen may restrict electron flow from the Cytb6f to PSI. Changes in the chloroplast architecture can influence the long-range diffusion of Pc inside the thylakoid lumen (Kirchhoff et al. 2011; Höhner et al. 2020). The shortening or elongation of the distance between PSII to the Cytb6f could proceed due to variations in the grana diameter (Höhner et al. 2020). In particular, plants grown at low light conditions reveal somewhat reduced grana diameter (Wood et al. 2019; Flannery et al. 2021) and the concomitant shortening of an average distance between PSII to the Cytb6f complexes localized in grana.
There have been made two important observations. First, the light-induced swelling of thylakoids releases the restrictions for the Pc movements inside the lumen, providing the acceleration of the long-range lateral diffusion of Pc (Kirchhoff et al. 2011). Second, the widening of the grana disk diameter, and concomitant elongation of the Pc diffusion pathway, can lead to a significant increase in the half-time of Pc movement inside the lumen from the grana-hosted Cytb6f complexes to PSI (Höhner et al. 2020). The latter implies that significant elongation of the Pc diffusion path slowed down the operation of Pc. It should be noted, however, that under a wide range of experimental conditions, the Pc-dependent electron transport from PSII to P700+ occurs more rapidly (t1/2 ≤ 300 μs) than the oxidation of PQH2 by the Cytb6f (τ1/2 ≥ 5–20 ms, Stiehl and Witt 1969; Haehnel 1984; Tikhonov et al. 1984; Harbinson and Hedley 1989; Laisk et al. 2016; Ptushenko et al. 2019; Höhner et al. 2020).
Architecture and overview of the Cytb 6 f structure
Electron carriers and catalytic sites
First three-dimensional structures of the Cytb6f have been first obtained by the X-ray analysis (at a resolution of 3.0–3.1 Å) of crystal samples from the thermophilic cyanobacterium Mastigocladus laminosus (PDB code 1FV5; Kurisu et al. 2003) and the green alga Chlamidomonas reinhardtii (PDB code 1Q90; Stroebel et al. 2003). The Cytb6f complexes from different photosynthetic organisms reveal similar architecture. These complexes are organized as the dimers of multisubunit monomers (Fig. 4a) peculiar to the Cyt bc family of electron transport complexes (for review, see Berry et al. 2000; Crofts 2004a; Cramer and Hasan 2016; Malone et al. 2019, 2021; Sarewicz et al. 2021). Each monomer consists of eight polypeptide subunits with 13 trans-membrane helixes. The monomers include four “large” subunits (16–31 kDa): the Rieske iron–sulfur protein (ISP), the Cyt b6 and Cyt f proteins, and subunit IV (subIV). Each multisubunit monomer reveals two prosthetic groups, chlorophyll a (Chl a) and β-carotene, associated with the subIV. Several “small” hydrophobic subunits (3.3–4.1 kDa) are arranged at the outside periphery of the monomer ensembles. Dimeric structure of the Cyt b6f complex provides the formation of a large protein-free intermonomer cavity (~ 30 Å × 25 Å × 15 Å) through which PQH2 and PQ can penetrate into the quinone-binding sites. The potential interactions between the electron transport chains localized in two monomers of the dimeric b6f complex have been discussed in the literature (for references, see Crofts et al. 2008; Nawrocki et al. 2019; Crofts 2021; and references therein). Below, I focus on the primary PQH2 oxidation processes at the Qo catalytic site located within a single multisubunit monomer.

The molecular architecture of the spinach dimeric Cytb6f complex reconstructed from the cryo-EM data (PDB code 6RQF; Malone et al. 2019). a Present the side view of the Cytb6f complex, demonstrating the protrusion of the external domains of Cyt b6 and Cyt f into the bulk phase of the thylakoid lumen. b and c show the trans-membrane and top (from the stromal side) views of the Cytb6f complex, and the locations of prosthetic groups and plastoquinone molecules, PQ1, PQ2 and PQ3. Figures were produced using Accelerys DV visualizer software package (http://www.accelrys.com)
The Cytb6f complex contains several electron carriers, which perform the catalytic functions associated with redox changes of PQH2 and PQ. Figure 4b and c depict the trans-membrane and top views of the native spinach Cytb6f complex (PDB code 6RQF; Malone et al. 2019), indicating the positions of electron carriers participating in PQH2 oxidation and PQ reduction at the catalytic sites Qo and Qi, respectively.Footnote 1 Each monomer ensemble contains the following carriers: the Fe2S2 cluster of the ISP, two hemes of the Cyt b6 (the low-potential heme b6L and the high-potential heme b6H), and an atypical heme cn positioned in close proximity to the high-potential heme b6H on the stromal side of the complex. A c-type heme f protrudes into the thylakoid lumen. Along with the redox cofactors, there are two Chl a molecules positioned inside the dimer complex.
Crystallization of the Cytb6f complexes with the quinone analogue inhibitors, tridecyl-stigmatellin (TDS) and NQNO (2n-nonyl-4-hydroxy-quinoline-N-oxide), revealed two sites for quinone binding: the Qo site (quinol oxidase) and the Qi site (quinone reductase) (Yamashita et al. 2007). The quinol-binding portal of the Qo site represents a hydrophobic cavity (~ 11 Å × 12 Å) covered inside by lipid molecules (Hasan and Cramer 2014; Cramer and Hasan 2016; Bhaduri et al. 2019). The quinone exchange portal of the catalytic site Qo is positioned near the Fe2S2 cluster of the ISP. One lobe of the Qo volume is oriented toward the Fe2S2 cluster while the other side extends toward heme b6L. TDS was found in the close vicinity of the Rieske protein, forming the hydrogen bond with the His residue ligating one of the Fe atoms of the Fe2S2 cluster. Figure 5 shows a fragment of the Qo site, which contains the inhibitor TDS located near the Fe2S2 cluster (PDB code 1Q90). Note that the position of the ring of the 2,3,5-trimethylbenzoquinol molecule (TMBQH2, the tail-less analog of PQH2), is nicely fitted to the TDS molecule resolved in the crystal structure of the Cytb6f. Both species, TMBQH2 and TDS, form the hydrogen bond (–O–H∙∙∙Nε<) with the His155 residue of the ISP.

The arrangement of an inhibitor tridecyl-stigmatellin (TDS) hydrogen-bonded to atom Nε of His155 in the crystal structure of the Cytb6f complex (PDB entry 1Q90, Stroebel et al. 2003). Green balls show the position of the head group atoms of plastoquinone
The second quinone-binding center (site Qi) is located on the stromal side of the Cytb6f, at the interface between an “atypical” heme cn and the large inter-protein quinone exchange cavity between hemes b6H and cn (Kurisu et al. 2003; Stroebel et al. 2003; Yamashita et al. 2007; Malone et al. 2019, 2021; Sarewicz et al. 2021, 2023). It is believed that heme cn participates in cyclic electron transport around PSI, mediating electron transfer from the acceptor side of PSI (via Fd) to PQ located in the Qi site (Strand et al. 2016, 2017; Schuller et. al., 2019).
According to the Mitchell’s Q cycle (Mitchell 1975, 1976; Berry et al. 2000; Crofts 2004a, 2021; Osyczka et al. 2005; Cramer et al. 2006, 2011; Mulkidjanian 2010), in the catalytic site Qo two electrons are extracted from PQH2 and directed to the high-potential and low-potential redox chains (Fig. 6a). The very idea that in the course of quinol oxidation two electrons are directed into separate electron transfer chains was suggested by Wikström and Berden, who had found that external oxidant (O2) induced the reduction of Cyt b in mitochondria in the presence of antimycin (Wikström and Berden 1972). In the Cytb6f complex, one electron is transferred to the Fe2S2 cluster of the ISP (reaction 1), another electron is directed to the low-potential heme b6L (reaction 2). Reduced ISP (ISPred) donates an electron to Pc and further to P700+ via the high-potential chain: ISPred → Cyt f → Pc → P700+. From the plastosemiquinone (PSQ·) formed after the first step of PQH2 oxidation, an electron reduces the low-potential heme b6L (Joliot and Joliot 1988). Reduced heme b6L donates an electron to the high-potential heme b6H positioned near the PQ-binding center Qi, which operates as the PQ reductase (Kramer and Crofts 1993; Crofts 2004c, 2021). At the Qi center, according to modified Q cycle, the PQ molecule accepts one electron from Cyt b6 and serves as the recipient of the second electron coming from the acceptor side of PSI through the cyclic electron transfer chain: PSI → Fd → FQR → cn → (PQ)i. Here, FQR and cn denote the illusive Fd-quinone reductase (Munekage et al. 2004) and c-type cytochrome (Kurisu et al. 2003; Stroebel et al. 2003), respectively. The fully reduced PQH2 molecule dissociates from Qi and then can bind to the vacant center Qo. Both reactions of the bifurcate oxidation of PQH2 proceed as the proton-coupled electron transfer (PCET) processes:

a A schematic diagram of a bifurcated (two-electron) oxidation of plastoquinol (PQH2) at the catalytic site Qo of the Cytb6f complex. Numbers 1 and 2 indicate the reactions of electron transfer from PQH2 to the Fe2S2 cluster (1) and from semiquinone PQH· to the low-potential heme b6L (2). Modified Fig. 2 from Ustynyuk and Tikhonov (2022). b A diagram of the midpoint redox potentials of electron carriers illustrating electron transfer along the high-potential (ISP, Cyt f, Pc, and P700) and low-potential redox (hemes b6L, b6H, and cn) branches of electron transfer in the Cytb6f complex. The redox potential levels depicted by red triangle and horizontal lines represent the average values taken from the literature for Chlamydomonas reinhardtii (Pierre et al. 1995; Zito et al. 1998; Alric et al. 2005; Nelson and Yocum 2006; Hasan et al. 2013a); blue triangle and horizontal lines correspond to cyanobacteria (Nakamura et al. 2011). Green circles and horizontal lines depict the redox potentials measured by low-temperature EPR spectroscopy in spinach chloroplasts (Szwalec et al. 2022)
Here, PSQ· denotes the semiquinone species (protonated or deprotonated, PQH· or PQ·−) formed after the first step of the bifurcated reaction. The term proton-coupled implies that the reactions (1) and (2) are tightly coupled with the proton transfer to appropriate proton-accepting groups (Mayer and Rhile 2004).
Thermodynamic reasons for the assignment of electron carriers to the high- and low-potential branches are based on measurements of the midpoint redox-potentials (Em) of these carriers. Figure 6b presents a diagram illustrating this point (for references, see (Tikhonov 2014, 2018; Malone et al. 2021)). The driving force for the first reaction of PQH2 oxidation by the ISPox is characterized by a high redox-potential of the redox pair PQH2/PQ· (Em ~ 450 mV). The lower value of Em of the redox pair ISPred/ISPox (Em ~ 300–320 mV; Nitscke et al. 1992) implies that electron transfer from PQH2 to ISPox is the up-hill (energy-accepting) process that would limit the overall rate of PQH2 oxidation (for more details see below Section "Oxidation of PQH2 by the Cytb6f complex, the Q cycle"). Further reactions of electron transfer along the high-potential branch to P700+ occur as the down-hill reactions (Fig. 6b).
The very essence of the second reaction (the oxidation of PSQ·) is that the plastosemiquinone radical PSQ· is a rather strong reductant capable of reducing electron carriers standing in the low-potential chain. The potential of the redox pair PSQ·/PQ is lower (Em ~ − 250 mV) (Rich and Bendall 1980) than the Em values of electron carriers of the low-potential branch (Joliot and Joliot 1988). In general, the Em values for the hemes the low-potential chain reported in the literature reveal a tendency of increasing in the line b6L → b6H → cn (Fig. 6b). This is in agreement with the traditional point of view on the Q cycle operation. It should be noted, however, that specific Em values of the hemes b6L, b6H and cn reported in the literature are sometimes scattered, depending on the system investigated and the methods used for determination of Em (Rich and Bendall 1980; Hurt and Hauska 1982, 1983; Clark and Hind 1983; Joliot and Joliot 1988; Furbacher et al. 1989; Pierre et al. 1995; Zito et al. 1998; Alric et al. 2005; Nakamura et al. 2011). In particular, recent redox titration of spinach chloroplasts (Szwalec et al. 2022) based on independent methods, optical spectroscopy and low-temperature EPR, revealed unexpected low value (Em ≈ − 111 mV) attributed to heme b6H, which was lower than the Em value of heme b6L (Em ≈− 73 mV). The authors conclude that this result may dismiss the long-standing assumption that heme b6L has lower Em value than heme b6H. This also implies that in the Cytb6f complex electron flow between hemes b6L and b6H may slow down electron flow along the low-potential chain. It is conceivable that this effect might be one of the factors contributing to the regulation of electron flow through the Cytb6f complex.
Plastoquinone molecules inside the Cytb 6 f complex and the enter/exit pathways
Crystal structures
The dimer structure of the Cytb6f provide the formation of a rather large cavity (~ 30 × 25 × 15 Å) through which PQH2 and PQ molecules enter into the quinone-binding centers (Cramer et al. 2006; Hasan et al. 2013c). The PQH2 binding portal Qo, where the PQ/PQH2 exchange occurs, lies inside the cavity covered by lipid molecules (about 23 potential sites of lipid binding per monomer; Hasan and Cramer 2014; Cramer and Hasan 2016). Figure 7 shows a fragment of the Qo site in the crystal structure of the Cytb6f complex (PDB code 1Q90; Stroebel et al. 2003), which involves heme b6L, Glu78, and the Fe2S2 cluster of the ISP surrounded by two His residues and several amino acids residues. The structure was supplemented by TMBQH2, the tailless analog of plastoquinol. The position of TMBQH2 near the ISP was determined as described in (Ustynyuk and Tikhonov 2022). The formation of the hydrogen bond between the –OH group of the quinol molecule and the atom Nε of the His residue, liganding one of the Fe atoms of the Fe2S2 cluster (His155 in Fig. 7), is considered as the preconditioning for the formation of substrate-enzyme complex (PQH2-His) at the catalytic site Qo (for references, see Crofts et al. 1983; Crofts 2004a, 2004b, 2004c, 2021; Mulkidjanian 2010; Cramer and Hasan 2016; Sarewicz et al. 2021, 2023). The atom Nε is assumed to be the primary recipient of the proton donated by quinol. Alternative model, suggesting that the primary acceptor of the proton may be a water molecule, has been suggested in (Postila et al. 2013; Barragan et al. 2016). The PQ binding site Qi is located in the stroma-exposed domain of Cytb6f, at the interface between heme cn and the inter-protein quinone exchange cavity (Fig. 4). The unique heme cn exists in the Cytb6f, but it is absent in the Cytbc1 complexes. According to the modified Q-cycle, heme cn may participate in cyclic route of electron flow around PSI, mediating the reduction of PQ bound to Qi (Kurisu et al. 2003; Stroebel et al. 2003; Munekage et al. 2004; Shikanai 2007).

A fragment of the crystal structure of the Cytb6f complex from Chlamidomonas reinhardtii (PDB entry 1Q90) used to illustrate the primary reactions of a bifurcated oxidation of quinol at the Qo site of the Cytb6f complex. Red and blue arrows show the directions of the electron and proton transfer reactions related to plastoquinol oxidation. Figures were produced using Accelerys DV visualizer software package (http://www.accelrys.com)
Cryo-EM structures
PQH2 molecules formed in PSII travel to the intermonomer cavity of the Cytb6f by lateral diffusion in the membrane. Penetration of PQH2 into the quinol-binding portal Qo is associated with its diffusion within the Cytb6f lipoprotein complex: it is likely that PQH2 molecules with flexible isoprenoid chains reach the quinone-binding catalytic sites by percolation through the intermonomer cavity and curved intraprotein pathway inside the Cytb6f complex. In first crystal structures of the Cytb6f complex, neither plastoquinol nor plastoquinone have been resolved within the Qo portal. Significant progress in understanding the structure and function of the native Cytb6f complex has been recently achieved with the use of the cryo-EM technique (Malone et al. 2019, 2021; Proctor et al. 2022; Sarewicz et al. 2023). The first native structure of the spinach Cytb6f complex, obtained without the crystallization procedure and in the lack of inhibitors (PDB code 6RQF; Malone et al. 2019), revealed three PQ molecules (designated as PQ1, PQ2 and PQ3) fixed at different positions inside the intermonomer cavity (Fig. 8). The benzene ring of PQ1 is adjacent to the heme b6L and Chl a; PQ2 is located near the hemes b6H and cn; PQ3 is situated between the hemes b6H and cn bound to different monomers of the dimeric Cytb6f complex.

The location of plastoquinone molecules in the stroma-oriented cavity of the dimeric Cytb6f complex from spinach (PDB code 6RQF) recovered by the cryo-EM (Malone et al. 2019). Arrows indicate the positions of two Fe2S2 clusters, hemes of Cyt f, the low- and high-potential hemes b6L and b6H, hemes cn, and two Chl a molecules. Symbols PQ1, PQ2, and PQ3 denote plastoquinone molecules positioned in the intermonomer cavity at a distance from the Fe2S2 clusters of the ISPs
In the first cryo-EM structure of the native spinach Cytb6f complex all plastoquinone molecules were resolved away from the catalytic center Qo (Malone et al. 2019). No doubt it might seem very surprising that neither PQ nor PQH2 molecules were found in the Qo portal. The mystery of the lost plastoquinones has been recently solved. The breakthrough in this field was made by Osyczka and collaborators (Sarewicz et al. 2023). They resolved the high-resolution cryo-EM structures of the spinach Cytb6f homodimer with the worm-like intra-protein pathway for traffic of endogenous plastoquinones, providing the passage of PQH2 to the catalytic center Qo and the exit of PQ to lipid domains. In each multisubunit monomer, the authors visualized three plastoquinone molecules arranged one after another (a tail-head–tail-head–tail-head arrangement). Such a traffic of quinones inside the Cytb6f complex never been considered before. The head group of one of three PQH2 molecules (PQ1) was positioned very close to the Fe2S2 cluster. A model developed by Sarewicz et al. (2023) suggests the one-way diffusion of quinones through the intra-protein channel during the catalytic cycle. According to the model, two plastoquinone molecules, PQ1 and PQ3, occupy the entry and exit moieties of the long intra-protein channel. PQ2 is located in the middle part of the channel. It has been proposed that the entering of PQ1 into the channel entrance involves a slip of its tail inside the protein cavity. It is likely that the re-arrangement of PQ1 inside the channel can be accompanied by a flip of its head group toward the ISP, providing the formation of the hydrogen bond between PQ1 and the ISP. The penetration of PQ1 into the quinone-conducting channel further proceeds toward the position of PQ2, initiating the sequence of events associated with the bifurcated oxidation of PQH2 at the Qo site. It has been found that the head group of PQ1 is proximal to the Fe2S2 cluster. It seems, however, that the position of the head group of PQ1 was not optimal for its bifurcated oxidation (~ 6.5 Å from His128). The authors propose that the catalytically active position of PQ1 is highly transient and the bifurcated reaction occurs when PQH2 travels between PQ1 and PQ2 positions. Oxidized PQ molecule moves to the next position in the chain (PQ3), from which it can travel to the bulk of the lipid domain of the thylakoid membrane (Fig. 9b).

Schematic routes of plastoquinone diffusion inside the Cytb6f complex. Traces of plastoquinone traffic are shown by double-line blue arrows. a PQH2 molecules reduced in PSII enter the intermonomer cavity. b The one-way diffusion of plastoquinone molecules (PQ1, PQ2, and PQ3) from the thylakoid membrane through the intraprotein channels toward the catalytic Qo centers of the spinach Cytb6f complex. The structures of the Cytb6f complex and the trace of the channel for plastoquinone diffusion were discovered by Sarewicz et al. (2023) using high-resolution cryo-EM (PDB code 6RQF)
An interesting observation reported by Malone et al. (2019) is that the phytyl tail of Chl a lie on the way of plastoquinone passage to Qo (Figs. 4c and 8). According to (Sarewicz et al. 2023), the phytyl tail of Chl a could occupy the intraprotein PQ-conducting channel. It has been found that the phytyl tail may have two conformations (Malone et al. 2019). This suggests that the phytyl tail might operate as a mechanical “gate”, providing the admission of PQH2 to the catalytic site Qo. The traffic of PQH2 through the intra-propein quinone-conducting channel would be controlled by conformational changes of the phytyl tail of Chl a. In one case, the phytyl tail restricts the PQH2 diffusion, preventing the access of PQH2 to the catalytic site Qo (Malone et al. 2019; Sarewicz et al. 2023). After the phytyl chain turn, the penetration of PQH2 to the Qo site becomes possible (Malone et al. 2019; Sarewicz et al. 2023).
Oxidation of PQH2 by the Cytb 6 f complex, the Q cycle
The oxidation of PQH2 at the Qo center starts with the transfer of the H atom from the −OH group of PQH2 to oxidized ISPox (Crofts 2004a, 2004b, 2004c; Crofts et al. 2013). One of the Fe ions of the Fe2S2 cluster is ligated by two His residues (Fig. 5). Structural data suggest that the Nε atom of deprotonated His (His155 in C. reinhardtii (Stroebel et al. 2003), His129 in M. laminosus (Kurisu et al. 2003), or His128 in spinach (Malone et al. 2019)) is the prime candidate for the role of the proton recipient from PQH2. The formation of the H-bond between the –OH group of PQH2 and the Nε atom of deprotonated His is considered as a pre-requisite for quinol oxidation. The existence of this bond has been demonstrated by spectroscopic methods (EPR, NMR, and ATR-FTIR) in Cyt bc1 complexes (Samoilova et al. 2002; Zu et al. 2003; Iwaki et al. 2005; Lin et al. 2006; Hsueh et al. 2010), which are akin to the Cyt b6f. The fully oxidized Fe2S2 cluster is the EPR silent diamagnetic species (the total spin S = 0) due to the antiferromagnetic coupling between two paramagnetic Fe3+ ions (S = 5/2). After the one-electron reduction of ISPox, the Fe2S2 cluster becomes paramagnetic and reveals the EPR signal characterized by S = 1/2 (Sarewicz et al. 2021; Ruuge and Tikhonov 2022).
Electron transfer from the reduced ISP (ISPred) to Cyt f (or Cyt c1 in the Cytbc1 complex) is associated with the large-scale conformational changes in the ISP. After the reduction of the ISP, its mobile domain, which contains the cluster Fe2S2, displaces from the plastoquinone-binding site and moves toward heme f. The long-range “tethered” diffusion of the mobile fragment enables electron transfer to heme f, which further reduces Pc. It is likely that the tethered diffusion of the ISP extrinsic domain does not limit the overall rates of the Cytb6f and Cytbc1 turnover. For example, in Rb. sphaeroides the binding/dissociation reactions and movements of ISP occur more rapidly (~ 30–60 μs) than the rate-limiting reactions in the high-potential redox chain (Crofts 2004b). A rapid movement of the Fe2S2 cluster away from the Qo site precludes the donation of the second electron from PSQ· to the high-potential branch, directing the PSQ· radical to reduce heme b6L.
The radical pairs PSQ·–Fe2S2· formed after the first step of quinol oxidation have been detected in Cytbc1 and Cytb6f complexes by the electron paramagnetic resonance (EPR) method (Sarewicz et al. 2013, 2017, 2018, 2021; Pietras et al. 2016; Bujnowicz et al. 2019). The low-temperature EPR signals detected at cryogenic temperatures were attributed to the reduced cluster Fe2S2· of the Rieske protein (a triplet with the central line at g = 1.95), the semiquinone radical PSQ· (a singlet line at g = 2.045), and a new EPR signal related presumably to the radical pair PSQ·–Fe2S2·. A semiquinone PSQ· is coupled to the reduced cluster Fe2S2· via spin–spin exchange interaction. The coupling energy, evaluated on the basis of EPR data, appears to be of a rather small value (~ 3.5 GHz or ~ 1 K). This suggests that the radical pair PSQ·-Fe2S2· to be an unstable transient structure. After the breakdown of the radical pair, the highly reactive semiquinone species PSQ· can donate an electron to heme b6L of the low-potential branch of electron transfer in the Cytb6f (Kramer and Crofts 1993). Further electron transfer along the low-potential chain (PSQ· → b6L → b6H → cn → PQi) would provide the reduction of PQ molecule (PQi) bound to the redox site Qi. According to a model of modified Q cycle in the Cytb6f, the PQi molecule is believed to receive a second electron from the acceptor side of PSI through the chain of CET around PSI (Munekage et al. 2004; Strand et al. 2016). Fully reduced PQH2 molecule releases from the site Qi and then can rebind to the vacant center Qo. Thus, due to the cyclic operation of the Cytb6f complex, associated with the PQH2 return from the Qi site to the catalytic center Qo, one turnover of this complex will provide enhanced stoichiometry of the trans-thylakoid proton transfer (H+/e− = 2), when two protons are translocated per one electron delivered from PQH2 to PSI through the high-potential chain (Mitchell 1975, 1976).
Electron transfer along the high-potential chain is associated with significant conformational changes in the Cytb6f complex. The Fe2S2 cluster of the ISP is separated from heme f by ~ 26 Å; this excludes direct electron transfer from the ISPred to Cyt f by the mechanism of quantum mechanical tunneling (Moser et al. 1997; Page et al. 1999). There is a number of experimental evidence that the mobile domain of the ISPred, which contains the Fe2S2 cluster, moves from the Qo site toward heme f by the mechanism of the tethered-diffusion. Electron transfer by domain movement was strongly documented by the X-ray analysis of the crystal structures of the Cytbc1 complex (Zhang et al. 1998). In the crystals of the Cytbc1 complex with some inhibitors, the positions of the mobile ISP domain containing the Fe2S2 cluster were resolved in different places of the complex, reflecting the flexibility of this domain. After the re-oxidation of the ISPred by Cyt f, the mobile extrinsic domain of the ISP with the oxidized cluster Fe2S2 returns to its initial (proximal) position at the Qo site. In the Cytb6f complex, the conformational mobility of the ISP subunit has long been a subject of debate. The hinge fragment and the extrinsic mobile domain of the ISP were disordered in the crystallographic structures of the Cytb6f, indicating a significant flexibility of the ISP mobile fragment (Baniulis et al. 2009; Hasan et al. 2013b). The disorder of the ISP extrinsic domain is usually considered as the evidence of its high conformational mobility. It has been suggested that the conformational mobility of the ISP is determined by its lipid environment (Hasan et al. 2013b; Hasan and Cramer 2014). There are indications that the shuttle movements of the flexible extrinsic domain of the ISP between the Qo site and heme f occur more rapidly than the electron transfer from PQH2 to Fe2S2 rapidly (Breyton 2000; Yan and Cramer 2003; de Vitry et al. 2004; Hasan et al. 2013b), and, therefore, these movements do not limit the overall rate of the Cytb6f turnover.
The breakdown of the unstable radical pair PSQ·–Fe2S2· would facilitate the oxidation of the radical PSQ· due to its movement inside the intra-protein cavity (Δl ~ 1 nm) toward heme b6L and the proton-accepting −COO− group of Glu78 (Fig. 10a). Molecular dynamics calculations performed by Cramer and collaborators (Hasan et al. 2014; Ness et al. 2019) confirm the possibility of a fast displacement of PQH· from the Qo-binding site (Δτ ~ 10 ns). If this is the case, reaction (2) would proceed almost simultaneously with reaction (1). Once the radical shifts toward b6L and Glu78, PSQ· becomes oxidized to PQ. A rapid oxidation of PSQ· (reaction 2) should preclude the donation of the second electron to the high-potential branch. Short life-time of PQH· would also reduce a probability of superoxide (O2·−) formation due to O2 interaction with PQH· (Mubarakshina et al. 2006; Ivanov et al. 2018).

a A fragment of the crystal structure of the spinach Cytb6f complex (PDB code 6RQF) involved into a bifurcated oxidation of PQH2 (HO-Q-OH) at the Qo site. Red and blue arrows designate the electron and proton transfer from the H(1) atom of plastoquinol to His128, and the electron and proton transfer from the H(2) atom of plastosemiquinone (HO-Q-O·) to heme b6L and the −COO− group of Glu78. b A tentative pathway of the H+ transfer from the protonated group −COOH of Glu78 to the bulk phase of the thylakoid lumen
Both steps of a bifurcated oxidation of PQH2 are tightly coupled to proton transfer to appropriate proton-accepting groups. One of the His residues of the ISP is considered as the primary recipient of a proton donated by PQH2. The −COO− group of highly conserved Glu78 (spinach numbering) may serve as the acceptor of the second proton donated by PQH· (−COO− + H+ →−COOH; Zito et al. 1998; Osyczka et al. 2006; Hasan et al. 2013c; Victoria et al. 2013). Glu78 stands in the position proximal to heme b6L. In the Cytb6f complex, the mobility of the −COO− group is limited due to a salt bridge between Glu78 и Arg87. This group is oriented toward a throat of the hydrophilic tunnel that forms a proton-conducting pathway for the proton liberated from plastosemiquinone and transferred to the thylakoid lumen. The proton transferred passes through the channel, which includes the proton-binding groups of Glu3 and Glu58 (Fig. 10b).
Concerning the second step of PQH2 oxidation, it is worth noting that the heme b6L and the −COO− group of Glu78 stand at a rather long distance (by ~ 6.5 Å) from the place where the radical PQH· appears after the first step of the PQH2 oxidation. The remoteness of the electron and proton acceptors from PQH· would preclude its oxidation, and only after a rapid displacement of PQH· toward heme b6L and Glu78 the oxidation of PQH· could be efficiently realized. The assumption about rapid movements of a semiquinone within the Qo portal, based on the analysis of kinetic data, has been proposed by Antony Crofts and collaborators, who studied the ubiquinol oxidation inside the Cytbc1 complex (Crofts 2004b, 2021; Crofts et al. 2017). This proposal was also supported by estimations performed within the framework of a simple model based on conventional kinetic approaches (Tikhonov 2014) and quantum chemical modeling of PQH2 oxidation at the Qo site (Ustynyuk and Tikhonov 2022).
Quantum chemical modeling of a bifurcated oxidation of PQH2
The density function theory (DFT) approach has been used to analyze quinol oxidation by Cytbc complexes (Shimizu et al. 2008; Frolov and Tikhonov 2009; Postila et al. 2013; Barragan et al. 2015, 2016; Husen and Solov’yov 2016; Ustynyuk et al. 2018; Ustynyuk and Tikhonov 2018, 2022). One of somewhat truncated quantum chemical models, used in (Ustynyuk and Tikhonov 2022) for modelling a bifurcated oxidation of PQH2 at the catalytic Qo site, contained four functional groups: 1) the Fe2S2 cluster surrounded by two His residues; 2) heme b6L; 3) Glu78, and 4) 2,3,5-trimethylbenzoquinol (TMBQH2), a tail-less analog of PQH2. Results of quantum chemical calculations are consistent with experimental data. Oxidized cluster Fe2S2 is diamagnetic (total spin S = 0) due to antiferromagnetic interaction of two paramagnetic ions, Fe3+(1) and Fe3+(2), with individual spins S = 5/2, demonstrating that two Fe ions in the oxidized and reduced states of the ISP have opposite projections of their spins (Noodleman et al. 1995, 2002; Siegbahn and Blomberg 1999; Sarewicz et al. 2021). Reduced cluster Fe2S2 is a paramagnetic species with the total spin S = 1/2. The oxidized heme b6L is also paramagnetic, this is consistent with experiment (Palmer 1985; Sarewicz et al. 2021). Calculated spin of the TMBQH· radical was determined as S = 1/2 (Ustynyuk and Tikhonov 2018, 2022).
The first step of PQH2 oxidation
Figure 11 reproduces the plot of the system energy versus the distance between the atom H(1) of TMBQH2 and the Nε atom of His155 (C. reinhardtii numbering, PDB code 1Q90). The two local minima correspond to the initial and the final positions of H(1), characterizing the formation of the hydrogen bond H(1)−Nε (\(R_{{{\text{H}} - {\text{N}}_{{\upvarepsilon }} }}\) = 1.11 Å). The oxidation of TMBQH2 can be considered as the proton coupled electron transport (PCET) process, when the electron is directed to the Fe3+(1) ion of the Fe2S2 cluster, while the proton is accepted by the Nε atom of His155 (Fig. 11a). This process needs the overcoming of the energy barrier ΔE≠ (Fig. 11b). After a rapid (nonadiabatic) transfer of a proton to Nε, the energy of the system increases by ΔE = 25 kJ mol−1. The rise of energy is followed by its decrease by ΔErel = 8 kJ mol−1, which occurs in the result of geometry “relaxation” followed the H atom transfer (for detail, see Ustynyuk and Tikhonov 2022). The overall change in the system energy can be evaluated as ΔE1 = ΔE—ΔErel ≈ 17 kJ mole−1. Similar values of the energy rise have been reported for the Cytbc1 complex (Crofts et al. 2000; Zu et al. 2003; Barragan et al. 2016). An increase in the system energy is consistent with experimental evidence that the first step of the bifurcated oxidation of quinol at the Qo site of the Cyt bc complexes is the endergonic (energy-accepting) process (Crofts 2004a, 2000c, 2021; Crofts et al. 2000, 2013). Thermodynamic analysis of the first electron transfer in Rb. sphaeroides, performed in terms of the Marcus-Brønsted equation, suggested that the overall reaction of an electron and a proton transfer from quinol to the ISP favored a proton first then electron sequence (for details, see Crofts et al. 2000; Zu et al. 2003; Crofts 2021). The overall free energy change in the reaction Hisox + QH2 → His(H+)red + QH· was estimated as ΔG ~ 9–10 kJ mol−1.

DFT modelling of the first stage of 2,3,5-trimethylbenzoquinol (TMBQH2) oxidation by the ISP. a Depicts a fragment of the Cytb6f structure (PDB code 1Q90) used as a model system for analyzing the H(1) transfer from TMBQH2 (the tail-less plastoquinol analog) to the ISP. b Shows the dependence of the system energy versus the distance between the H(1) atom donated by TMBQH2 and the Nε atom of His155 (PDB entry 1Q90). The energy profile was reconstructed on the basis of data presented in (Ustynyuk and Tikhonov 2022)
The second step of quinol oxidation
The endergonic nature of the first step of the quinol oxidation suggests that the semiquinone product SPQ· requires its rapid removal to ensure sufficiently high rate of the forward reaction of electron transfer. The semiquinone species TMBQH is further oxidized to TMBQ; heme b6L acts as the electron acceptor, the −COO−group of Glu78 fulfills the role of the proton recipient (Fig. 12a). The question arises: is it possible that the radical TMBQH formed in the vicinity of the Fe2S2 cluster might efficiently donate an electron to heme b6L without its movement toward heme b6L and Glu78? The distances between TMBQH positioned near the Fe2S2 cluster and the participants of the second reaction (heme b6L and the −COO− group of Glu78) are too long to provide efficient oxidation of TMBQH without its shift toward heme b6L. DFT computations revealed that the direct oxidation of TMBQH by remote heme b6L and the proton transfer to the −COO− group (~ 6 Å) would be strongly restricted due to a high energy barrier (ΔE≠ ~ 270 kJ mole−1; Ustynyuk and Tikhonov 2022). However, as the radical TMBQH get closer to Glu78 and heme b6L, its oxidation becomes possible, because the reaction becomes exergonic. Figure 12b demonstrates that an energy of the model system decreases with shortening the distance between TMBQH and Glu78. The energy of the system further decreases with the transfer of the proton H(2) of TMBQH from its initial position, corresponding to minimum of energy in Fig. 12b, to the −COO− group of Glu78 (Fig. 12c, ΔE2 ≈ − 100 kJ mole−1).

DFT modelling of the second stage of a bifurcated oxidation of plastoquinol. a Depicts a fragment of the crystal structure of the Cytb6f complex (PDB code 1Q90) used as a model system for the analysis of the radical TMBQH oxidation by heme b6L. b demonstrates how an energy of the model system changes with shortening the distance between TMBQH and Glu78. The distance between the H(2) atom of TMBQH and the nearest atom O of the –COO− group of Glu78 is used as the parameter characterizing the shift of TMBQH toward Glu78. c shows changes in the energy of the model system upon the transfer of the proton H(2) of TMBQH from its initial position, corresponding to minimum of energy in b, to the −COO− group of Glu78. The energy profile was reconstructed on the basis of data presented in (Ustynyuk and Tikhonov 2022)
The outcomes of DFT calculations support the notion that the first step of the bifurcated reaction is the energy-accepting process which determines the overall rate of PQH2 oxidation. A rapid movement of plastosemiquinone toward heme b6L and the –COO− group of Glu78 would accelerate its oxidation, thereby facilitating the overall rate of PQH2 oxidation. In favor of a high mobility of plastoquinone inside the intraprotein cavity of the portal Qo may serve the results of molecular dynamics simulations, which predict that PQH2 can move throw the cavity (about 5-7 Å) within a few ns (Hasan et al. 2014).
Energetics and kinetics of the PQH2/PQ turnover inside the Qo portal
The cycle of PQH2/PQ turnover includes: the PQH2 enter into the quinol-binding portal Qo, the PQH2 oxidation per se, and the release of PQ from the portal Qo. The steric constraints in the Cyt b6f structure could limit diffusion-controlled processes associated with the PQH2 turnover, imposing the limitations on the rate of electron transfer from PQH2 to Cyt f. The influence of diffusional restrictions in the Qo portal on the quinol turnover has been clearly demonstrated in Cramer’s laboratory (Ness et al. 2019). Using genetic modifications of subIV (Pro105Ala and Pro112Ala) in cyanobacteria Synechococcus sp. PCC 7002, the authors were able to narrow the entrance into the portal Qo, thereby creating an obstacle for the penetration of PQH2 to the quinol-binding site. In the result of this manipulation, they observed a marked deceleration of the Cyt f reduction (average half-times 6.7 ± 1.3 ms → 20.7 ± 5.2 ms) and a two-fold slowing down in the rate of cell growth.
The redox steps of PQH2 turnover inside the Cytb6f complex are the basic events that determine the overall rate of the intersystem electron transport. The uphill electron transfer from PQH2 to ISPox is assumed to determine the rate of the two-electron quinol oxidation at the Qo site (Crofts 2004a, 2004b, 2021). The energy uptake (ΔE1 > 0) during this reaction would be regained due to the energy-donating step of the plastosemiquinone (PSQ·) oxidation (ΔE2 < 0), because PSQ· is a strong electron donor capable of reducing heme Cyt b6L. The overall energy balance should be favorable for PQH2 oxidation (ΔE1 + ΔE2 < 0). From the physical point of view, the coupling between the energy-accepting and energy-donating reactions (1) and (2) could be realized, provided both reactions occur as concerted processes (Blumenfeld and Tikhonov 1994; Snyder et al. 2000; Osyczka et al. 2004, 2005; Zhu et al. 2007; Reece and Nocera 2009). A simple kinetic model suggests that the rate constant k2 of electron transfer from PSQ· to heme b6L may be about k2 ~108 s−1 (Tikhonov 2014). According to molecular dynamics simulations for the Cytbc1 (Crofts et al. 2017), it is feasible that semiquinone can rapidly move (~ 4 Å/ns) in the cavity of the hydrophobic portal Qo. It is highly likely that the dissociation of the radical pair and semiquinone movements occur rapidly, in the range < 10 μs. Since the oxidation of PSQ· would proceed much more rapidly than the first step of PQH2 oxidation by the ISP (τ1/2 ≥ 4–20 мc), we could suggest that both steps of PQH2 oxidation might be virtually considered as synchronous processes.
Note that plastosemiquinones may also serve as the electron donors for superoxide radical generation in the Cytbc complexes that might proceed via the side channel of SPQ· oxidation due to electron transfer to molecular oxygen and the formation of harmful superoxide radicals (Cape et al. 2006, 2007; Baniulis et al. 2013, 2016; Bujnowicz et al. 2019; Sarewicz et al. 2021). The removal of semiquinone species due to their rapid oxidation by Cyt b6L would minimize the production of reactive oxygen species (ROS). Rapid disappearance of chemically active semiquinone radicals would diminish a probability of O2 reduction by PSQ·, thereby precluding the formation of ROS (for review, see Asada 2006; Mubarakshina et al. 2006; Halliwell and Gutteridge 2007; Ivanov et al. 2018). Relatively short life-time of PSQ· may explain why it is difficult to detect semiquinone radicals in the Cytbc1 and Cytb6f complexes by EPR (Sarewicz et al. 2017, 2021). Note that the specific rate of superoxide formation in the Cytb6f complexes is higher by an order of magnitude than in the Cytbc1 complex (Baniulis et al. 2013). It has been proposed that the retention of PSQ· due to the steric effects of the Chl phytyl tail should stimulate the superoxide production in the Cytb6f.
Summing up, we can state that: (1) the second reaction of PQH2 oxidation occurs much more rapidly (by a factor of ~ 103) than the first reaction (for references, see Crofts 2021); (2) the intermediate complex ISPH·-PSQ· is an unstable structure; after the radical pair break, the tethered diffusion of the extrinsic domain of the reduced ISPred toward heme f occurs; (3) electron transfer from PSQ· to heme b6L should be stimulated by migration of PSQ· toward heme Cyt f to shorten the distance between the electron donor and acceptor. Using the Moser–Dutton ruler for the rate of electron tunnelling between redox centers (Moser et al. 1997; Page et al. 1999), expanded by Crofts (2004b) for proton-coupled electron transfer reactions, we could evaluate the rate constant k1 of PQH2 oxidation within the framework of a simple kinetic model as k1 ~ 40–170 s−1; these values reasonably agree with experimental data on electron transfer from PQH2 to P700+ (Tikhonov 2018).
Regulation of the Cytb 6 f functions in chloroplasts
The pH-dependent control of the Cytb 6 f turnover
The light-induced acidification of the lumen (pHin↓) is one of the major regulators of the intersystem electron flow. The feedback control of electron transport provides optimal functioning of photosynthetic apparatus, preventing an excessive acidification of the lumen and the over-excitation of PSII (Rumberg and Siggel 1969; Tikhonov et al. 1981, 1984; Nishio and Whitmarsh 1993; Schönknecht et al. 1995; Kramer et al. 1999; Jahns et al. 2002). A decrease in pHin decelerates the oxidation of PQH2 at the Qo site and attenuates the activity of PSII due to an enhancement of heat dissipation of light energy in the light-harvesting antenna of PSII (Rees et al. 1989; Müller et al. 2001; Li et al. 2009).
The ISP operates as a switch controlling the rate of the intersystem electron flow (for illustration, see Fig. 13). Electron transfer from PQH2 to the ISP is governed by a “proton-gated” affinity mechanism, determined by the ability of proton binding to the His group of the ISP (Brandt 1996; Link 1997, 1999). The intrinsic mobile fragment of the ISP is positioned in the lumen-oriented domain of the enzyme (the Qo site of the Cytb6f), and connected with the bulk of the lumen via two intra-protein proton-conductive “trails” (Tikhonov 2014, 2018). The oxidized ISP (ISPox) is characterized by pKox ~ 6–6.5 (Finazzi 2002; Soriano et al. 2002). At pHin > pKox, the proton-binding His residue of the ISP (e.g., His155 in C. reinhardtii or His128 in spinach) is deprotonated, being able of accepting a proton from PQH2. Increased activity of hydrogen ions inside the lumen (e.g., at pHin < 6.2) will induce the protonation of the functional His residue of the ISP. The reduction of the ISP induces an increase in the affinity of the ISP to a proton; the pKred value of His increases with the reduction of ISP up to pKred ≈ 8.3 − 8.9 (Zu et al. 2003; Iwaki et al. 2005; Lin et al. 2006; Hsueh et al. 2010; Lhee et al. 2010). In illuminated chloroplasts, the relationship pHin ≤ pKred holds true. The protonated His is unable to accept a proton; therefore, the acidification of the lumen would impede the oxidation of PQH2. Thus, the back-pressure of hydrogen ions from the lumen would retard the proton dissociation from the ISP(H+), thereby slowing down the oxidation of PQH2 and decelerating electron flow through the Cytb6f. The oxidation of the ISP by Cyt f would lead to a decrease in pKISP, promoting the proton release from the ISP into the lumen.

A diagram illustrating the redox-dependent protonation/deprotonation events in the ISP protein of the Cytb6f complex. Blue and red lines show tentative pH-dependences for the reduced and oxidized forms of the ISP. Dashed line depicts the pH-dependence of electron flow through the Cytb6f complex as calculated according to formula (3) with the model parameters pKox = 6.2 and pKred = 8.7 (see text for explanations)
The above reasonings allow to figure out the pH-dependence of the intersystem of electron flow through the Cytb6f (Jb6f). The overall electron flux should be determined by two factors: (i) a probability p(ISPox) of finding the oxidized ISPox in deprotonated state, and (ii) a probability of finding ISP in reduced and protonated state, p(ISP(H+)red. The latter factor implies that the reduction of ISP by PQH2 occurs simultaneously with the ISP protonation (the PCET reaction). Obviously, the steady-state electron flux through the Cytb6f will be proportional to the product of the two probabilities, Jb6f ~ p(ISPox) × p(ISP(H+)red. Simple calculations lead to the following expression:
The model predicts the bell-shape pH-dependence of Jb6f. In Fig. 13, green dashed line presents the function Jb6f (pH) calculated for pKox = 6.2 and pKred = 8.7 (for detail, see Tikhonov 2014, 2018). This curve adequately describes the experimental pH-dependence of the rate of electron flow through the Cytb6f complex in chloroplasts (Hope et al. 1994; Hope 2000).
The pH-dependent regulation of electron flow through the Cytb6f lies in the basis of the so-called photosynthetic control phenomenon (Rumberg et. al., 1968; West and Wiskich 1968; Schönknecht et al. 1995; Kramer et al. 1999; Foyer et al. 2012; Tikhonov 2012, 2013; Colombo et al. 2016). The term photosynthetic control implies that the rate of photosynthetic electron transport depends on metabolic state of chloroplasts, which is determined by the adenylate status of chloroplasts (the ATP/ADP ratio) (for references, see Tikhonov 2014, 2018).
In the context of pH-dependent control of PQH2 oxidation in chloroplasts, it necessary to note that the lateral heterogeneity of lamellar membranes and uneven distribution of the ATP synthase complexes may lead to a pH gradient in the lumen from the granal to the stromal thylakoids, with more significant acidification of the granal lumen. A critical analysis of experimental data and the results of computer modelling (Tikhonov and Vershubskii 2014; Vershubskii et al. 2017) support the notion that the long-range diffusion of protons within the lumen and obstructed diffusion of mobile electron carriers (PQH2 and Pc) could influence the lateral profiles of pH along the thylakoid membranes. The model predicts significant alkalization of the inter-thylakoid gap and the establishment of nonuniform lateral profiles of ΔpH under the photophosphorylation conditions. As it was suggested by Kirchhoff et al. (2017), the partial decoupling of the proton concentrations between the lumen compartments of granal and stromal thylakoids might have important implications for independent regulation of LEF and CEF.
Another mechanism of pH-dependent down-regulation of the intersystem electron flow is accomplished by attenuating PSII turnover caused by the lumen acidification known as the non-photochemical quenching, NPQ (for references, see Rees et al. 1989; Lazar 1999; Müller et al. 2001; Li et al. 2009; Jahns and Holzwarth 2012; Demmig-Adams et al. 2012; Ruban et al. 2012; Rochaix 2014; Colombo et al. 2016). The NPQ mechanism is realized by the light-induced enhancement of thermal dissipation of energy in the light-harvesting antenna of PSII. Both down-regulation mechanisms (the pH-dependent deceleration of Cytb6f turnover and generation of NPQ) are characterized by close pK values (~ 6.0–6.5), providing similar contributions to the pH-dependent attenuation of electron flow from PSII to PSI.
Plastoquinone pool capacity
PQH2/PQ traffic in the thylakoid membrane over-crowded with protein complexes (about 70–80% protein in spinach) is one of the factors that could influence the rate of electron transport between PSII and PSI (Kirchhoff et al. 2000). The diffusion-controlled turnover of PQH2 and the overall rate of the intersystem electron transport may depend on the relative size of the photo-reducible plastoquinone pool (Kurreck et al. 2000). A high content of plastoquinone molecules would enhance the connectivity between spatially separated PSII and Cytb6f complexes (Haehnel 1984; Siggel et al. 1972; Tikhonov and Vershubskii 2017). A relative capacity of the PQH2/PQ pool may be determined by the plant growth conditions and their physiological state (McCauley and Melis 1986; Suslichenko and Tikhonov 2019; Flannery et al. 2021; Suslichenko et al. 2022). The regulatory feedbacks, e.g., the activation/deactivation of PSI and PSII complexes, activation of the CBC reactions, and the pH-dependent control of the Cytb6f turnover, will modulate dynamics of PQH2/PQ redox transients (Buchanan 1980; Foyer et al. 2012; Rochaix 2014). In chloroplasts, variability of the photo-reducible plastoquinone pool capacity is provided by plastoglobules, the lipid storing particles, which serve as the reservoirs contributing to the plastoquinone pool (Pralon and Kessler 2016). Plastoglobules are structurally and functionally associated with the thylakoid membranes: they are attached to stromal thylakoids and contain about 70–75% of total plastoquinone. An increase in the relative size of this pool would stimulate the connectivity between spatially separated PSII and Cytb6f complexes, thereby supporting high rates of the intersystem electron transfer (Rumberg et al. 1968; Stiehl and Witt 1969; Siggel et al. 1972; Haehnel 1984; Tikhonov and Vershubskii 2017). Enhanced size of the photo-reducible PQH2/PQ pool is characteristic of plants grown in low-light (LL) conditions (Suslichenko and Tikhonov 2019). Therefore, the LL-acclimated plants with increased size of the plastoquinone pool in chloroplasts would support efficient electron transport even with the light attenuation. Otherwise, the reduction of the PQH2/PQ pool in high light-grown plants should prevent their too high photochemical activity, thereby avoiding the oxidative stress at strong light.
Sub-localization of the Cytb 6 f complexes in thylakoid membranes
Standing at the crossroad of electron transport pathways, the Cytb6f complex performs a role of a hub which directs electrons to noncyclic or cyclic routes of electron flow (Munekage et al. 2004; Shikanai 2007; Joliot and Johnson 2011; Hertle et al. 2013; Tikhonov 2014; Strand et al. 2016; Malone et al. 2021). In chloroplasts, one of the structure-dependent mechanisms of short-term regulation of electron transport (in a timescale of seconds to minutes) may be associated with a redistribution of electron transport complexes between the granal and stromal thylakoids (for review and references, see Ruban and Johnson 2015; Wood et al. 2019). Changes in plant acclimation conditions, e.g., spectral composition and light intensity, may influence the composition and lateral distribution of photosynthetic complexes, including the Cytb6f (Boardman 1977; Anderson et al. 1988, 2012; Flannery et al. 2021). Sub-localization of the Cytb6f complexes in granal and stromal domains is dynamic, depending on the functional state of plants and architecture of chloroplasts (Vallon et al. 1991; Johnson et al. 2014; Dumas et al. 2016; Wood et al. 2018, 2019; Flannery et al. 2021; Hepworth et al. 2021). In particular, Wood et al. (2019) reported about rapid (t1/2 ~ 10 min) reversible changes in grana size: WL of low-intensity (LL) reduced size of grana in Arabidopsis and spinach, while WL of high-intensity (HL) increased grana size. The dynamics of thylakoid stacking is primarily determined by phosphorylation of the light-harvesting complex II (LHCII) (Wood et al. 2018, 2019). The relative abundance of the Cytb6f varied upon the long-term (weeks) acclimation of plants to LL and HL growth light intensity, positively correlating with changes in PSII activity. In HL-grown plants, an increase in CEF has been observed (Flannery et al. 2021; Benkov et al. 2023).
The re-modelling of the chloroplast lamellar system may regulate the partition of electrons between LEF and CEF pathways. However, the question about sub-localization of the Cytb6f and the rate of the intersystem electron transport remains a matter of discussions. There is no consensus about the light-induced re-localization of the Cytb6f between the granal and stromal domains of the thylakoid lamellas, although certain correlations between the grana size and the abundance of the Cytb6f complexes have been observed (Höhner et. al., 2020; Flannery et al. 2021). As noted above, in the Arabidopsis mutants with extremely wide grana (up to 1,600 nm in diameter) an average time of plastoquinol diffusion between the PSII to Cytb6f complexes markedly increased, but it remained independent of granum diameter close to native (Höhner et al. 2020). The re-localization of Cytb6f complexes from the granal to stromal domains of the chloroplast membranes could enhance the CEF capacities. The re-distribution of the Cytb6f might be induced by changes in the distance between adjacent membranes in stacked piles of granal thylakoids (Kirchhoff et al. 2017).
State transitions
The Cytb6f complex is involved in regulation of the light energy partitioning between the light-harvesting antennas of PSI and PSII, using the state transitions mechanism of the redistribution of mobile light-harvesting complexes between PSII and PSI (Allen 1981, 1992; Lemeille and Rochaix 2010; Minagawa 2011; Rochaix et al. 2012). A macromolecular device, which receives a signal from the reduced PQH2 pool and induces the state I → state II transition, is inherent to the Cytb6f complex and specific protein subunits bound to it. The signal transducer, which actuates the LHCII kinase, is specific for the quinone bound and its redox state (Vener et al. 1997, 1998; Zito et al. 1999; Finazzi et al. 2001; Dumas et al. 2017). Putative molecular mechanism of the kinase activation due to PQH2-induced structural changes in the Cytb6f complex has been suggested by Hasan et al. (2013a). There are the catalytic and regulatory domains of the kinase with two Cys residues, which are essential for the enzyme activity. The redox state of these residues may be regulated by Fd and thioredoxin through the membrane-bound thiol oxidoreductases (Lennartz et al. 2001; Motohashi and Hisabori 2006; Dietz and Pfannschmidt 2011). The over-reduction of the acceptor side of PSI will cause the reduction of Cys residues, thereby inducing the “State II → State I” transition, upon which a PSI activity will lessen.
Phosphorylation of the mobile light-harvesting subunits of PSII is a reversible process. After a decrease in the concentration of PQH2, the dephosphorylation of phosphorylated complexes occurs due to the LHCII phosphatase TAP/PPH1 activity (state II → state I transition; Puthiyaveetil et al. 2016). Dephosphorylated light-harvesting subunits rebind to PSII antenna, and chloroplasts return to the initial state I. Finally, we must emphasize the regulatory role of the Cytb6f complex associated with signaling and mediating gene expression in the plant cell (Depege et al. 2003; Bellafiore et al. 2005; Puthiyaveetil et al. 2016).
Concluding remarks
Standing between PSII and PSI, the Cytb6f complex provides the intersystem electron transport and plays the pivoting role in regulation of electron transport in oxygenic photosynthesis. The bifurcated oxidation of PQH2 at the Qo site is one of the clue events that determine the rate of electron transfer from PSII to PSI. Experimental studies and theoretical analysis of the bifurcated oxidation of PQH2 suggest that the overall rate of PQH2 turnover is determined by the energy-accepting (endergonic) reaction of electron transfer from PQH2 to the ISP. The further reaction of plastosemiquinone (PSQ·) oxidation is the energy-donating (endergonic) process, the rate of which should increase upon its movement toward the low-potential heme b6L within the intraprotein cavity that includes the Qo site. The movement of PSQ· closer to the heme would allow about a 103-fold increase in the rate of its oxidation (for references, see Crofts 2021). The bifurcated oxidation of PQH2 is tightly coupled to the proton release into the lumen. This process is controlled by the intra-thylakoid pHin, demonstrating the slowing down of PQH2 oxidation with the lumen acidification (Tikhonov et al. 1981, 1984; Kramer et al. 1999).
The Cytb6f complex is involved to directing electrons to alternative routes of electron transport (Munekage et al. 2004; DalCorso et al. 2008; Strand et al. 2016; Zhao et al. 2020). The Cytb6f complexes localized in grana mediate linear electron transport (LET) from PSII to PSI, while the complexes localized in stroma-exposed domains of the thylakoid membranes participate in cyclic electron transport (CET) around PSI. Structural changes and/or metabolic shifts in chloroplasts would influence a balance between LET and CET, supporting optimal functioning of photosynthetic apparatus under variable growth conditions (Yamory and Shikanai 2016).
The Cytb6f complex participate in partitioning the light energy absorbed between the light-harvesting antennas of PSI and PSII (state transitions), using the Stt7/STN7 kinase, which is inherent to the Cytb6f. Modulating the photochemical activities of PSI and PSII, these transitions provide optimal functioning of ETC under variable metabolic conditions. The feedback regulation of the redox state of the PQH2/PQ pool, caused by changes in activities of the PSI, PSII and Cytb6f complexes, may be involved in the retrograde signaling in the plant cell (Allen and Pfannschmidt 2000; Fernández and Strand (2008); Foyer et al. 2012; Mielecki et al. 2020).
Abbreviations
- CBC:
-
The Calvin–Benson cycle
- cryo-EM:
-
Cryogenic electron microscopy
- Cyt:
-
Cytochrome
- Cytb 6 f :
-
The Cyt b6f complex
- DFT:
-
Density functional theory
- EPR:
-
Electron paramagnetic resonance
- ETC:
-
Electron transport chain
- ISP:
-
The iron–sulfur protein
- Fd:
-
Ferredoxin
- FNR:
-
Ferredoxin-NADP-reductase
- NDH-1:
-
NADH dehydrogenase-like complex type-1
- PCET:
-
Proton-coupled electron transfer
- PSI and PSII:
-
Photosystem I and photosystem II, respectively
- Pc:
-
Plastocyanin
- PQ, PSQ·, and PQH2 :
-
Plastoquinone (oxidized), plastosemiquinone, and plastoquinol (completely reduced) forms of plastoquinone; respectively
- P700 :
-
Primary electron donor in PSI
- ROS:
-
Reactive oxygen species
- TDS:
-
Tridecyl-stigmatellin
- TMBQH2 :
-
2,3,5-Trimethylbenzoquinol
- WL:
-
White light
References
Albertsson P-Å (2001) A quantitative model of the domain structure of the photosynthetic membrane. Trends Plant Sci 6:349–354
Allen JF, Bennett J, Steinback KE, Arntzen CJ (1981) Chloroplast protein phosphorylation couples plastoquinone redox state to distribution of excitation energy between photosystems. Nature 291:25–29
Alric J, Pierre Y, Picot D, Rappaport F (2005) Spectral and redox characterization of the heme ci of the cytochrome b6f complex. Proc Natl Acad Sci USA 102:15860–15865
Allen JF (1992) Protein phosphorylation in regulation of photosynthesis. Biochim Biophys Acta 1098:275–335
Allen JF, Pfannschmidt T (2000) Balancing the two photosystems: photosynthetic electron transfer governs transcription of reaction centre genes in chloroplasts, Philos. Trans R Soc Lond B Biol Sci 355:1351–1359
Anderson B, Anderson JM (1980) Lateral heterogeneity in the distribution of chlorophyll-protein complexes of the thylakoid membranes of spinach chloroplasts. Biochim Biophys Acta 593:427–440
Armbruster U, Labs M, Pribil M, Viola S, Xu W, Scharfenberg M, Hertle AP, Rojahn U, Jensen PE, Rappaport F, Joliot P, Dörmann P, Wanner G, Leister D (2013) Arabidopsis CURVATURE THYLAKOID1 proteins modify thylakoid architecture by inducing membrane curvature. Plant Cell 25:2661–2678
Anderson JM, Chow WS, Goodchild DJ (1988) Thylakoid membrane organization in sun/shade acclimation. Aust J Plant Physiol 15:11–26
Anderson JM, Horton P, Kim EH, Chow WS (2012) Towards elucidation of dynamic structural changes of plant thylakoid architecture, Philos. Trans R Soc Lond B Biol Sci 367:3515–3524
Asada K (2006) Production and scavenging of reactive oxygen species in chloroplasts and their functions. Plant Physiol 141:391–396
Baniulis D, Yamashita E, Whitelegge JP, Zatsman AI, Hendrich MP, Hasan SS, Ryan CM, Cramer WA (2009) Structure-function, stability, and chemical modification of the cyanobacterial cytochrome b6f complex from Nostoc sp. PCC 7120. J Biol Chem 284:9861–9869
Baniulis D, Hasan SS, Stofleth JT, Cramer WA (2013) Mechanism of enhanced superoxide production in the cytochrome b6f complex of oxygenic photosynthesis. Biochemistry 52:8975–8983
Baniulis D, Hasan SS, Miliute I, Cramer WA (2016) Mechanisms of superoxide generation and signaling in cytochrome bc complexes. In: Cramer W, Kallas T (eds) Cytochrome complexes: evolution, structures, energy transduction, and signaling. Springer, Dordrecht, pp 397–417
Benkov MA, Suslichenko IS, Trubitsin BV, Tikhonov AN (2023) Effects of plant acclimation on electron transport in chloroplast membranes of Cucumis sativus and Cucumis melo, Biochemistry (Moscow). Supplement Series a: Membrane and Cell Biology 17:92–105
Barragan AM, Crofts AR, Schulten K, Solov’yov IA (2015) Identification of ubiquinol binding motifs at the Qo-site of the cytochrome bc1 complex. J Phys Chem B 119:433–447
Barragan AM, Schulten K, Solov’yov IA (2016) Mechanism of the primary charge transfer reaction in the cytochrome bc1 complex. J Phys Chem B 120:11369–11380
Bellafiore S, Barneche F, Peltier G, Rochaix J-D (2005) State transitions and light adaptation require chloroplast thylakoid protein kinase STN7. Nature 433:892–895
Bendall DS, Manasse RS (1995) Cyclic phosphorylation and electron transport. Biochim Biophys Acta 1229:23–38
Berry EA, Guergova-Kuras M, Huang LS, Crofts AR (2000) Structure and function of cytochrome bc complexes. Annu Rev Biochem 69:1005–1075
Bhaduri S, Zhang H, Erramilli S, Cramer WA (2019) Structural and functional contributions of lipids to the stability and activity of the photosynthetic cytochrome b6f lipoprotein complex. J Biol Chem 294:17758–17767
Bhowmick A, Hussein R, Bogacz I et al (2023) Structural evidence for intermediates during O2 formation in photosystem II. Nature. https://doi.org/10.1038/s41586-023-06038-z
Blumenfeld LA, Tikhonov AN (1994) Biophysical Thermodynamics of intracellular processes. molecular machines of the living cell. Springer, New York
Boardman NK (1977) Comparative photosynthesis of sun and shade plants. Annu Rev Plant Physiol 28:355–377
Boyer PD (1997) The ATP synthase: a splendid molecular machine. Annu Rev Biochem 66:717–749
Brandt U (1996) Bifurcated ubihydroquinone oxidation in the cytochrome bc1 complex by proton-gated charge transfer. FEBS Lett 387:1–6
Breyton C (2000) Conformational changes in the cytochrome b6f complex induced by inhibitor binding. J Biol Chem 275:13195–13201
Buchanan BB (1980) Role of light in the regulation of chloroplast enzymes. Annu Rev Plant Physiol 31:341–374
Bujnowicz Ł, Sarewicz M, Borek A, Kuleta P, Osyczka A (2019) Suppression of superoxide production by a spin-spin coupling between semiquinone and the Rieske cluster in Cytochrome bc1. FEBS Lett 593:3–12
Buchert F, Mosebach L, Gäbelein P, Hippler M (2020) PGR5 is required for efficient Q cycle in the cytochrome b6f complex during cyclic electron flow. Biochem J 477:1631–1650
Cape JL, Bowman MK, Kramer DM (2006) Understanding the cytochrome bc complexes by what they don’t do. The Q-cycle at 30. Trends Plant Sci 11:46–55
Cape JL, Bowman MK, Kramer DM (2007) A semiquinone intermediate generated at the Qo site of the cytochrome bc1 complex: importance for the Q-cycle and superoxide production. Proc Natl Acad Sci USA 104:7887–7892
Clark RD, Hind G (1983) Spectrally distinct cytochrome b-563 components in a chloroplast cytochrome b-f complex: interaction with a hydroxyquinoline N-oxide. Proc Natl Acad Sci USA 80:6249–6253
Colombo M, Suorsa M, Suorsa RF, Ferrari R, Tadini L, Barbato R, Pesaresi P (2016) Photosynthesis control: an underrated short-term regulatory mechanism essential for plant viability. Plant Signal Behav 11:e1165382
Cramer WA, Zhang H, Yan J, Kurisu G, Smith JL (2006) Transmembrane traffic in the cytochrome b6f complex. Annu Rev Biochem 75:769–790
Cramer WA, Hasan SS (2016) Structure-function of the cytochrome b6f lipoprotein complex. In: Cramer WA, Kallas T (eds) Cytochrome complexes: evolution, structures, energy transduction, and signaling. Springer, Dordrecht, pp 177–207
Cramer WA, Hasan SS, Yamashita E (2011) The Q cycle of cytochrome bc complexes: a structure perspective. Biochim Biophys Acta 1807:788–802
Crofts AR (2004a) The cytochrome bc1 complex: function in the context of structure. Annu Rev Physiol 66:689–733
Crofts AR (2004b) Proton-coupled electron transfer at the Qo-site of the bc1 complex controls the rate of ubihydroquinone oxidation. Biochim Biophys Acta 1655:77–92
Crofts AR (2004c) The Q-cycle: a personal perspective. Photosynth Res 80:223–243
Crofts AR (2021) The modified Q-cycle: a look back at its development and forward to a functional mode. Biochim Biophys Acta 1862:148417
Crofts AR, Meinhardt SW, Jones KR, Snozzi M (1983) The role of the quinone pool in the cyclic electron-transfer chain of Rhodopseudomonas sphaeroides: a modified Q-cycle mechanism. Biochim Biophys Acta 723:202–218
Crofts AR, Guergova-Kuras M, Kuras R, Ugulava N, Li J, Hong S (2000) Proton-coupled electron transfer at the Qo site: what type of mechanism can account for the high activation barrier? Biochim Biophys Acta 1459:456–466
Crofts AR, Holland JT, Victoria D, Kolling DRJ, Dikanov SA, Gilbreth R, Lhee S, Kuras R, Kuras MG (2008) The Q cycle reviewed: how well does a monomeric mechanism of the bc1 complex account for the function of a dimeric complex? Biochim Biophys Acta 1777:1001–1019
Crofts AR, Hong S, Wilson C, Burto R, Victoria D, Harrison C, Shulten K (2013) The mechanism of ubihydroquinone oxidation at the Qo-site of the cytochrome bc1 complex. Biochim Biophys Acta 1827:1362–1377
Crofts AR, Rose SW, Burton RL, Desi AV, Kenis PJA, Dikanov SA (2017) The Q-cycle mechanism of the bc1 complex: a biologist’s perspective on atomic studies. J Phys Chem 121:3701–3717
DalCorso G, Pesaresi P, Masiero S, Aseeva E, Schunemann D, Finazzi G, Joliot P, Barbato R, Leister D (2008) A complex containing PGRL1 and PGR5 is involved in the switch between linear and cyclic electron flow in Arabidopsis. Cell 132:273–285
De Vitry C, Ouyang Y, Finazzi G, Wollman F-A, Kallas T (2004) The chloroplast Rieske iron–sulfur protein: at the crossroad of electron transport and signal transduction. J Biol Chem 279:44621–44627
Dekker JP, Boekema EJ (2005) Supramolecular organization of thylakoid membrane proteins in green plants. Biochim Biophys Acta 1706:12–39
Demmig-Adams B, Cohu CM, Muller O, Adams WW (2012) Modulation of photosynthetic energy conversion efficiency in nature: from seconds to seasons. Photosynth Res 113:75–88
Depege N, Bellafiore S, Rochaix JD (2003) Role of chloroplast ̀ protein kinase Stt7 in LHCII phosphorylation and state transition in Chlamydomonas. Science 299:1572–1575
Dietz K-J, Pfannschmidt T (2011) Novel regulators in photosynthetic redox control of plant metabolism and gene expression. Plant Physiol 155:1477–1485
Dumas L, Chazaux M, Peltier G, Johnson X, Alric J (2016) Cytochrome b6f function and localization, phosphorylation state of thylakoid membrane proteins and consequences on cyclic electron flow. Photosynth Res 129:307–320
Dumas L, Zito F, Blangy S, Auroy P, Johnson X, Peltier G, Alric J (2017) A stromal region of cytochrome b6f subunit IV is involved in the activation of the Stt7 kinase in Chlamydomonas. Proc Natl Acad Sci USA 114:12063–12068
Edwards G, Walker D (1983) C3, C4: mechanisms, and cellular and environmental regulation, of photosynthesis. University of California Press, Berkeley
Fernández AP, Strand A (2008) Retrograde signaling and plant stress: plastid signals initiate cellular stress responses. Curr Opin Plant Biol 11:509–513
Finazzi G, Zito F, Barbagallo RP, Wollman F-A (2001) Contrasted effects of inhibitors of cytochrome b6f complex on state transitions in Chlamydomonas reinhardtii: the role of Qo site occupancy in LHCII kinase activation. J Biol Chem 276:9770–9774
Finazzi G (2002) Redox-coupled proton pumping activity in cytochrome b6f, as evidenced by the pH dependence of electron transfer in whole cells of Chlamydomonas reinhardtii. Biochemistry 41:7475–7482
Flannery SE, Hepworth C, Wood WHJ, Pastorelli F, Hunter CN, Dickman MJ, Jackson PJ, Johnson MP (2021) Developmental acclimation of the thylakoid proteome to light intensity in Arabidopsis. Plant J 105:223–244
Foyer CH, Neukermans J, Queval G, Noctor G, Harbinson J (2012) Photosynthetic control of electron transport and the regulation of gene expression. J Exp Bot 63:1637–1661
Frolov AE, Tikhonov AN (2009) The oxidation of plastoquinol by a cytochrome b6f complex: a density functional theory study. Russian J Phys Chem A 83:506–508
Furbacher PN, Girvin ME, Cramer WA (1989) On the question of interheme electron transfer in the chloroplast cytochrome b6 in situ. Biochemistry 28:8990–8998
Gross EL (1993) Plastocyanin: structure and function. Photosynth Res 37:103–116
Gu L, Grodzinski B, Han J, Marie T, Zhang Y-J, Song YC, Sun Y (2022) Granal thylakoid structure and function: explaining an enduring mystery of higher plants. New Phytol 236:319–329
Haehnel W (1976) The reduction kinetics chlorophyll aI as indicator for proton uptake between the light reactions in chloroplasts. Biochim Biophys Acta 440:506–521
Haehnel W, Propper A, Krause H (1980) Evidence for complexed plastocyanin as the immediate electron donor of P-700. Biochim Biophys Acta 593:384–399
Haehnel W (1984) Photosynthetic electron transport in higher plants. Annu Rev Plant Physiol 35:659–693
Halliwell B, Gutteridge JMC (2007) Free Radicals in biology and medicine, 5th edn. Oxford University, Oxford
Harbinson J, Hedley CL (1989) The kinetics of P-700+ reduction in leaves: a novel in situ probe of thylakoid functioning. Plant, Cell and Environ 12:357–369
Hasan SS, Cramer WA (2012) On rate limitations of electron transfer in the photosynthetic cytochrome b6f complex. Phys Chem Chem Phys 14:13853–13860
Hasan SS, Cramer WA (2014) Internal lipid architecture of the hetero-oligomeric cytochrome b6f complex. Structure 22:1008–1015
Hasan SS, Yamashita E, Cramer WA (2013a) Transmembrane signaling and assembly of the cytochrome b6f-lipidic charge transfer complex. Biochim Biophys Acta 1827:1295–1308
Hasan SS, Stofleth JY, Yamashita E, Cramer WA (2013b) Light-induced conformational changes within the cytochrome b6f complex of oxygenic photosynthesis. Biochemistry 52:2649–2654
Hasan SS, Yamashita E, Baniulis D, Cramer WA (2013c) Quinone-dependent proton transfer pathways in the photosynthetic cytochrome b6f complex. Proc Natl Acad Sci USA 110:4297–4302
Hasan SS, Proctor EA, Yamashita E, Dokholyan NV, Cramer WA (2014) Traffic within the cytochrome b6f lipoprotein complex: gating of the quinone portal. Biophys J 107:1620–1628
Hepworth C, Wood WHJ, Emrich-Mills TZ, Proctor MS, Casson S, Johnson MP (2021) Dynamic thylakoid stacking and state transitions work synergistically to avoid acceptor-side limitation of photosystem I. Nat Plants 7:87–98
Hertle AP, Blunder T, Wunder T, Pesaresi P, Pribil M, Armbruster U, Leister D (2013) PGRL1 is the elusive ferredoxin-plastoquinone reductase in photosynthetic cyclic electron flow. Mol Cell 49:511–523
Höhner R, Pribil M, Herbstová M, Lopez LS, Kunz H-H, Li M, Wood M, Svoboda M, Puthiyaveetil S, Leister L, Kirchhoff H (2020) Plastocyanin is the long-range electron carrier between photosystem II and photosystem I in plants. Proc Natl Acad Sci USA 117:15354–15362
Hope AB, Valente P, Matthews DB (1994) Effects of pH on the kinetics of redox reactions in and around the cytochrome bf complex in an isolated system. Photosynyh Res 42:111–120
Hope AB (2000) Electron transfers amongst cytochrome f, plastocyanin and photosystem I: kinetics and mechanisms. Biochim Biophys Acta 1456:5–26
Hsueh K-L, Westler WM, Markley JL (2010) NMR investigations of the Rieske protein from Thermus thermophilus support a coupled proton and electron transfer mechanism. J Am Chem Soc 132:7908–7918
Hurt E, Hauska G (1982) Identification of the polypeptides in the cytochrome B6/f complex from spinach chloroplasts with redox-center-carrying subunits. J Bioenerg Biomembr 14:405–424
Hurt EC, Hauska G (1983) Cytochrome B6 from isolated cytochrome B6f complexes. Evidence for two spectral forms with different midpoint potentials. FEBS Lett 153:413–419
Husen P, Solov’yov IA (2016) Spontaneous binding of molecular oxygen at the Qo-site of the bc1 complex could stimulate superoxide formation. J Am Chem Soc 138:12150–12158
Ivanov BN, Borisova-Mubarakshina MM, Kozuleva MA (2018) Formation mechanisms of superoxide radical and hydrogen peroxide in chloroplasts, and factors determining the signalling by hydrogen peroxide. Funct Plant Biol 45:102–110
Iwai M, Takizawa K, Tokutsu R, Okamuro A, Takahashi Y, Minagawa J (2010) Isolation of the elusive supercomplex that drives cyclic electron flow in photosynthesis. Nature 464:1210–1213
Iwaki M, Yakovlev G, Hirst J, Osyczka A, Dutton PL, Marshall D, Rich P (2005) Direct observation of redox-linked histidine bc1 complex by ATR-FTIR spectroscopy. Biochemistry 44:4230–4237
Jahns P, Holzwarth AR (2012) The role of the xanthophyll cycle and of lutein in photoprotection of photosystem II. Biochim Biophys Acta 1817:182–193
Jahns P, Graf M, Munekage Y, Shikanai T (2002) Single point mutation in the Rieske iron–sulfur subunit of cytochrome b6/f leads to an altered pH dependence of plastoquinol oxidation in Arabidopsis. FEBS Lett 519:99–102
Joliot P, Joliot A (1988) The low-potential electron-transfer chain in the cytochrome b6f complex. Biochim Biophys Acta 933:319–333
Joliot P, Joliot A (2002) Cyclic electron transfer in plant leaf. Proc Natl Acad Sci USA 99:10209–10214
Joliot P, Johnson GN (2011) Regulation of cyclic and linear electron flow in higher plants. Proc Natl Acad Sci USA 108:13317–13322
Johnson JE, Berry JA (2021) The role of Cytochrome b6f in the control of steady-state photosynthesis: a conceptual and quantitative model. Photosynth Res 148:101–136
Junge W, Nelson N (2015) ATP synthase. Annu Rev Biochem 83:631–657
Johnson MP, Vasilev C, Olsen JD, Hunter CN (2014) Nanodomains of cytochrome b6f of photosystem II complexes in spinach grana thylakid membranes. Plant Cell 26:3051–3061
Kirchhoff H, Horstmann S, Weis E (2000) Control of the photosynthetic electron transport by PQ diffusion in microdomains in thylakoids of higher plants. Biochim Biophys Acta 1459:148–168
Kirchhoff H, Mukherjee U, Galla HJ (2002) Molecular architecture of the thylakoid membrane: lipid diffusion space for plastoquinone. Biochemistry 41:4872–4882
Kirchhoff H, Hall C, Wood M, Herbstová M, Tsabari O, Nevo R, Charuvi D, Shimoni E, Reich Z (2011) Dynamic control of protein diffusion within the granal thylakoid lumen. Proc Natl Acad Sci USA 108:20248–20253
Kirchhoff H, Li M, Puthiyaveetil S (2017) Sublocalization of cytochrome b6f complexes in photosynthetic membranes. Trends Plant Sci 22:574–582
Kramer DM, Crofts AR (1993) The concerted reduction of the high- and low-potential chains of the bf complex by plastoquinol. Biochim Biophys Acta 1183:72–84
Kramer DM, Sacksteder CA, Cruz JA (1999) How acidic is the lumen? Photosynth Res 60:151–163
Kurisu G, Zhang H, Smith JL, Cramer WA (2003) Structure of the cytochrome b6f complex of oxygenic photosynthesis: tuning the cavity. Science 302:1009–1014
Kurreck J, Schodel R, Renger G (2000) Investigation of the plastoquinone pool size and fluorescence quenching in thylakoid membranes and photosystem II (PS II) membrane fragments. Photosynth Res 63:171–182
Laisk A, Oja V, Eichelmann H (2016) Kinetics of plastoquinol oxidation by the Q-cycle in leaves. Biochim Biophys Acta 1857:819–830
Laughlin TG, Bayne AN, Trempe J-F, Savage DF, Davies KM (2019) Structure of the complex I-like molecule NDH of oxygenic photosynthesis. Nature 566:411–414
Laughlin TG, Savage DF, Davies KM (2020) Recent advances on the structure and function of NDH-1: The complex I of oxygenic photosynthesis. Biochim Biophys Acta 1861:148254
Lazar D (1999) Chlorophyll a fluorescence induction. Biochim Biophys Acta 1412:1–28
Lemeille S, Rochaix J-D (2010) State transitions at the crossroad of thylakoid signaling pathways. Photosynth Res 106:33–46
Lennartz K, Plucken H, Seidler A, Westhoff P, Bechtold N, Meierhoff K (2001) HCF164 encodes a thioredoxin-like protein involved in the biogenesis of the cytochrome b6f complex in Arabidopsis. Plant Cell 13:2539–2551
Lhee S, Kolling DRJ, Nair SK, Dukatov SA, Crofts AR (2010) Modifications of protein environment of the [2Fe-2S] cluster of the bc1 complex: effects on the biophysical properties of the Rieske iron–sulfur protein and on the kinetics of the complex. J Biol Chem 285:9233–9248
Li Z, Wakao S, Fischer BB, Niyogi KK (2009) Sensing and responding to excess light. Annu Rev Plant Biol 60:239–260
Lin I-J, Chen Y, Fee JA, Song J, Westler WM, Markley JL (2006) Rieske protein from Thermus thermophilus: 15N NMR titration study demonstrates the role of iron-ligated histidines in the pH dependence of the reduction potential. J Am Chem Soc 128:10672–10673
Link TA (1997) The role of the “Rieske” iron sulfur protein in the hydroquinone oxidation (Qp) site of the cytochrome bc1 complex: the “proton-gated affinity change” mechanism. FEBS Lett 412:257–264
Link TA (1999) The structures of Rieske and Rieske-type proteins. Adv Inorg Chem 47:83–157
Malone LA, Qian P, Mayneord GE, Hitchcock A, Farmer DA, Thompson RF, Swainsbury DJK, Ranson NA, Hunter CN, Johnson MP (2019) Cryo-EM Structure of the spinach cytochrome b6f complex at 3.6 Å resolution. Nature 575:535–539
Malone LA, Proctor MS, Hitchcock A, Hunter CN, Johnson MP (2021) Cytochrome b6f – Orchestrator of photosynthetic electron transfer. Biochim Biophis Acta 1862:148380
Mamedov M, Govindjee G, Nadtochenko V, Semenov AYu (2015) Primary electron transfer processes in photosynthetic reaction centers from oxygenic organisms. Photosynth Res 125:51–63
Mayer JM, Rhile IJ (2004) Thermodynamics and kinetics of proton-coupled electron transfer: stepwise vs. concerted pathways. Biochim Biophys Acta 1655:51–58
McCauley SW, Melis A (1986) Quantification of plastoquinone photoreduction in spinach chloroplasts. Photosynth Res 8:3–16
Mielecki J, Gawroński P, Karpiński S (2020) Retrograde signaling: understanding the communication between organelles. Int J Mol Sci 21:6173. https://doi.org/10.3390/ijms21176173
Minagawa J (2011) State transitions—the molecular remodeling of photosynthetic supercomplexes that controls energy flow in the chloroplast. Biochim Biophys Acta 1807:897–905
Mitchell P (1969) Chemiosmotic coupling and energy transduction. Theor Exp Biophys 2:159–216
Mitchell P (1975) The protonmotive Q cycle: a general formulation. FEBS Lett 59:137–139
Mitchell P (1976) Possible molecular mechanisms of the protonmotive function of cytochrome systems. J Theor Biol 62:327–367
Mitchell P (2011) Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biochim Biophys Acta 1807:1507–1538
Moser CC, Page CC, Chen X, Dutton PL (1997) Biological electron tunnelling through native protein media. J Biol Inorg Chem 2:393–398
Motohashi K, Hisabori T (2006) HCF164 receives reducing equivalents from stromal thioredoxin across the thylakoid membrane and mediates reduction of target proteins in the thylakoid lumen. J Biol Chem 281:35039–35047
Mubarakshina M, Khorobrykh S, Ivanov B (2006) Oxygen reduction in chloroplast thylakoids results in production of hydrogen peroxide inside the membrane. Biochim Biophys Acta 1757:1496–1503
Munekage Y, Hashimoto M, Miyake C (2004) Cyclic electron flow around photosystem I is essential for photosynthesis. Nature 429:579–582
Munekage Y, Hojo M, Meurer J, Endo T, Tasaka M, Shikanai T (2002) PGR5 is involved in cyclic electron flow around photosystem I and is essential for photoprotection in Arabidopsis. Cell 110:361–371
Munekage YN, Genty B, Peltier G (2008) Effect of PGR5 impairment on photosynthesis and growth in Arabidopsis thaliana. Plant Cell Physiol 49:1688–1698
Mulkidjanian AY (2010) Activated Q-cycle as a common mechanism for cytochrome bc1 and cytochrome b6f complexes. Biochim Biophys Acta 1797:1858–1868
Müller P, Li X-P, Niyogi KK (2001) Non-photochemical quenching. A response to excess light energy. Plant Physiol 125:1558–1566
Nawrocki WJ, Bailleul B, Picot D, Cardol P, Rappaport F, Wollman F-A, Joliot P (2019) The mechanism of cyclic electron flow. Biochim Biophys Acta 1860:433–438
Nakamura A, Suzawa T, Kato Y, Watanabe T (2011) Species dependence of the redox potential of the primary electron donor P700 in photosystem I of oxygenic photosynthetic organisms revealed by spectoelectrochemistry. Plant Cell Physiol 52:815–823
Nelson N, Yocum CF (2006) Structure and function of photosystems I and II. Annu Rev Plant Biol 57:521–565
Ness J, Naurin S, Effinger K, Stadnytskyi V, Ibrahim IM, Puthiyaveetil S, Cramer WA (2019) Structure-based control of the rate limitation of photosynthetic electron transport. FEBS Lett 593:2103–2111
Nishio JN, Whitmarsh J (1993) Dissipation of the proton electrochemical potential in intact chloroplasts: II. The pH gradient monitored by cytochrome f reduction kinetics. Plant Physiol 101:89–96
Nitscke W, Joliot P, Leibl U, Rutherford AW, Hauska G, Müller A, Riedel A (1992) The pH dependence of the redox midpoint of the 2Fe2S cluster from cytochrome b6f complex (the ‘Rieske centre’). Biochim Biophys Acta 1102:266–268
Noodleman L, Peng CY, Case DA, Mouesca J-M (1995) Orbital interactions, electron delocalization, and spin coupling in iron–sulfur clusters. Coor Chem Rev 144:199–244
Noodleman L, Lovell T, Liu T, Himo F, Torres RA (2002) Insights into properties and energetics of iron–sulfur proteins from simple clusters to nitrogenase. Curr Opin Chem Biol 6:259–273
Osyczka A, Moser CC, Daldal F, Dutton PL (2004) Reversible redox energy coupling in electron transfer chains. Nature 427:607–612
Osyczka A, Moser CC, Dutton PL (2005) Fixing the Q cycle. Trends Biochem Sci 30:176–182
Osyczka A, Zhang H, Mathe C, Rich PR, Moser CC et al (2006) Role of the PEWY glutamate in hydroquinone-quinone oxidation-reduction catalysis in the Qo site of cytochrome bc1. Biochemistry 45:10492–10503
Page CC, Moser CC, Chen X, Dutton PL (1999) Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 402:47–52
Palmer G (1985) The electron paramagnetic resonance of metalloproteins. Biochem Soc Trans 13:548–560
Pierre Y, Breyton C, Kramer D, Popot J-L (1995) Purification and characterization of the cytochrome b6f complex from Chlamydomonas reinhardtii. J Biol Chem 270:29342–29349
Pietras R, Sarewicz M, Osyczka A (2016) Distinct properties of semiquinone species detected at the ubiquinol oxidation Qo of cytochrome bc1 and their mechanistic implications. J R Soc Interface 13:20160133
Ponamarev MV, Cramer WA (1998) Perturbation of the internal water chain in cytochrome f of oxygenic photosynthesis: loss of concerted reduction of cytochromes f and b. Biochemistry 37:17199–17208
Postila PA, Kaszuba K, Sarewicz M, Osyczka A, Vattulainen I, Róg T (2013) Key role of water in proton transfer at the Qo-site of the cytochrome bc1 complex predicted by atomistic molecular dynamics simulations. Biochim Biophys Acta 1827:761–768
Pralon T, Kessler F (2016) Plastoglobules: lipid droplets at the thylakoid membrane. In: Kirchhoff H (ed) Chloroplasts: current research and future trends. Caister Academic Press, Norfolk, pp 171–186
Pribil M, Labs M, Leister D (2014) Structure and dynamics of thylakoids in land plants. Environ Bot 65:1955–1972
Pribil M, Sandoval-Ibáñez O, Xu W, Sharma A, Labs M, Liu Q, Galgenmüller C, Schneider T, Wessels M, Matsubara S, Jansson S, Wanner G, Leister D (2018) Fine-tuning of photosynthesis requires CURVATURE THYLAKOID1- mediated thylakoid plasticity. Plant Physiol 176:2351–2364
Proctor MS, Malone LA, Farmer DA, Swainsbury DJK, Hawkings FR, Pastorelli F, Emrich-Mills TZ, Siebert CA, Hunter CN, Johnson MP, Hitchcock A (2022) Cryo-EM structures of the Synechocystis sp. PCC 6803 cytochrome b6f complex with and without the regulatory etP subunit. Biochemical J 479:1487–1503
Ptushenko VV, Zhigalova TV, Avercheva OV, Tikhonov AN (2019) Three phases of energy-dependent induction of P+700 and Chl a fluorescence in Tradescantia fluminensis leaves. Photosynth Res 139:509–522
Puthiyaveetil S, Kirchhoff H, Höhner R (2016) Structural and functional dynamics the thylakoid membrane system. In: Kirchhoff H (ed) Chloroplasts: current research and future trends. Caister Academic Press, Norfolk, pp 59–87
Rees D, Young A, Noctor G, Britton G, Horton P (1989) Enhancement of the pH-dependent dissipation of excitation energy in spinach chloroplasts by light activation: correlation with the synthesis of zeaxanthin. FEBS Lett 256:85–90
Reece SY, Nocera DG (2009) Proton-coupled electron transfer in biology: results from synergetic studies in natural and model systems. Annu Rev Biochem 78:673–699
Rich PR, Bendall DS (1980) The redox potentials of the B-type cytochromes of higher plant chloroplasts. Biochim Biophys Acta 591:153–161
Rochaix J-D (2014) Regulation and dynamics of the light-harvesting system. Annu Rev Plant Biol 65:287–309
Rochaix J-D, Lemeille S, Shapiguzov A, Samol I, Fucile G, Willig A, Goldschmidt-Clermont M (2012) Protein kinases and phosphatases involved in the acclimation of the photosynthetic apparatus to a changing light environment. Philos Trans R Soc B 367:3466–3474
Romanovsky YuM, Tikhonov AN (2010) Molecular energy transducers of the living cell. Proton ATP synthase: a rotating molecular motor. Phys Usp 53:893–914
Rozak PR, Seiser RM, Wacholtz WF, Wise RR (2002) Rapid, reversible alterations in spinach thylakoid appression upon changes in light intensity. Plant Cell Environ 25:421–429
Ruban AV, Johnson MP, Duffy CDP (2012) The photoprotective molecular switch in the photosystem II antenna. Biochim Biophys Acta 1817:167–181
Ruban AV, Johnson MP (2015) Visualizing the dynamic structure of the plant photosynthetic membrane. Nat Plants 3:15161
Ruuge EK, Tikhonov AN (2022) Electron paramagnetic resonance study of the regulatory mechanisms of light phases of photosynthesis in plants. Biophysics 67:406–412
Rumberg B, Reinwald E, Schroder H, Siggel U (1968) Correlation between electron flow, proton translocation and phosphorylation in chloroplasts. Naturwissenschaften 55:77–79
Rumberg B, Siggel U (1969) pH changes in the inner phase of the thylakoids during photosynthesis. Naturwissenschaften 56:130–132
Ryzhikov SB, Tikhonov AN (1988) Regulation of electron transfer in photosynthetic membranes of higher plants. Biophysics 33:642–646
Sacksteder CA, Kanazawa A, Jacoby ME, Kramer DM (2000) The proton to electron stoichiometry of steady-state photosynthesis in living plants: a proton-pumping Q cycle is continuously engaged. Proc Natl Acad Sci USA 97:14283–14288
Samoilova RI, Kolling D, Uzawa T, Iwasaki T, Crofts AR, Dikanov S (2002) The interaction of the Rieske iron–sulfur protein with occupants of the Qo-site of the bc1 complex, probed by 1D and 2D electron spin echo envelope modulation. J Biol Chem 277:4605–4608
Sarewicz M, Dutka M, Pintscher S, Osyczka A (2013) Triplet state of the semiquinone-Rieske cluster as an intermediate of electronic bifurcation catalyzes by cytochrome bc1. Biochemistry 52:6388–6395
Sarewicz M, Bujnowicz Ł, Osyczka A (2018) Generation of semiquinone-[2Fe-2S]+ spin-coupled center at the Qo site of cytochrome bc1 in redox-poised, illuminated photosynthetic membranes from Rhodobacter capsulatus. Biochim Biophys Acta 1859:145–153
Sarewicz M, Bujnowicz Ł, Bhaduri S, Singh SK, Cramer WA, Osyczka A (2017) Metastable radical state, nonreactive with oxygen, is inherent to catalysis by respiratory and photosynthetic cytochromes bc1/b6f. Proc Natl Acad Sci USA 114:1323–1328
Sarewicz M, Pintscher S, Pietras R, Borek A, Bujnowicz Ł, Hanke G, Cramer WA, Finazzi G, Osyczka A (2021) Catalytic reactions and energy conservation in the cytochrome bc1 and b6f complexes of energy-transducing membranes. Chem Rev 121:2020–2108
Sarewicz M, Szwalec M, Pintscher S, Indyka P, Rawski M, Pietras R, Mielecki B, Koziej Ł, Jaciuk M, Glatt S, Osyczka A (2023) High-resolution cryo-EM structures of plant cytochrome b6f at work. Sci Adv 9:1–12
Schöttler MA, Tóth SZ, Boulouis A, Kahlau S (2015) Photosynthetic complex stoichiometry dynamics in higher plants: biogenesis, function, and turnover of ATP synthase and the cytochrome b6f complex. J Exp Bot 66:2373–2400
Schönknecht G, Neimanis S, Katona E, Gerst U, Heber U (1995) Relationship between photosynthetic electron transport and pH gradient across the thylakoid membrane in intact leaves. Proc Natl Acad Sci USA 92:12185–12189
Shikanai T (2007) Cyclic electron transport around photosystem I: genetic approaches. Annu Rev Plant Biol 58:199–217
Shikanai T (2016) Chloroplast NDH: A different enzyme with a structure similar to that of respiratory NADH dehydrogenase. Biochim Biophys Acta 1857:1015–1022
Shimizu M, Katsuda N, Katsurada T, Mitani M, Yoshioka Y (2008) Mechanism on two-electron oxidation of ubiquinol at the Qp site in Cytochrome bc1 complex: B3LYP study with broken symmetry. J Phys Chem 112:15116–15126
Schuller JM, Birrell JA, Tanaka H, Konuma T, Wulfhorst H, Cox N, Schuller SK, Thiemann J, Lubitz W, Sétif P, Ikegami T, Engel BDK, Nowaczyk MM (2019) Structural adaptations of photosynthetic complex I enable ferredoxin-dependent electron transfer. Science 363:257–260
Siegbahn PEM, Blomberg MRA (1999) Density functional theory of biologically relevant metal centers. Annu Rev Phys Chem 50:221–249
Siggel U, Renger G, Stiehl HH, Rumberg B (1972) Evidence for electronic and ionic interaction between electron transport chains in chloroplasts. Biochim Biophys Acta 256:328–335
Snyder CH, Gutierrez-Cirlos EB, Trumpower BL (2000) Evidence for a concerted mechanism of ubiquinol oxidation by the cytochrome bc1 complex. J Biol Chem 275:13535–13541
Soriano GM, Ponamarev MV, Tae G-S, Cramer WA (1996) Effect of the interdomain basic region of cytochrome f on its redox reactions in vivo. Biochemistry 35:14590–14598
Soriano GM, Guo L-W, de Vitry C, Kallas T, Cramer WA (2002) Electron transfer from the Rieske iron–sulfur protein (ISP) to cytochrome f in vitro. Is a guided trajectory of the ISP necessary for competent docking? J Biol Chem 277:41865–41871
Staehelin LA (2003) Chloroplast structure: from chlorophyll granules to supramolecular architecture of thylakoid membranes. Photosynth Res 76:185–196
Stiehl HH, Witt HT (1969) Quantitative treatment of the function of plastoquinone in photosynthesis. Z Naturforsch B 24:1588–1598
Strand DD, Fisher N, Kramer DM (2016) Distinct energetics and regulatory functions of the two major cyclic electron flow pathways in chloroplasts. In: Kirchhoff H (ed) Chloroplasts: current research and future trends. Caister Academic Press, Norfolk, pp 89–100
Strand DD, Fisher N, Kramer DM (2017) The higher plant plastid NAD(P)H dehydrogenase-like complex (NDH) is a high efficiency proton pump that increases ATP production by cyclic electron flow. J Biol Chem 292:11850–11860
Stroebel D, Choquet Y, Popot J-L, Picot D (2003) An atypical heam in the cytochrome b6f complex. Nature 426:413–418
Suslichenko IS, Tikhonov AN (2019) Photo-reducible plastoquinone pools in chloroplasts of Tradescentia plants acclimated to high and low light. FEBS Lett 593:788–798
Suslichenko IS, Trubitsin BV, Vershubskii AV, Tikhonov AN (2022) The noninvasive monitoring of the redox status of photosynthetic electron transport chains in Hibiscus rosa-sinensis and Tradescantia leaves. Plant Physiol Biochem 185:233–243
Szwalec M, Bujnowicz Ł, Sarewicz M, Osyczka A (2022) Unexpected heme redox potential values implicate an uphill step in cytochrome b6f. J Phys Chem B 26:9771–97801
Takizawa K, Cruz JA, Kanazawa A, Kramer DM (2007) The thylakoid proton motive force in vivo. Quantitative, non-invasive probes, energetics, and regulatory consequences of light-induced pmf. Biochim Biophys Acta 1767:1233–1244
Tikhonov AN (2012) Energetic and regulatory role of proton potential in chloroplasts. Biochem Mosc 77:956–974
Tikhonov AN (2013) pH-Dependent regulation of electron transport and ATP synthesis in chloroplasts. Photosynth Res 116:511–534
Tikhonov AN (2014) The cytochrome b6f complex at the crossroad of photosynthetic electron transport pathways. Plant Physiol Biochem 81:163–183
Tikhonov AN (2018) The cytochrome b6f complex: biophysical aspects of its functioning in chloroplasts. In: Harris JR, Boekema EJ (eds) Membrane protein complexes: structure and function, subcellular biochemistry, vol 87. Springer, Singapore, pp 287–328
Tikhonov AN, Vershubskii AV (2014) Computer modeling of electron and proton transport in chloroplasts. BioSystems 121:1–21
Tikhonov AN, Vershubskii AV (2017) Connectivity between electron transport complexes and modulation of photosystem II activity in chloroplasts. Photosynth Res 133:103–114
Tikhonov AN, Khomutov GB, Ruuge EK, Blumenfeld LA (1981) Electron transport control in chloroplasts. Effects of photosynthetic control monitored by the intrathylakoid pH. Biochim Biophys Acta 637:321–333
Tikhonov AN, Khomutov GB, Ruuge EK (1984) Electron transport control in chloroplasts. Effects of magnesium ions on the electron flow between two photosystems. Photobiochem Photobiophys 8:261–269
Tremmel IG, Kirchhoff H, Weis E, Farquhar GD (2003) Dependence of plastoquinol diffusion on the shape, size, and density of integral thylakoid proteins. Biochim Biophys Acta 1603:97–109
Ustynyuk LY, Trubitsin BV, Tikhonov AN (2018) DFT modeling of the first step of plastoquinol oxidation by the iron-sulfur protein of the cytochrome b6f complex. Mendeleev Commun 28:170–172
Ustynyuk LYu, Tikhonov AN (2018) The cytochrome b6f complex: DFT modeling of the first step of plastoquinol oxidation by the iron-sulfur protein. J Organomet Chem 867:290–299
Ustynyuk LYu, Tikhonov AN (2022) Plastoquinol oxidation: rate-limiting stage in the electron transport chain of chloroplasts. Biochem Mosc 87:1084–1097
Vallon O, Bulte L, Dainese P, Olive J, Bassi R, Wollman F-A (1991) Lateral redistribution of cytochrome b6/f complexes along thylakoid membranes upon state transitions. Proc Natl Acad Sci USA 88:8262–8266
Vener AV, Kan PJM, Rich PR, Ohad I, Andersson B (1997) Plastoquinol at the quinol oxidation site of reduced cytochrome bf mediates signal transduction between light and protein phosphorylation: thylakoid protein kinase deactivation by a single-turnover flash. Proc Natl Acad Sci USA 94:1585–1590
Vener AV, Ohad I, Andersson B (1998) Protein phosphorylation and redox sensing in chloroplast thylakoids. Curr Opin Plant Biol 1:217–223
Vershubskii AV, Trubitsin BV, Priklonskii VI, Tikhonov AN (2017) Lateral heterogeneity of the proton potential along the thylakoid membranes of chloroplasts. Biochim Biophys Acta 1859:388–401
Victoria D, Burton R, Crofts AR (2013) Role of the –PEWY-glutamate in catalysis at the Qo-site of the Cyt bc1 complex. Biochim Biophys Acta 1827:365–386
Walker JE (2013) The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans 41:1–16
West KR, Wiskich JT (1968) Photosynthetic control by isolated pea chloroplasts. Biochem J 109:527–532
Wikström MKF, Berden JA (1972) Oxidoreduction of cytochrome b in the presence of antimycin. Biochim Biophys Acta 283:403–420
Williams RJP (1988) Proton circuits in biological energy interconversions. Ann Rev Biophys Chem 17:71–97
Wood WHJ, MacGregor-Chatwin C, Barnett SFH, Mayneord GE, Huang X, Hobbs JK, Hunter CN, Johnson MP (2018) Dynamic thylakoid stacking regulates the balance between linear and cyclic photosynthetic electron transfer. Nat Plants 4:116–127
Wood WHJ, Barnett SFH, Flannery S, Hunter CN, Johnson MP (2019) Dynamic thylakoid stacking is regulated by LHCII phosphorylation but not its interaction with PSI. Plant Physiol 188:2152–2166
Yadav KNS, Semchonok DA, Nosek L, Kouřil R, Fucile G, Boekema EJ, Eichacker LA (2017) Supercomplexes of plant photosystem I with cytochrome b6f, light-harvesting complex II and NDH. Biochim Biophys Acta 1858:12–20
Yamashita E, Zhang H, Cramer WA (2007) Structure of the cytochrome b6f complex: Quinone analogue inhibitors as ligands of heme cn. J Mol Biol 370:39–52
Yamory W, Shikanai T (2016) Physiological functions of cyclic electron transport around photosystem I in sustaining photosynthesis and plant growth. Ann Rev Plant Biology 67:81–106
Yan J, Cramer WA (2003) Functional insensitivity of the cytochrome b6f complex to structure changes in the hinge region of the Rieske iron–sulfur protein. J Biol Chem 278:20925–20933
Zhang ZL, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung L-W, Crofts AR, Berry EA, Kim S-H (1998) Electron transfer by domain movement in cytochrome bc1. Nature 392:677–684
Zhao L-S, Huokko T, Wilson S, Simpson DM, Wang Q, Ruban AV, Mullineaux Y-Z, Zhang CW, Liu L-N (2020) Structural variability, coordination and adaptation of a native photosynthetic machinery. Nat Plants 6:869–882
Zhu J, Egawa T, Yeh S-R, Yu L, Yu C-A (2007) Simultaneous reduction of iron–sulfur protein and cytochrome bL during ubiquinol oxidation in cytochrome bc1 complex. Proc Natl Acad Sci USA 104:4864–4869
Zito F, Finazzi G, Joliot P, Wollman FA (1998) Glu78, from the conserved PEWY sequence of subunit IV, has a key function in the cytochrome b6f turnover. Biochemistry 37:10395–10403
Zito F, Finazzi G, Delosme R, Nitschke W, Picot D, Wollman F-A (1999) The Qo site of cytochrome b6f complexes controls the activation of the LHCII kinase. EMBO J 18:2961–2969
Zu Y, Manon M-J, Couture MM-J, Kolling DRJ, Crofts AR, Eltis LD, Fee JA, Hirst J (2003) The reduction potentials of Rieske clusters: the importance of the coupling between oxidation state and histidine protonation state. Biochemistry 42:12400–12408
Acknowledgements
This article is dedicated to the memory of Vladimir Shuvalov, an outstanding scientist who made fundamental contributions to elucidation of the mechanisms of electron transfer in photosynthetic systems. I would like to express my special acknowledgment to Dr. L.Yu. Ustynyuk for her decisive contribution to DFT modeling of a bifurcated oxidation of plastoquinol by the Cytb6f complex. I am deeply grateful to Drs. E.K. Ruuge, G.B. Khomutov, B.V. Trubitsin, and A.V. Vershubskii for fruitful collaboration in our earlier experimental and theoretical studies of regulatory processes in chloroplasts. I also thank the Reviewers for usefull and constructive comments.
Funding
This work was partly supported by the Russian Science Foundation (Grant 21-74-20047).
Author information
Authors and Affiliations
Contributions
ANT: design, planning the work, data processing, writing of the manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors declare no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by the author.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tikhonov, A.N. The cytochrome b6f complex: plastoquinol oxidation and regulation of electron transport in chloroplasts. Photosynth Res 159, 203–227 (2024). https://doi.org/10.1007/s11120-023-01034-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-023-01034-w