
Abstract
This chapter presents an overview of structural properties of the cytochrome (Cyt) b 6 f complex and its functioning in chloroplasts. The Cyt b 6 f complex stands at the crossroad of photosynthetic electron transport pathways, providing connectivity between Photosystem (PSI) and Photosysten II (PSII) and pumping protons across the membrane into the thylakoid lumen. After a brief review of the chloroplast electron transport chain, the consideration is focused on the structural organization of the Cyt b 6 f complex and its interaction with plastoquinol (PQH2, reduced form of plastoquinone), a mediator of electron transfer from PSII to the Cyt b 6 f complex. The processes of PQH2 oxidation by the Cyt b 6 f complex have been considered within the framework of the Mitchell’s Q-cycle. The overall rate of the intersystem electron transport is determined by PQH2 turnover at the quinone-binding site Qo of the Cyt b 6 f complex. The rate of PQH2 oxidation is controlled by the intrathylakoid pHin, which value determines the protonation/deprotonation events in the Qo-center. Two other regulatory mechanisms associated with the Cyt b 6 f complex are briefly overviewed: (i) redistribution of electron fluxes between alternative (linear and cyclic) pathways, and (ii) “state transitions” related to redistribution of solar energy between PSI and PSII.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
10.1 Introduction
Photosynthesis is one of the most important processes in nature. Photosynthetic organisms of oxygenic type (cyanobacteria, algae, higher plants), using the solar energy, assimilate carbon dioxide and produce molecular oxygen. The energy of light quanta absorbed by the pigment-protein complexes of photosystem I (PSI) and photosystem II (PSII) is converted into the energy of separated charges in photoreaction centers (Blankenship 2002; Cardona et al. 2012; Müh et al. 2012; Ruban 2012; Mamedov et al. 2015). The light-driven actuation of photoreaction centers initiates electron transfer along the photosynthetic electron transport chain (ETC). Two electrons extracted from the water molecule split by the water-oxidizing complex (WOC) of PSII, are passed to the terminal electron acceptor of PSI, NADP+. PSI complex delivers electrons to NADP+ through ferredoxin (Fd) and ferredoxin-NADP-oxidoreductase (FNR): PSI → Fd → FNR → NADP+. The cytochrome (Cyt) b 6 f complex is another multisubunit complex, which plays a crucial role in photosynthetic electron transport, because it stands at the crossroad of electron transport pathways between PSII and PSI. These photosystems are interconnected via the membrane-bound Cyt b 6 f complex and mobile electron carriers, plastoquinone (PQ), plastocyanin (Pc): H2O → PSII → PQ → b 6 f → Pc → PSI. Electron transfer from H2O to NADP+ is accompanied by alkalization of stroma (the volume between the chloroplast envelope and thylakoids) and acidification of the intra-thylakoid lumen (the internal volume of thylakoids). The light-induced uptake of protons from the bulk of stroma and the proton release in the lumen lead to generation of the trans-thylakoid difference in electrochemical potentials of hydrogen ions, \( \varDelta {\tilde{m}}_{{\mathrm{H}}^{+}} \) (often termed as the proton-motive force), which serves as the driving force for operation of the ATP synthase complex CF0 ‐ CF1(ADP + Pi → ATP + H2O) (Mitchell 1966; Blankenship 2002; Junge and Nelson 2015). The macroergic products of the light-induced stages of photosynthesis, ATP and NADPH, are used mainly in biosynthetic reactions of the Calvin-Benson cycle (CBC) (Edwards and Walker 1983; Blankenship 2002).
In this Chapter, the structural and functional properties of the Cyt b 6 f complex are considered in the context of its interaction with PQH2 (the double-reduced form of PQ) and feedback regulation of electron transport in chloroplasts. The reaction of PQH2 oxidation by the Cyt b 6 f complex represents the “bottle-neck” link in the ETC between PSII and PSI, which virtually determines the overall rate of the intersystem electron transport in chloroplasts. The redox state of the PQ pool plays the pivoting role in regulation of photosynthetic processes, because it serves the role of a peculiar “sensor” (for review, see Pesaresi et al. 2010) that triggers the short-term and long-term mechanisms of photosynthetic apparatus (PSA) response to varying environmental conditions (Kramer et al. 2004; Cruz et al. 2007; Eberhard et al. 2008; Demmig-Adams et al. 2012; Horton 2012; Rochaix 2014; Puthiyaveetil et al. 2016). The flexibility of PSA functioning in chloroplasts is achieved by cooperation of several feedback mechanisms of electron transport control. These regulatory mechanisms include different events: (i) pH-dependent control of PQH2 oxidation by the Cyt b 6 f complex, (ii) optimization of the light quanta partitioning between PSI and PSII (“state transitions”) triggered by the Cyt b 6 f complex, and (iii) redistribution of electron fluxes through alternative pathways of electron transport. All these mechanisms are associated with the functioning of the Cyt b 6 f complex.
10.2 Photosynthetic Electron Transport Chain
The multisubunit electron-transport complexes are embedded into lamellar membranes of thylakoids, closed vesicles situated under the chloroplast envelope. Figure 10.1 depicts a scheme of basic electron transport pathways in chloroplasts. The peculiarities of functioning photoreaction centers of PSI, PSII, and the Cyt b 6 f complex are briefly considered below.

A scheme of electron transport pathways in chloroplasts and the arrangement of protein complexes (Photosystem I, Photosystem II, Cyt b 6 f, FNR, and NDH) in the thylakoid membrane. Two electrons extracted from the water molecule in Photosystem II are transferred to plastoquinone (PQ), reducing PQ to plastoquinol (PQH2). Electrons from PQH2 are transferred via the Cyt b 6 f complex to reduce plastocyanin (Pc). Photosystem I oxidizes Pc on the lumenal side of the thylakoid membrane and reduces ferredoxin (Fd) on the stromal side of the membrane. Reduced Fd molecules donate electrons to NADP+. Reduced and protonated NADPH molecules are consumed in the Calvin-Benson cycle. Electron transport processes are accompanied by accumulation of hydrogen ions in the thylakoid lumen (Modified from Figure 1 in Tikhonov 2014)
10.2.1 Photosystems I and II
Photosystem I
The multisubunit pigment-protein PSI complex catalyzes electron transfer from Pc (or Cyt c 6 in cyanobacteria) on the lumenal side of thylakoids to Fd (or flavodoxin) located in stroma (Brettel 1997; Jordan et al. 2001; Fromme et al. 2001; Nelson and Yocum 2006; Shelaev et al. 2010; Mamedov et al. 2015). Reduced ferredoxin (Fd−) passes an electron to FNR, which provides the two-electron reduction of NADP+ to NADPH. The core domain of the PSI complex contains a special pair of chlorophyll (Chl) a molecules (Chl1A and Chl1B) located at the interface of subunits PsaA and PsaB, which form the primary electron donor termed P700. The light-induced excitation of P700 induces charge separation in PSI: excited center \( {\mathrm{P}}_{700}^{\ast } \) donates an electron to the primary electron acceptor (Chl2A or Chl2B). On the acceptor side of PSI, electron carriers are arranged as two quasi-symmetrical cofactor branches, which consist of two Chl a molecules (Chl2A and Chl3A in A-branch; Chl2B and Chl3B in B-branch) and one phylloquinone molecule (A1A or A1B, respectively). The two branches converge at the acceptor FX (one of three [FeS]4 clusters of PSI, FX, FA, and FB). There are experimental evidences in favor of preferential role of A-branch in electron transfer on the acceptor side of PSI (for references, see Mamedov et al. 2015). From reduced FX the electron is transferred to Fd via the redox centers FA and FB (FX → FA → FB → Fd). Reduced Fd molecules deliver electrons to NADP+ via FNR (Fig. 10.1).
Oxidized center \( {\mathrm{P}}_{700}^{+} \) accepts an electron from reduced Pc (Pc−), which, in turn, accepts an electron from the Cyt b 6 f complex. Pc serves the role of the electron transfer shuttle, which, moving within the lumen, connects electronically the Cyt b 6 f complex and PSI. Lateral diffusion of Pc− inside the lumen, and further donation of an electron from Pc− to \( {\mathrm{P}}_{700}^{+} \), do not limit electron transport between PSII and PSI. Oxidation of Pc− by \( {\mathrm{P}}_{700}^{+} \)occurs more rapidly (t 1/2 < 200 μs, at ambient temperatures) than electron transfer from PSII to Pc via the PQ pool and Cyt b 6 f complex (t 1/2 ≥ 4 – 20 ms) (Stiehl and Witt 1969; Witt 1979; Haehnel 1984).
Photosystem II
PSII contains the primary electron donor P680 and the water-oxidizing complex (WOC). Two electrons extracted from H2O in WOC (H2O → 1/2O2 + 2H+ + 2e−) are used to reduce PQ to PQH2. Transfer of electrons from H2O to PQ molecules proceeds as the result of consecutive one-electron reactions induced by light quanta absorbed by the light-harvesting antenna of PSII. A special pair of Chl a molecules embedded into the core of the PSII complex form the primary electron donor in PSII termed P680 (for review, see Cardona et al. 2012, Müh et al. 2012). There are two branches of electron cofactors on the acceptor side of PSII, A-branch and B-branch. Excited redox center \( {\mathrm{P}}_{680}^{\ast } \) donates an electron to the primary electron acceptor of A-branch, Chl a molecule termed ChlD1, which, in its turn, passes the electron to the secondary acceptor pheophytin (Phe): \( {\mathrm{P}}_{680}^{\ast}\to {\mathrm{Chl}}_{\mathrm{D}1}\to {\mathrm{P}\mathrm{he}}_{\mathrm{D}1} \). Reduced Phe donates the electron to the primary plastoquinone PQA tightly bound to PSII (\( {\mathrm{Phe}}^{-}{\mathrm{PQ}}_{\mathrm{A}}{\mathrm{PQ}}_{\mathrm{B}}\to \mathrm{Phe}\kern0.28em {\mathrm{PQ}}_{\mathrm{A}}^{-}{\mathrm{PQ}}_{\mathrm{B}} \)). \( {\mathrm{PQ}}_{\mathrm{A}}^{-} \) reduces the secondary plastoquinone \( {\mathrm{PQ}}_{\mathrm{B}} \) bound to PSII (\( {\mathrm{PQ}}_{\mathrm{A}}^{-}{\mathrm{PQ}}_{\mathrm{B}}\to {\mathrm{PQ}}_{\mathrm{A}}{\mathrm{PQ}}_{\mathrm{B}}^{-} \)). The second electron donated by \( {\mathrm{P}}_{680}^{\ast } \) to \( {\mathrm{PQ}}_{\mathrm{A}} \) provides the double-electron reduction of \( {\mathrm{PQ}}_{\mathrm{B}} \) (\( {\mathrm{PQ}}_{\mathrm{A}}^{-}{\mathrm{PQ}}_{\mathrm{B}}^{-}\to {\mathrm{PQ}}_{\mathrm{A}}{\mathrm{PQ}}_{\mathrm{B}}^{=} \)), which is followed by protonation of \( {\mathrm{PQ}}_{\mathrm{B}}^{=} \) due to the uptake of two protons from stroma (\( {\mathrm{PQ}}_{\mathrm{B}}^{=}+2{\mathrm{H}}_{\mathrm{out}}^{+}\to {\mathrm{PQ}}_{\mathrm{B}}{\mathrm{H}}_2 \)). Reduced secondary plastoquinone, \( {\mathrm{PQ}}_{\mathrm{B}}{\mathrm{H}}_2 \), dissociates from PSII in exchange for a new oxidized PQ molecule. Diffusing in the membrane, the hydrophobic PQH2 molecule reaches the Cyt b 6 f complex. After binding of PQH2 to the Qo-center of this complex, the PQH2 molecule oxidizes, donating two electrons to appropriate electron acceptors of the Cyt b 6 f complex and releasing two protons, which finally migrate into the bulk phase of the thylakoid lumen. On the donor side of PSII, decomposition of water molecules in the WOC is accompanied by the release of protons into the lumen. The overall balance of electron and proton transport processes in PSII is the following: (i) two electrons extracted from one H2O molecule are used to reduce PQ to PQH2, and (ii) two protons are taken up from stroma and two protons apper in the lumen per one PQH2 molecule formed (\( {\mathrm{H}}_2\mathrm{O}+\mathrm{PQ}+2{\mathrm{H}}_{\mathrm{out}}^{+}\to 1/2{\mathrm{O}}_2+{\mathrm{PQH}}_2+2{\mathrm{H}}_{\mathrm{in}}^{+} \)).
10.2.2 The Role of Cytochrome b 6 f Complex in the Pathway Between Photosystems I and II
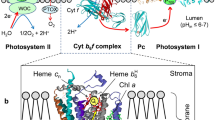
The Cyt b 6 f complex (plastoquinol:plastocyanin oxidoreductase) is organized as the hetero-oligomeric protein complex, which mediates electron transfer between PSII and PSI by oxidizing PQH2 and reducing Pc. Three-dimentional structures of this complex were initially obtained at a resolution of 3.0–3.1 Å from the thermophilic filamentous cyanobacterium Mastigocladus laminosus (PDB entry 1VF5; Kurisu et al. 2003), and the green alga Chlamydomonas reinhardtii (PDB entry 1Q90; Stroebel et al. 2003), in the presence of the quinone analogue inhibitor, tri-decyl-stigmatellin (TDS). The crystal structures of the Cyt b 6 f complexes from M. laminosus and C. reinhardtii are similar. The Cyt b 6 f complex is organized as the functional dimer of multisubunit monomers (Fig. 10.2). Dimeric organization of the Cyt b 6 f complex is similar, in general, to that of the Cyt bc 1 complex of the Cyt bc family (Xia et al. 1997, 2013; Iwata et al. 1998; Berry et al. 2000; Crofts 2004a; Cramer et al. 2006, 2011). Each multisubunit monomer of the functional dimer of the Cyt b 6 f complex consists of eight polypeptide subunits with 13 trans-membrane helixes, including four “large” (16–31 kDa) polypeptide subunits (petA, B, C, and D): the iron-sulfur protein (ISP) Rieske, the Cyt b 6 and Cyt f proteins, and subunit IV (Fig. 10.3). “Small” (3.3–4.1 kDa) hydrophobic subunits (petG, L, M, and N), each of them containing one trans-membrane helix, are arranged at the outside periphery of the monomer ensembly of petA, B, C, and D subunits.

The side view of the dimeric Cyt b 6 f complex from Chlamydomonas reinhardtii (PDB entry 1Q90, Stroebel et al. 2003). Figure was produced using Accelerys DV visualizer software package (http://www.accelrys.com) (Modified from Figure 2 in Tikhonov 2014)

Overview of structure and function of the Cyt b 6 f complex from Chlamydomonas reinhardtii (PDB entry 1Q90, Stroebel et al. 2003). The view is perpendicular to the membrane plane. Colour code of main polypeptides: cyan, Cyt f; grey, Cyt b 6; purple, the iron-sulfur protein; blue, subunit IV. Cofactors: red, hemes \( {b}_6^{\mathrm{L}} \), \( {b}_6^{\mathrm{H}} \), and c i, as indicated; orange, heme f; Fe atoms are shown as dark red spheres; green, Chl a. For true colours of subunits and cofactors see the online version of this Chapter. Plastoquinone binding site Qo is positioned between heme \( {b}_6^{\mathrm{L}} \)and the [Fe2S2] cluster of the iron-sulfur protein. Plastoquinone binding site Qi is placed between hemes \( {b}_6^{\mathrm{H}} \)and c i (Modified from Figure 3 in Tikhonov 2014. Figure was produced using Accelrys DV visualizer software package (http://www.accelrys.com))
The catalytic functions of the Cyt b 6 f complex are provided by four redox centers bound to the “large” subunits: the Rieske iron-sulfur cluster [Fe2S2], two hemes of the Cyt b 6 (the low-potential heme \( {b}_6^{\mathrm{L}} \) and the high-potential heme \( {b}_6^{\mathrm{H}} \)), and heme f of the Cyt f. These cofactors are involved in the electron transfer reactions within the Cyt b 6 f complex. Furthermore, there are three unusual prosthetic groups: (1) Chl a, (2) β -carotene, and (3) a unique heme c i (Stroebel et al. 2003), which is often termed as heme c n because of its location on the “negative” side of the membrane (Kurisu et al. 2003). Heme c i(c n) is bound covalently to the Cyt b 6 f complex in close proximity to the high-potential heme \( {b}_6^{\mathrm{H}} \). The Cyt b 6 and Cyt f subunits are functionally analogous to the Cyt b 6 and Cyt c 1 proteins in the mitochondrial and bacterial Cyt bc 1 complexes (Xia et al. 1997, 2013; Iwata et al. 1998; Berry et al. 2000; Crofts 2004a). The Cyt b 6 f complex contains the binding centers for PQH2 and PQ molecules. Crystallization of the Cyt b 6 f complexes with quinone analogue inhibitors, TDS and NQNO (2n–nonyl-4-hydroxy-quinoline-N-oxide) revealed two sites for quinone binding: the Qo-center (quinol oxidase) and the Qi-center (quinone reductase) (Yamashita et al. 2007). The quinone exchange cavity of the site Qo is positioned near the [Fe2S2] cluster of the ISP. Site Qi is located on the stromal side of the complex, at the interface between heme c i and the large inter-protein quinone exchange cavity.
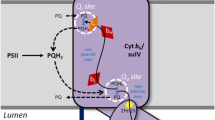
Oxidation of PQH2 occurs at the catalytic center Qo, which is situated in the cavity at the interface between the Cyt b 6 subunit and the ISP. This center is oriented towards the lumenal side of the thylakoid membrane. The oxidation of PQH2 in the Qo -site is accompanied by dissociation of two protons, which finally appear in the aqueous bulk phase of the thylakoid lumen. According to the Q-cycle mechanism first suggested by Peter Mitchell (1976), the two-electron oxidation of PQH2 in the Qo -center is a bifurcated reaction (Berry et al. 2000; Crofts 2004a, b; Osyczka et al. 2004, 2005; Mulkidjanian 2005; Cramer et al. 2006, 2011; Crofts et al. 2013; Xia et al. 2013). One electron is directed from PQH2 to a high-potential chain, the other electron travels along a low-potential chain (Fig. 10.3). The first electron comes to Pc through the high-potential redox chain, which consists of the ISP and Cyt f (PQH2 → ISP → Cyt f → Pc). The second electron is directed to reduce PQ at the Qi -site on the stromal side, traveling through the low- and high-potential hemes \( {b}_6^{\mathrm{L}} \) and \( {b}_6^{\mathrm{H}}:{\left({\mathrm{PQH}}^{\cdotp}\right)}_{\mathrm{o}}\to {b}_6^{\mathrm{L}}\to {b}_6^{\mathrm{H}}\to {\left(\mathrm{PQ}\right)}_{\mathrm{i}} \). Here, (PQH•)o denotes the semiquinone form of plastoquinone formed upon the bifurcated oxidation of PQH2 at the Qo-site, (PQ)i denotes the PQ molecule bound to the quinone-binding site Qi located in the stromal part of the Cyt b 6 f complex.
In the Qi-center, after two successive steps of PQ reduction to PQ=, two protons are taken up from stroma (\( \mathrm{PQ}+2{\mathrm{e}}^{\hbox{--} }+2{\mathrm{H}}_{\mathrm{out}}^{+}\to {\mathrm{PQH}}_2 \)). The protonated (electrically neutral) PQH2 molecule dissociates from the Qi-site and now it can bind to the vacant Qo-site to be oxidized on the lumenal side of the Cyt b 6 f complex. Due to the round trip of one electron in the Q-cycle, the proton pumping activity of the Cyt b 6 f complex increases by a factor of 2. Taking into account the overall balance of PQ turnover in the Q-cycle, we see that two hydrogen ions are pumped into the lumen per one electron (H+/e− = 2) transferred from PQH2 to \( {\mathrm{P}}_{700}^{+} \) via the high-potential chain (\( \mathrm{ISP}\to \mathrm{Cyt}\kern0.50em f\to \mathrm{Pc}\to {\mathrm{P}}_{700}^{+} \)). Note that the unique heme c i may be an adaptation of photosynthetic organisms for cyclic route of electron flow around PSI. Electrons from the acceptor side of PSI may return to the Qi-center of the Cyt b 6 f complex via Fd, FNR, and/or an atypical heme c i (Kurisu et al. 2003; Stroebel et al. 2003; Munekage et al. 2004; Joliot and Joliot 2005; Shikanai 2007).
10.2.3 The Cytochrome b 6 f Complex and Alternative Pathways of Electron Transport
The Cyt b 6 f complex is the participant of cyclic electron transport around PSI. On the acceptor side of PSI, apart from the mainstream pathway of electron flow to the CBC (the so-called “linear” electron flow, LEF), the electron flux may be diverted to cyclic routes around PSI. Cyclic electron flow around PSI (CEF1) is important for fine-tuning of the energy and redox balance in chloroplasts. There is general consensus that CEF1 helps to sustain the ratio ATP/NADPH = 3/2 required for optimal functioning of the CBC (for review, see Bendall and Manasse 1995; Allen 2003; Cruz et al. 2007; Johnson 2011; Strand et al. 2016). The light-induced transfer of four electrons from two H2O molecules to two NADP+ molecules driven by the tandem operation of PSII and PSI (\( 2{\mathrm{H}}_2\mathrm{O}+2{\mathrm{NADP}}^{+}\to {\mathrm{O}}_2+2\mathrm{NADPH} \)) is accompanied by pumping of 12 protons into the thylakoid lumen. This number of protons is insufficient for synthesis of three ATP molecules required to provide the ratio ATP/NADPH = 3/2. Actually, in chloroplasts the stoichiometric ratio n = H+/ATP for the ATP synthase reaction (ADP + Pi + nHin → ATP + nHout) is higher than n = 4 (Seelert et al. 2000; Junge and Nelson 2015; Turina et al. 2016). Therefore, 12 protons translocated inside the thylakoid per two NADPH molecules formed is insufficient to maintain the required ratio ATP/NADPH = 3/2. Additional pumping of protons coupled to operation of CEF1 could contribute to generation of \( \varDelta {\tilde{m}}_{{\mathrm{H}}^{+}} \), providing synthesis of “extra” ATP molecules. Thus, the synergism of two pathways, LEF and CEF1, helps to maintain the ratio ATP/NADPH = 3/2 required for CO2 fixation in the CBC. Even a relatively small contribution of CEF1 to generation of ΔpH may be enough to supplement ATP formation, providing thus the well-balanced ATP/NADPH ratio. CEF1 is also important for effective responses of PSA to fluctuations of light intensity, thereby avoiding the risk of photodamage to chloroplasts (Suorsa et al. 2012; Kono and Terashima 2014; Kono et al. 2014; Yamori and Shikanai 2016).
Most of CEF1 pathways involve the Cyt b 6 f complex (Fig. 10.1). Recent genetic and biochemical studies clarified the physiological role of CEF1 and helped to elucidate the participation of different chloroplast proteins in CEF1 (for review, see Shikanai 2007).There are several routes of electron flow on the acceptor side of PSI, when electrons from PSI may be delivered to different channels (for recent review, see Strand et al. 2016). Electrons from PSI can be recycled to PQ from either reduced Fd (Fd−) or NADPH (“short” and “long” rounds, respectively). There are two “short’ pathways of CEF1 related to electron transfer from Fd− to PQ molecule bound to the Qi-center mediated by the ferredoxin-plastoquinone-reductase (FQR) and FNR, without the participation of NADPH (pathways 1 and 2 depicted in Fig. 10.1). The “long” pathway of CEF1 involves the formation of NADPH and return of electrons through the chloroplast NAD(P)H-dehydrogenase (NDH): PSI → Fd → FNR → NADPH → NDH → PQ (Fig. 10.1, pathway 3).
One of the “short” routes of CEF1 implies the participation of the elusive FQR complex: PSI → Fd → FQR → PQ → b 6 f (Bendall and Manasse 1995). It has been demonstrated that the products of two genes, PGR5 (proton gradient regulation) and PGRL1 (PGR5–like protein 1), may be involved into Fd-dependent CEF1 in eukaryotes (Munekage et al. 2002, 2004, 2008; Shikanai 2007; DalCorso et al. 2008; Suorsa et al. 2012; Hertle et al. 2013). Plants deficient in one of these proteins show disturbed CEF1, suggesting that PGR5 and PGRL1 may be considered as the components of FQR. The exact role of these proteins in CEF1 was unclear until it was demonstrated that PGRL1 accepted electrons from Fd− in a PGR5-dependent manner and reduced PQ (Hertle et al. 2013). These observations serve as compelling evidence that PGRL1 operates as the elusive FQR protein. It is interesting to note that PGRL1 is the redox regulated protein, its activity requires a Fe-containing cofactor and six redox-active cysteine residues. PGR5 is used for electron transfer from Fd− to PGRL1. Both proteins, PGR5 and PGRL1, are necessary for PSA protection against photodamage induced by rapid fluctuations of ambient light (Munekage et al. 2008; Suorsa et al. 2012).
Another “short” route of Fd-dependent CEF1 implies the return of electrons from PSI to PQ through the FNR complex. The formation of a supercomplex FNR ‐ b 6 f (Zhang et al. 2001; Benz et al. 2010) may facilitate the direct electron transfer to the Qi-site of the Cyt b 6 f complex (Fig. 10.1, pathway 2). However, the nature of the immediate electron donor to PQ molecule at the Qi-site is under debate (Shikanai 2007; DalCorso et al. 2008; Iwai et al. 2010; Johnson 2011). There are reasons to believe that an atypical heme c i positioned on the stromal side of the Cyt b 6 f complex may serve as the immediate electron donor to PQ (Kurisu et al. 2003; Stroebel et al. 2003; Alric et al. 2005; Cramer et al. 2006, 2011; Hasan et al. 2013a). The PQ molecule bound to the Qi-center may be reduced to PQH2 by electrons coming from different chains. One electron comes from the high-potential heme \( {b}_6^{\mathrm{H}} \), whereas the second electron will arrive from PSI (Fig. 10.1, pathways 1 and 2). The reduced PQH2 molecule dissociates from the Qi-site and then can return to the Qo-center, participating in the next cycle of PQH2 turnover.
In the “long” (NADPH-dependent) route of CEF1, the NDH complex returns electrons from NADPH (and/or NADH) to the intersystem ETC (Fig. 10.1, pathway 3). Genetic and biochemical data give unequivocal evidence for participation of the chloroplast NDH in the “long” route of CEF1 (Burrows et al. 1998; Endo et al. 1998; Shikanai et al. 1998; Joët et al. 2001; Shikanai 2016). There are indications that the NDH and PSI complexes can form a supercomplex (NDH-PSI) in higher plants (Peng et al. 2008, 2009) and cyanobacteria (Kubota et al. 2009). The location of NDH in the stromal lamellae close to PSI as well as an elevated content of NDH in the bundle sheath cells of C4 plants with high levels of CEF1 (Kubicki et al. 1996) support the notion of NDH participation in CEF1.
10.2.4 Lateral Heterogeneity of Thylakoid Membranes
The participation of the Cyt b 6 f complex in CEF1 depends on its location in the thylakoid membrane. The arrangement of membrane-embedded electron transport complexes and mobile electron carriers with respect to stromal and granal thylakoids is shown schematically in Fig. 10.4. It is well-known fact that PSI, PSII, and ATP synthase (CF0 ‐ CF1) complexes are distributed nonuniformly over the membranes of granal and stromal thylakoids (Albertsson 2001; Staehelin 2003; Dekker and Boekema 2005). Stacked thylakoids of grana are enriched with PSII; most PSI and CF0 ‐ CF1 complexes are localized in the unstacked domains of stroma-exposed thylakoids, grana margins, and grana end membranes. The Cyt b 6 f complexes are spread almost uniformly along the thylakoid membranes (Anderson 1982). About 55% of the Cyt b 6 f complexes are localized in appressed membranes of grana, and about 45% of complexes are distributed over the stromal lamellae, in the margins and grana end membranes. Although significant amounts of PSI, PSII, and Cyt b 6 f complexes are laterally segregated in the thylakoid membrane, most of them are in close contact. The content of different electron-transport complexes and their ratio (PSII/b 6 f/PSI) are variable, being sensitive to the plant growth conditions (for review, see Anderson et al. 1988; Lichtenthaler and Babani 2004; Eberhard et al. 2008; Schöttler et al. 2015; Puthiyaveetil et al. 2016). The amounts of PQ and Pc are higher than that of PSI or PSII. The relative capacity of the PQ pool, related to PSII, was estimated as PQ/PSII ~ 10 times (Stiehl and Witt 1969; Witt 1979; Haehnel 1984).

A scheme of the possible arrangement of the electron transport and ATP synthase complexes in the stromal and granal domains of the thylakoid membrane (Modified from Figure 4 in Tikhonov 2014)
It is important to note that there are two population of the Cyt b 6 f complex, that differ from each other with respect to their location in the stromal and granal domains of thylakoid membranes. Under the normal physiological conditions, grana-exposed thylakoids are assembled in the form of grana consisting of tightly packed thylakoid disks. Because of steric restrictions, direct contacts of granal Cyt b 6 f complexes with the FNR complexes will be excluded. This circumstance suggests that only the stroma-exposed fraction of Cyt b 6 f complexes may participate in the CEF1 reactions (Joliot and Joliot 2005, 2006). Although significant amounts of PSI and PSII complexes are laterally segregated, most of them are in close contact with the Cyt b 6 f complexes (Albertsson 2001). The proximal location of the Cyt b 6 f complexes to PSII in granal domains of thylakoid membranes should facilitate electron transfer between PSII and the Cyt b 6 f complexes due to a short distance for PQH2 diffusion in the lipid phase of the membrane (Kirchhoff 2013, 2014). This circumstance may shed a light on the nature of the rate-limiting step in the intersystem chain of electron transport, considered below.
10.2.5 The Rate-Limiting Step in the Intersystem Chain of Electron Transport
The rate of PQ turnover is determined by several events: PQ reduction to \( {\mathrm{PQ}\mathrm{H}}_2\left({\mathrm{PQ}}_{\mathrm{B}}+2{\mathrm{e}}^{\hbox{--} }+2{\mathrm{H}}_{\mathrm{out}}^{+}\to {\mathrm{PQ}}_{\mathrm{B}}{\mathrm{H}}_2\right) \), dissociation of PQH2 from PSII, its diffusion towards the Cyt b 6 f complex, and oxidation of PQH2 at the Qo-site. The peculiarities of chloroplast architecture raise the question as to whether or not the lateral and transverse diffusion of PQH2 molecules within the thylakoid membrane would restrict the intersystem electron transfer. Which stage of PQH2 oxidation determines the rate of PQH2 turnover, either the lateral diffusion of PQH2 in the membrane between spatially separated electron transport complexes or PQH2 oxidation after its binding to the Cyt b 6 f complex? There are indications that the lateral diffusion of PQH2 may restrain electron transfer from PSII to PSI under certain conditions (Lavergne and Joliot 1991; Kirchhoff 2008, 2014). Diffusion of PQH2 within the membrane may be retarded due to obstructed diffusion of PQH2 through the lipid domains, over-crowded with densely packed protein complexes. In the meantime, as noted above, the distribution of Cyt b 6 f complexes among PSII complexes located in granal thylakoids minimizes the average distance traversed by PQ molecules, providing rapid turnover of the PQ shuttle between the Cyt b 6 f and PSII complexes.
In earlier work (Stiehl and Witt 1969), these authors scrutinized in detail the PQ turnover in spinach chloroplasts by optical methods. Redox transients of PQH2 were measured by monitoring absorption changes in the UV region. It was demonstrated that PQH2 delivered electrons to \( {\mathrm{P}}_{700}^{+} \) (via intermediates) with the half-time t 1/2 ≈ 15 − 17.5 ms. Similar times were obtained by Haehnel for Cyt f and \( {\mathrm{P}}_{700}^{+} \) reduction (Haehnel 1973, 1976a, b). Electron transfer from the Cyt b 6 f complex to \( {\mathrm{P}}_{700}^{+} \)occured much more rapidly than PQH2 oxidation: t 1/2 ≈ 35 − 350 μs for electron transfer from Cyt f to Pc, and t 1/2 ≈ 20 − 200 μs for electron transfer from Pc to \( {\mathrm{P}}_{700}^{+} \)(Haehnel 1984; Hope 2000). These results unequivocally demonstrated that PQH2 interaction with the Cyt b 6 f complex to be the rate-limiting event in the chain of electron transport processes between PSII and PSI.
There is strong evidence that within a wide range of experimental conditions (pH, ionic strength, and temperature) PQH2 formation and its diffusion in the membrane do not limit the intersystem electron transport. The light-induced reduction of PQ in PSII \( \left({\mathrm{PQ}}_{\mathrm{B}}+2{\mathrm{e}}^{\hbox{--} }+2{\mathrm{H}}_{\mathrm{out}}^{+}\to {\mathrm{PQ}}_{\mathrm{B}}{\mathrm{H}}_2\right) \), dissociation of PQBH2 from PSII (PQBH2 → PQH2) and PQH2 diffusion to the Cyt b 6 f complex occur more rapidly than PQH2 interaction with the Cyt b 6 f complex (Haehnel 1976a; Tikhonov et al. 1984). This statement can be illustrated by the experimental data presented in Fig. 10.5. In chloroplasts pre-illuminated with the far-red light (λ max = 707 nm) exciting predominantly PSI, the PQ pool and most of P700 centers become oxidized because of negligible injection of electrons from PSII to the intersystem ETC. In response to a short flash of white light exciting both photosystems, electrons donated by PSII are used to reduce oxidized centers \( {\mathrm{P}}_{700}^{+} \) (Fig. 10.5a). In the particular case of Mg2+-depleted chloroplasts, the reduction of \( {\mathrm{P}}_{700}^{+} \) proceeds after a well-resolved lag-phase, the duration of which (Δτ) involves the time of PQH2 formation and diffusion across and along the thylakoid membrane towards the Cyt b 6 f complex. The half-time of \( {\mathrm{P}}_{700}^{+} \) reduction, which is determined mainly by electron transfer from PQH2 bound to the Cyt b 6 f complex, is markedly higher than the lag-phase Δτ (Fig. 10.5a). This observation demonstrates that PQH2 oxidation at the Qo-site of the Cyt b 6 f complex represents the “bottle-neck” in the ETC between PSII and PSI, which controls the overall rate of the intersystem electron transport. Actually, the interaction of Pc− with \( {\mathrm{P}}_{700}^{+} \) usually occurs much more rapidly (t 1/2 ≤ 200 μs, at ambient temperatures) than electron transfer from PQH2 to Pc via the Cyt b 6 f complex (Stiehl and Witt 1969; Witt 1979; Haehnel 1984). The duration of the lag-phase Δτ is sensitive to Mg2+-induced structural changes in chloroplasts. At physiological concentrations of MgCl2 (2–4 mM), Δτ is significantly shorter than in Mg2+-depleted chloroplasts, although the addition of MgCl2 had no marked effect on τ 1/2 (Tikhonov et al. 1984). Since the lag-phase Δτ is always shorter (Δτ ≤ 4 ms) than the half-time of \( {\mathrm{P}}_{700}^{+} \) reduction (t 1/2 ≈ 18 – 20 ms), one can conclude that the formation of PQH2 and its diffusion to the Cyt b 6 f complex occur more rapidly than PQH2 oxidation at the Qo-site.

Panel A shows the kinetics of P700 redox transients induced by a short pulse (t 1/2 = 7 μs) of white light given on the background of a continuous far-red light (λ max = 707 nm). Panels B and C illustrate the influence of pH and temperature on kinetic parameters Δt and τ 1/2 for P700 transients (see panel A for definition) (Modified figures adopted from Tikhonov (2014, panel A) and Tikhonov et al. (1984, panels B and C))
A significant difference between kinetic parameters Δτ and τ 1/2 was observed over a wide range of pH (Fig. 10.5b) and temperature (Fig. 10.5c). These observations provide clear evidence that the overall rate of the intersystem electron transfer is determined mainly by PQH2 oxidation at the Qo-site, a rate that decelerates with the lumen acidification and a decrease in temperature. Short times of PQH2 diffusion from PSII to the Cyt b 6 f complex may be accounted for, at least partly, by the close neighbourhood of these complexes in the granal domains of the thylakoid membrane. Note that the temperature dependence of \( {\mathrm{P}}_{700}^{+} \)reduction in isolated bean chloroplasts reveals the characteristic “break” at ≈20°C. Below this temperature (≤20°C), kinetic parameter τ 1/2 (the half-time of \( {\mathrm{P}}_{700}^{+} \) reduction) strongly depends on the temperature; at higher temperatures (≥20°C), τ 1/2 is almost independent of temperature. As it was demonstrated in earlier works (Tikhonov et al. 1980, 1983), this peculiarity of the temperature dependence of τ 1/2 (as well as the rate of ATP synthesis) strongly correlates with thermo-induced structural changes detected in the lipid phase of the thylakoid membrane with the lipid-soluble spin-probes (nitroxide radicals). One can speculate, therefore, that thermoinduced changes in the lipid phase of the thylakoid membrane can affect the rate of PQ turnover. For instance, “solidification” of the lipid bilayer with lowering the temperature may reduce the rate of the PQH2 ‐ b 6 f complex formation, and/or would cause the slowing down of PQH2 oxidation due to decelerated release of protons into the lumen, which is considered as the prerequisite for PQH2 oxidation by the Cyt b 6 f complex.
The notion that a relatively high mobility of PQH2 in the thylakoid membrane finds support from computer simulation of PQ diffusion, suggests that PQH2 could travel farther than 290 nm in 10 ms (Tremmel et al. 2003). This estimate is in agreement with experimental data, demonstrating that electron transfer between PSII to PSI is not limited by PQH2 migration along the thylakoid membrane. High rates of PQH2 diffusion in the thylakoid membrane suggest that the rate of PQ turnover in chloroplasts is determined predominantly by the events associated with the PQH2 penetration to the Qo-cavity and its oxidation within the Cyt b 6 f complex (for review, see Tikhonov 2013, 2014).
Summing up the above reasonings, one can conclude that the light-induced reduction of PQ to PQH2 in PSII occurs much more rapidly than PQH2 oxidation by the Cyt b 6 f complex. It is noteworthy, however, that experimental data for partial reactions of electron transfer within the Cyt b 6 f complex are often scattered, depending on the system investigated and its metabolic state. For instance, the post-illumination reduction of Cyt f in different species of intact leaves was characterized by half-times ranging from 20 to 28 ms for a wide range of light intensities (up to 2800 of photons μmol m−2s−1, Kramer et al. 1999). Alternatively, several authors reported a more rapid turnover of the Cyt b 6 f complex in leaves (Harbinson and Hedley 1989; Laisk et al. 2005). Relatively short apparent times of Cyt f and Cyt b reduction (t 1/2 ≈ 3 – 6 ms) are typical of intact C. reinhardtii cells (Soriano et al. 1996; Ponamarev and Cramer 1998) and the cyanobacterium Synechococcus sp. PCC 7002 (Yan and Cramer 2003). Dispersion of kinetic data might be explained by several reasons, e.g., due to differences between the species and variable stoichiometry between PSII, Cyt b 6 f, and PSI complexes (Schöttler et al. 2015; Puthiyaveetil et al. 2016). Variability of electron capacities of redox partners on the donor and acceptor sides of the Cyt b 6 f complex may also influence the kinetic behaviour of the system, exaggerating or underestimating the contributions of rapid and slow phases of electron transport processes (for recent discussion of this point, see Tikhonov 2016). Nevertheless, the rate of PQH2 oxidation in the Cyt b 6 f complex comprises the rate-limiting step in the chain of electron transport between PSII and PSI.
Let us now consider another aspect of PQH2 interaction with the Cyt b 6 f complex related to variability of the rates of PQH2 oxidation and \( {\mathrm{P}}_{700}^{+} \) reduction. Speaking of kinetic peculiarities of PQH2 interaction with the Cyt b 6 f complex, one has to take into account connectivity between spatially separated electron transport complexes via the mobile electron carriers, PQ and Pc (this scenario is depicted symbolically in Fig. 10.6a). Spatially separated PSII and Cyt b 6 f complexes can interact with each other due to rapid diffusion of PQH2 and PQ molecules within the thylakoid membrane and fast diffusion of Pc molecules in the lumen. An apparent rate of electron transfer from PQH2 to PSI is sensitive to the redox status of the ETC. With the rise of PQH2 concentration, the probability of formation of the substrate-enzyme complex PQH2 ‐ b 6 f increases, thereby accelerating the overall rate of electron flow from PSII to \( {\mathrm{P}}_{700}^{+} \). This point can be illustrated by the data presented in Fig. 10.6b, which shows that the initial rate of \( {\mathrm{P}}_{700}^{+} \) reduction (\( {R}_{{\mathrm{P}}_{700}} \)) in response to light pulses of various duration increases with the rise of a number of electrons per P700 (N e) injected from PSII into the intersystem ETC (Tikhonov et al. 1980). Saturation of kinetic parameter \( {R}_{{\mathrm{P}}_{700}} \) at sufficiently high numbers of electrons injected (N e > 2) implies that the proton-coupled electron transfer (PCET) events, taking place within the Cyt b 6 f complex after PQH2 binding to the Qo-center, determine the rate of PQH2 turnover. A similar result was reported by Haehnel (1973) who observed acceleration of \( {\mathrm{P}}_{700}^{+} \) reduction with an increase in the number of consecutive light flashes illuminating spinach chloroplasts. Analysis of experimental data on the redox transients of P700 within the framework of a mathematical model also suggests that even significant attenuation of PSII activity (for instance, due to generation of NPQ) may not cause drastic reduction of electron flow through PSI during the action of a sufficiently strong continuous actinic light (Tikhonov and Vershubskii 2017). This result can be explained by “electronic” and/or “excitonic” connectivity between different PSII units (Siggel et al. 1972; Tikhonov and Ruuge 1979; Haehnel 1982; Stirbet 2013). At sufficiently strong actinic light, the overall flux of electrons between PSII and PSI would maintain at a high level even upon the attenuation of PSII activity, provided the rate-limiting step of electron transfer is beyond the stage of PQH2 formation (Tikhonov and Vershubskii 2017). This is because the PQH2 pool serves as the redox buffer, which can accumulate electron equivalents capable of reducing \( {\mathrm{P}}_{700}^{+} \) via the Cyt b 6 f complex and Pc.

Panel A illustrates the connectivity between different electron transport complexes by means of mobile electron carriers, plastoquinone (PQ) and plastocyanin (Pc); ripple-type arrows (termed “spillover”) symbolize the excitonic mechanism of interaction between the light harvesting complexes of PSII (LHCII), after (Tikhonov and Vershubskii 2017). Panel B shows the dependence of the initial rate of post-illumination reduction of \( {\mathrm{P}}_{700}^{+} \)in bean chloroplasts versus an average number of electrons (per P700) injected into the intersystem electron transport chain in response to light pulses of different duration. Kinetic data used in plot B were compiled from Figs. 7 and 8 presented in (Tikhonov et al. 1980). Open and closed circles correspond to measurements at 20 °C and 29 °C, respectively
10.3 Plastoquinol Interaction with the Cytochrome b 6 f Complex
10.3.1 Q-Cycle
Oxidation of PQH2 occurs after its penetration into the quinone-exchange cavity and binding to the Qo-center positioned on the lumenal side of the Cyt b 6 f complex. One can say that two reagents, PQH2 and oxidized ISP (ISPox), form the “enzyme-substrate” complex (ES-complex). According to the earlier models of quinol oxidation, electron transfer from the quinol molecule to the ISPox proceeds only after the quinol deprotonation reaction (QH2 → QH− + H+).Footnote 1 The “proton-gated affinity change” (Link 1997) and the “proton-gated charge transfer” (Brandt 1996; Brandt and Okun 1997) mechanisms imply that it is the anion form of the quinol (QH−) that binds to the Qo-center and then donates electron donor to oxidized redox center of the Rieske protein (ISPox). Crofts and collaborators suggested that the formation of the ES-complex does not need the dissociation of a proton from QH2, but involves the dissociated form of the ISPox (Crofts 2004a, b; Crofts et al. 2013). The ES-complex QH2 ‐ ISPox is stabilized by the hydrogen bond between the -OH group of the quinol molecule and the imidazolate ring of deprotonated ISPox (Fig. 10.7). The redox center of the ISP contains the [Fe2S2] cluster, one of the Fe atoms of which is ligated by two His residues (His136 and His155 in C. reinhardtii). In the oxidized state, this cluster is diamagnetic (spin S = 0) due to the antiferromagnetic coupling between two Fe3+ ions. In the reduced state, the [Fe2S2] cluster becomes paramagnetic (spin S = 1/2) and, therefore, it can be detected by the electron paramagnetic resonance method at cryogenic temperatures (Zhang et al. 1996; Soriano et al. 2002).

Schematic representation of electron and proton transfer reactions upon the two-electron oxidation of quinol at the Qo-site of the Cyt b 6 f complex from Chlamydomonas reinhardtii (PDB entry 1Q90, Stroebel et al. 2003). Figure was produced using Accelerys DV visualizer software package (http://www.accelrys.com)
The bifurcated reaction of PQH2 oxidation proceeds as two concerted reactions of proton-coupled electron transfer (PCET): (i) PQH2 → SQ + e− + H+, and (ii) SQ → PQ + e− + H+, where SQ denotes the semiquinone form of PQ or ubiquinone (UQ) (Berry et al. 2000; Cramer et al. 2006, 2011; Hasan et al. 2013a; Crofts 2004a, b; Crofts et al. 2013; Snyder et al. 2000; Cape et al. 2007). The term “proton-coupled” means that reactions (i) and (ii) are tightly coupled to proton transfer from PQH2 and SQ to appropriate proton-accepting groups, respectively. The term “concerted” implies that both reactions (i) and (ii) occur simultaneously (or almost simultaneously) (Osyczka et al. 2004, 2005; Zhu et al. 2007).
Plastoquinol Oxidation Reactions
The pictorial scheme of events associated with the bifurcated (two-electron) oxidation of PQH2 within the framework of the Mitchellian Q-cycle is shown in Fig. 10.8. The turnover of PQH2 molecules in the Qo-center starts with the first reaction of electron transfer. Oxidized iron-sulfur cluster of the ISP (\( {\mathrm{ISP}}_{\mathrm{ox}} \)) serves as the primary electron acceptor in the high-potential redox chain reduced by PQH2:
This reaction is the PCET process, in which the electron and proton transfer proceed as tightly coupled events. Structural data suggest that the Nε atom of the histidine residue, which ligates the [Fe2S2] cluster of the ISP extrinsic domain of the Cyt b 6 f complex (His155 in C. reinhardtii or His129 in M. laminosus), is the prime candidate for the role of the recipient accepting the proton from PQH2 (Fig. 10.7). This residue of the ISP is within hydrogen-bonding distance from the –OH group of the quinol. The formation of the hydrogen bond between this group and the Nε atom of the neighboring histidine is considered as an essential prerequisite for the first reaction (10.1) of quinol oxidation (Hsueh et al. 2010; Lhee et al. 2010). The notion about the formation of such a hydrogen bond finds strong support from spectroscopic (EPR, NMR, and ATR-FTIR) studies of the bc 1 complex (Samoilova et al. 2002; Zu et al. 2003; Iwaki et al. 2005; Lin et al. 2006; Hsueh et al. 2010; Lhee et al. 2010), which is akin to the b 6 f complex.

A sketch illustrating plastoquinone turnover in the Cyt b 6 f complex within the framework of the Q-cycle model
The plastosemiquinone molecule formed as the result of the first PCET reaction (10.1) will be in the neutral form (PQH•). In the second PCET reaction (10.2), PQH• donates an electron to the low-potential heme \( {b}_6^{\mathrm{L}} \)of the low-potential branch of the Cyt b 6 f complex:
In chloroplasts, the key role in the proton migration from PQH• to the lumen belongs to the proton-accepting carboxyl group of glutamate (Glu78). Glu 78 is part of a highly conserved PEWY sequence of subunit IV, which participates in the formation of the quinone-binding pocket of the Qo-center. The proton-accepting group of Glu78 is positioned between the PQH2 molecule and heme \( {b}_6^{\mathrm{L}} \) (Fig. 10.7). This group may serve as the primary recipient of the proton from PQH• (Zito et al. 1998). It has been suggested, on the basis of structural and mutagenesis data for the Cyt bc 1 complex, that Glu in the PEWY-span accepts a proton from the neutral semiquinone, and delivers it by rotation of the carboxylic side chain to the proton exit channel, leaving the anionic form of SQ in the Qo-site (Victoria et al. 2013). Mutagenesis of Glu78 is known to impair turnover of the Cyt b 6 f complex in C. reinhardtii (Zito et al. 1998; Finazzi 2002). A similar effect of the impediment to UQH2 oxidation has been described for the Cyt bc 1 complex (Victoria et al. 2013).The proton from the carboxyl group of Glu78 migrates to the lumen through one of putative proton-conducting pathways (Hasan et al. 2013c; Tikhonov 2014).
The reduced ISPred passes an electron to heme f located in the peripheral domain of the Cyt f protein. The ISP occupies a cleft between the large and small domains of Cyt f (Fig. 10.3). A key step of electron transfer from the [Fe2S2] cluster of the ISP to the Cyt f heme involves the large-scale conformational changes within the Cyt b 6 f complex. A distance between the redox cluster [Fe2S2] docked nearby the Qo-center and heme f is too long (≈ 26 Å) in order to provide physiologically rapid direct transfer of electrons from the ISPred to Cyt f by the mechanism of quantum mechanical tunneling (Page et al. 1999). However, a high mobility of the extrinsic domain of the ISP containing the redox cluster [Fe2S2] should facilitate electron transfer between the ISPred and oxidized Cyt f. It is likely that after the reduction of ISP its extrinsic domain moves from the proximal position in the vicinity of the Qo-site towards the distal position close to the heme f, thereby providing electron transfer from the ISPred to Cyt f. This case is cartooned as the transition from the state (1) to state (2) shown in Fig. 10.8.
There are several lines of evidence for a high mobility of the extrinsic domain of the ISP containing the redox center [Fe2S2] within the whole ensemble of the Cyt b 6 f complex (Breyton 2000; Heimann et al. 2000; Soriano et al. 2002; Roberts et al. 2002; Yan and Cramer 2003; de Vitry et al. 2004). One of the convincing arguments in favor of the cluster [Fe2S2] mobility, which paves the way for electronic communication between the ISP and Cyt f, may be considered from the results of the X-ray data on the Cyt b 6 f complex from M. laminosus (Hasan et al. 2013b). This conclusion stems from extensive crystallographic disorder of the ISP extrinsic domain indicating its conformational flexibility. The ISP disorder has been observed in the Cyt b 6 f complex supplemented with anionic lipids. This indicated that the electric charges on the lipid headgroups may influence motion of the ISP extrinsic domain within the Cyt b 6 f complex. Structural data first obtained for a variety of Cyt bc 1 complexes of different origin present the apt evidence for the long-range movements of the flexible domain of the ISP containing the Rieske [Fe2S2] cluster (for review, see Darrouzet et al. 2001; Xia et al. 2013). Two different positions of the [Fe2S2] cluster relative to heme c 1 have been found in the X-ray crystallographic structures in the presence or absence of various inhibitors, suggesting the large-scale movement of the extrinsic domain of the ISP containing the [Fe2S2] cluster (Iwata et al. 1998; Kim et al. 1998; Zhang et al. 1998; Esser et al. 2006).
The long-range “tethered” diffusion of the [Fe2S2] cluster towards heme f enables electron transfer from ISPred to Cyt f and further to plastocyanin. After oxidation and deprotonation of the \( {\mathrm{ISP}}_{\mathrm{red}}\left({\mathrm{ISP}}_{\mathrm{red}}^{\overline{\bullet}}{\mathrm{H}}^{+}+f\to {\mathrm{ISP}}_{\mathrm{ox}}+{f}^{-}+{\mathrm{H}}_{\mathrm{in}}^{+}\right) \), its mobile extrinsic domain returns from the distant (Fig. 10.8, panel 2) to the proximal position near the Qo-site (Fig. 10.8, panel 3). Note that the movements of the ISP extrinsic domain between the proximal (Qo-site) and distal positions (heme f in the Cyt b 6 f complex or heme c 1 in the Cyt bc 1 complex) are more rapid than the overall process of quinol oxidation. A mutagenesis study of the ISP in C. reinhardtii (de Vitry et al. 2004) indicated that the protein-linking domain of the chloroplast ISP was much more flexible than its counterpart in mitochondria. This was explained by the greater flexibility of the polyglycine hinge in the Cyt b 6 f complex than of the polyalanine hinge in the Cyt bc 1 complex (Yan and Cramer 2003).
The destiny of the second electron donated by PQH2 differs from that of the first electron. After the removal of the first electron and proton from PQH2 in the Qo-center (the first PCET reaction), the extrinsic domain of the ISP moves towards heme f. Displacement of the redox center [Fe2S2] away from the Qo-site precludes the thermodynamically favorable donation of the second electron from SQ to the high-potential branch, directing the SQ radical to reduce the low-potential heme \( {b}_6^{\mathrm{L}} \). After electron transfer from the plastosemiquinone PQH• to the low-potential heme \( {b}_6^{\mathrm{L}} \)(reaction 2), the electron is transferred to the high-potential heme \( {b}_6^{\mathrm{H}} \)and further to heme c i, towards the PQ binding site Qi on the stromal side of the Cyt b 6 f complex, reducing (PQ)i to its plastosemiquinone anion-radical species \( {\left({\mathrm{PQ}}^{\underset{\_}{\bullet }}\right)}_{\mathrm{i}} \) (Fig. 10.8, panel 2).Footnote 2 After the second turn of PQH2 turnover, PQH2 forms in the Qi-center, \( {\left({\mathrm{PQ}}^{\underset{\_}{\bullet }}\right)}_{\mathrm{i}}+{\mathrm{e}}^{-}+2{\mathrm{H}}_{\mathrm{out}}^{+}\to {\left({\mathrm{PQ}\mathrm{H}}_2\right)}_{\mathrm{i}} \), and then dissociates into the membrane (Fig. 10.8, panels 3 and 4).
Taking into account that two protons appear in the lumen per one H2O molecule oxidized in the WOC of PSII (H+/e− = 1) and four protons are pumped into the lumen per two electrons transferred to PSI through the Cyt b 6 f complex (H+/e− = 2), the overall stoichiomentry of proton and electron transport is H+/e− = 3. This means that three hydrogen ions will appear into the lumen per one electron transferred from PSII to PSI. Thus, the operation of the Q-cycle enhances the proton/electron stoichiometry, increasing the efficiency of proton pumping into the lumen, as was first predicted by Peter Mitchell who put forward his brilliant idea of the Q-cycle as early as 1976 (Mitchell 1976). It is note worthy to stress that two-thirds of the trans-thyalkoid pH difference (ΔpH) in oxygenic photosynthesis originates from the proton pumping by the Cyt b 6 f complex.
Proton Exit Pathways
Oxidation of PQH2 occurs as the proton-coupled reactions, in which two protons migrate into the aqueous bulk phase of the lumen. Each of two steps of the PQH2 oxidation reaction is accompanied by deprotonation. Analysis of the crystal structure of the Cyt b 6 f complex from C. reinhardtii (PDB entry 1Q90) revealed two putative pathways for proton relase from the PQH2 molecule oxidized at the Qo-site (Fig. 10.9). The side residue of His ligating the [Fe2S2] cluster of the ISP (His155 in C. reinhardtii, or His129 in M. laminosus) may serve the role of the immediate recipient of the first proton donated by PQH2. Being bound to this His residue of the extrinsic domain of ISP, the proton extracted from PQH2 starts traveling along the exit route to the aqueous bulk phase of the lumen. After oxidation of protonated ISPred by Cyt \( f\left({\mathrm{ISP}}_{\mathrm{red}}^{\overline{\bullet}}{\mathrm{H}}^{+}+f\to {\mathrm{ISP}}_{\mathrm{ox}}+{f}^{-}+{\mathrm{H}}_{\mathrm{in}}^{+}\right) \) its affinity for a proton decreases and the proton releases from the ISP, migrating to the lumen through the intra-protein proton-conducting channel containing water molecules. The structural and biochemical data indicate that the water molecules of the proton-conductive chain, in addition to the surrounding amino acid residues, play an important role in Cyt f functioning (Ponamarev and Cramer 1998; Sainz et al. 2000). Interruption of the internal water chain, which forms a path for proton migration, impairs the operation of the Cyt b 6 f complex: electron transport from the ISP to Cyt f decelerates, and the concerted reduction of Cyt b 6 and Cyt f is lost. An alternative point of view on the nature of the primary recipient of the proton donated by quinol has been suggested by Postila et al. (2013), who scrutinized the problem on the basis of atomic molecular dynamics simulations for the Cyt bc 1 complex. They suggested that water molecules positioned in suitable places of the Qo-site nearby the UQH2 molecule could act as the primary proton-receiving partners in the two-electron oxidation of UQH2.

Side view of the monomeric fragment the Cyt b 6 f complex from Chlamydomonas reinhardtii (PDB entry 1Q90, Stroebel et al. 2003), which demonstrate putative exit pathways for protons dissociating from the Qo-site to the thylakoid lumen. The traces of the proton-conducting pathways (shown by green and brown spheres) were found in collaboration with B.V.Trubitsin, using the program Caver for detection of possible channels for traveling mobile molecules inside protein structures (Chovancová et al. 2012). The program output was visualized using PyMOL Molecular Graphics System
Oxidation of semiquinone PQH• formed after the first PCET reaction is also accompanied by the proton release into the lumen (\( {\mathrm{PQH}}^{\bullet }+{b}_6^{\mathrm{L}}\left(\mathrm{ox}\right)\to \mathrm{PQ}+{b}_6^{\mathrm{L}}\left(\mathrm{red}\right)+{\mathrm{H}}_{\mathrm{in}}^{+} \)). The proton released from PQH• migrates to the lumen through the second putative proton-conducting channel (Tikhonov 2013, 2014). As noted above, it is likely that PQH• donates a proton to the neighboring acidic group (−COO−) of Glu78 positioned in subunit IV between the quinone binding center Qo and heme \( {b}_6^{\mathrm{L}} \) (Fig. 10.7). From Glu78 the proton migrates to the lumen. The crystal structure of the Cyt b 6 f complex from M. laminosus (Hasan et al. 2013c) suggests that the proton-accepting group of Glu3 located in subunit G may attract a proton from Glu78 and then release it to the aqueous bulk phase of the lumen.
Proton Entry Pathways
At the PQ binding Qi-center located on the “negative” (stromal) side of the Cyt b 6 f complex, reduced PQ molecules undergo protonation through the proton-conductive pathways oriented towards the aqueous bulk phase of stroma. Refined crystal structure (2.70 Å) of the Cyt b 6 f complex from M. laminosus (Hasan et al. 2013c) revealed a unique short pathway for proton transfer to the Qi-site, which involves two residues of the Cyt b 6 protein (\( {\mathrm{H}}_{\mathrm{out}}^{+}\left(\mathrm{aq}\right)\to \mathrm{Asp}20\to \arg 207\to {\mathrm{Q}}_{\mathrm{i}} \)). The Asp20 side chain is located on the surface of the Cyt b 6 f complex; therefore, it has direct access to the aqueous phase. Both steps of the proton transfer within the Cyt b 6 protein (Asp20 → Arg207 and Arg207 → Qi) are mediated by hydrogen bonds. Arg207 has access to (PQ)i as well as heme c i. Another putative way of proton transfer may be mediated by the surface residues Lys24. In addition to these proton transfer pathways, the acidic side chains of Glu29 and Asp35 (subunit IV) may be involved in proton transfer from stroma to the PQ molecule, reduced at the Qi-center (Hasan et al. 2013c).
10.3.2 Kinetics of Plastoquinol Oxidation
The elucidation of the mechanism of PQH2 oxidation is a key to understanding the nature of the rate-limiting step in the intersystem ETC. Molecular machinery of quinol oxidation in the Cyt complexes of bc type have been scrutinized in a number of original works and related review articles (see Berry et al. 2000; Crofts 2004a, b; Crofts et al. 1999a, b, 2000, 2006, 2013; Mulkidjanian 2005; Xia et al. 2013). There are several events that have been considered to explain the nature of the rate-limiting step of quinol oxidation: (i) the formation of the quinol-ISP complex (Mulkidjanian 2005), (ii) deprotonation of the neutral quinol to the anionic form (\( {\mathrm{PQH}}_2\to {\mathrm{PQH}}^{-}+{\mathrm{H}}_{\mathrm{in}}^{+} \)) and the formation of the “enzyme-substrate” complex (PQH−/ISPox) followed by PQH− oxidation (Brandt and Okun 1997; Link 1997; Rich 2004), and (iii) constrained diffusion of the ISP mobile domain between the PQH2 binding site Qo and Cyt f (or Cyt c 1) (Izrailev et al. 1999; Crofts et al. 1999a, b; Darrouzet et al. 2001). Structural, kinetic and thermodynamic aspects of PQH2 oxidation at the Qo-site are considered below.
Structural and kinetic data suggest that the round-trip movements of the mobile domain of the ISP between the Qo-site and heme f could determine only partly the turnover rate of the Cyt b 6 f complex. A flexible “detail” of the ISP machinery (a “hinge”) provides restricted tethered diffusion of the extrinsic domain containing the redox [Fe2S2] cluster. However, the flip-flop motions of the [Fe2S2] cluster between the proximal and distal positions are rapid as compared to the rate of PQH2 oxidation; therefore, they should not limit the overall rate of PQ turnover. According to (Yan and Cramer 2003; de Vitry et al. 2004), electron transfer from the ISPred to heme f may occur more rapidly (t 1/2 ≤ 2 − 4 ms) than the overall rate of PQH2 oxidation (t 1/2 ≥ 4 − 20 ms, Stiehl and Witt 1969; Haehnel 1984). This implies that the rate of PQH2 oxidation should be determined predominantly by the processes associated with the formation of the substrate-enzyme complex PQH2/ISPox and intrinsic reactions related to PQH2 oxidation within the Cyt b 6 f complex.
Analyzing experimental data on ubiquinol (UQH2) oxidation in the bc 1 complex in the purple photosynthetic bacterium Rhodobacter sphaeroides, Crofts and collaborators concluded that the activated step of UQH2 oxidation was in a reaction step after the formation of the UQH2/ISPox complex (Hong et al. 1999). The pH-dependence of the rate of UQH2 oxidation reflected the pK of the oxidized ISP (ISPox) and requirement for the deprotonated form of ISPox in formation of the UQH2/ISPox complex. Conclusive argument in favor of the first electron transfer reaction as the rate-limiting step of quinol oxidation was based on experimental data in strains with mutations in the ISP that lowered its redox potential E m. The overall rate of UQH2 oxidation was determined by the driving force for the first electron transfer, \( \varDelta {G}_1^0=- zF\left({E}_{\mathrm{m}\left(\mathrm{ISP}\right)}-{E}_{\mathrm{m}\left({\mathrm{QH}}_2/\mathrm{SQ}\right)}\right) \), but was almost independent of the driving force for the second electron transfer, \( \varDelta {G}_2^0=- zF\left({E}_{\mathrm{m}\left({b}_{\mathrm{L}}\right)}-{E}_{\mathrm{m}\left(\mathrm{SQ}/\mathrm{Q}\right)}\right) \) (Crofts 2004b; Crofts et al. 2013). This strongly suggests that the stage of electron transfer from the quinol molecule to ISPox is the rate-limiting step of bifurcated oxidation of quinol.
Electron transfer from quinol to the redox center of ISPox is tightly coupled to the concerted proton transfer to an appropriate proton-accepting group. As noted above, the Nε atom of the histidine residue liganding to the Fe1 atom the [Fe2S2] cluster is the prime candidate for the role of the primary recipient of the proton donated by PQH2. Figure 10.10a illustrates the scenario of protonation/deprotonation events associated with the reduction/oxidation of the ISP. The affinity of the ISP for protons depends on its redox state (Zu et al. 2003; Iwaki et al. 2005; Lin et al. 2006; Hsueh et al. 2010; Lhee et al. 2010). The deprotonated state of ISPox is likely an obligatory prerequisite for the first step of PQH2 oxidation (Fig. 10.10a); therefore, the pK ox value of the ISPox will determine the pH-dependence of the rate of this reaction. In photosynthetic systems of oxygenic type, the pK value of functional proton-accepting groups of ISPox is characterized by pK ox ≈ 6 − 6.5 (Finazzi 2002; Soriano et al. 2002). Ex facte the affinity of the ISP for a proton should increase with the acquisition of the negative charge by the redox cluster [Fe2S2]. That is why pK ox increases to pK red ≈ 8.3 − 8.9 after the ISP reduction. This means that under the normal physiological conditions (pHin ≤ pHout ≤ 8) the ISPred will keep tightly the proton accepted from PQH2. After oxidation of the reduced and protonated form of the ISP, its affinity to the proton decreases (Fig. 10.10a), and the proton will dissociate into the lumen via a proton-conducting channel (\( {\mathrm{ISP}}_{\mathrm{red}}^{\overline{\bullet}}{\mathrm{H}}^{+}+\mathrm{Cyt}\kern0.28em {f}_{\mathrm{ox}}\to {\mathrm{ISP}}_{\mathrm{ox}}+\mathrm{Cyt}\kern0.28em {f}_{\mathrm{red}}+{\mathrm{H}}_{\mathrm{in}}^{+} \)).

A diagram illustrating redox-dependent protonation/deprotonation of the ISP protein (A), and pH-dependence of PQH2 oxidation by isolated cytochrome b 6 f complex (B). Open circles shown in panel B are taken from Fig. 4 in (Hope et al. 1994); solid line represents the result of simulation according to Eq. (10.3) for parameters pK ox = 6.2 and pK red = 8.7 (see text for explanations) (Modified from Figure 9 in Tikhonov 2014)
Let us consider the pH-dependence of PQH2 oxidation rate. At any given pH, the rate of PQH2 oxidation will be controlled by two factors: (i) a probability of finding the oxidized ISPox in deprotonated state, p(ISPox), the value of which is determined by pK ox, and (ii) a probability of finding reduced ISP in protonated state, \( p\left({\mathrm{ISP}}_{\mathrm{red}}^{\bullet }{\mathrm{H}}^{+}\right) \), determined by the pK red value. Therefore, the overall rate of PQH2 oxidation, k 1, will be proportional to the product \( p\left({\mathrm{ISP}}_{\mathrm{ox}}\right)\times p\left({\mathrm{ISP}}_{\mathrm{red}}^{\bullet }{\mathrm{H}}^{+}\right) \). Elementary calculations lead to the following term (Tikhonov 2014):
Equation (10.3) predicts the bell-shape pH-dependence of the rate constant \( {k}_1 \), which reflects the pK ox and pK red values of the ISP. Figure 10.10b shows that formula (10.3) adequately describes experimental pH-dependence of the rate of PQH2 oxidation by the Cyt b 6 f complex if pK ox = 6.2 and pK red = 8.7 (Hope et al. 1994).
10.3.3 Energy Profiles of Electron Transport Reactions in the Q o -Site
The energy profile of bifurcated oxidation of PQH2 can shed additional light on the nature of this reaction. Thermodynamic aspects of PQH2 oxidation can be illustrated by the comparison of the redox potential profiles for electron carriers of the high-potential (PQH2 → ISP → f → Pc → P700) and the low-potential (\( {\mathrm{PQH}}^{\bullet}\to {b}_6^{\mathrm{L}}\to {b}_6^{\mathrm{H}}\to {c}_{\mathrm{i}}\to {\left(\mathrm{PQ}\right)}_{\mathrm{i}} \)) redox chains shown in Fig. 10.11. The overall change in Gibbs free energy upon the two-electron oxidation of PQH2 is negative. As the result of complete turnover of PQH2 in the Q-cycle, one electron extracted from PQH2 reaches one of \( {\mathrm{P}}_{700}^{+} \) centers, and this is the energy-favorable (down-hill) processes. However, the fact of the mater is that the first step of PQH2 oxidation (electron transfer \( {\mathrm{PQH}}_2\overset{e^{-}}{\to }{\mathrm{ISP}}_{\mathrm{ox}} \) with the formation of PQH•) is the energy-uphill process, although the overall reaction of two-electron oxidation of PQH2 is thermodynamically favorable process (Fig. 10.11). By analogy with the Cyt bc 1 complex (for references, see Hong et al. 1999; Crofts et al. 2000; Crofts 2004a, b), it is safe to suggest that in the Cyt b 6 f complex this stage of electron transfer proceeds through the energy barrier, which decelerates the overall reaction of PQH2 oxidation. Plastosemiquinone PQH• formed in the result of the first electron transfer is a strong reductant capable of reducing the low-potential heme \( {b}_6^{\mathrm{L}} \). High reducing activity of PQH• is a general property of semiquinone species, because most of the redox couples SQ/Q are characterized by relatively low values of their redox potentials (Clark 1960). Further reactions of electron transfer along the low-potential and high-potantial chains proceed spontaneously with the loss of free energy.

A diagram of the midpoint redox-potentials of electron carriers of the high-potential (ISP, Cyt f, Pc, and P700) and low-potential (hemes \( {b}_6^{\mathrm{L}} \), \( {b}_6^{\mathrm{H}} \), and c i) branches of electron transport in the Cyt b 6 f complex. The levels of the midpoint potentials shown here are the averaged values taken from the literature for Chlamydomonas reinhardtii (Alric et al. 2005; Hasan et al. 2013a; Nelson and Yocum 2006; Pierre et al. 1995; Zito et al. 1998)
Let us compare the midpoint potentials of one-electron redox couples PQH2/SQ and SQ/PQ (\( {E}_{\mathrm{m}\left({\mathrm{PQH}}_2/\mathrm{SQ}\right)} \) and \( {E}_{\mathrm{m}\left(\mathrm{SQ}/\mathrm{PQ}\right)} \)) involved in electron transport along the high- and low-potential chains (Fig. 10.11). For the redox couple PQH2/PQ, the mid-point potential \( {E}_{\mathrm{m}\left({\mathrm{PQH}}_2/\mathrm{PQ}\right)} \) can be measured directly, since PQH2 and PQ are relatively stable species. However, direct determination of \( {E}_{\mathrm{m}\left({\mathrm{PQH}}_2/\mathrm{SQ}\right)} \) and \( {E}_{\mathrm{m}\left(\mathrm{SQ}/\mathrm{PQ}\right)} \) is problematic, because semiquinones are extremely reactive species. These potentials may be evaluated indirectly (Mitchell 1976; Chobot et al. 2008) using the relationship (10.4):
According to Gill and Tuteja (2010) and Bleier and Dröse (2013), semiquinone species transiently formed in the Qo-centers of the Cyt bc 1 and Cyt b 6 f complexes are able to reduce molecular oxygen O2 to superoxide radical, \( {\mathrm{O}}_2^{\overline{\bullet}} \). The standard redox potential relative to 105 Pa of O2 is \( {E}_{0\left({\mathrm{O}}_2^{\overline{\bullet}}/{\mathrm{O}}_2\right)}=-330\kern0.28em \mathrm{mV} \); the redox potential relative to 1 M O2 equals to −160 mV (Wood 1988). This implies a rather low value for \( {E}_{\mathrm{m}\left(\mathrm{SQ}/\mathrm{Q}\right)} \). Assuming that \( {E}_{\mathrm{m}\left({\mathrm{Q}}^{\overline{\bullet}}/\mathrm{Q}\right)}\approx -280\kern0.28em \mathrm{mV} \) is sufficient to provide generation of superoxide radicals in mitochondria, Snyder et al. (2000) inferred that \( {E}_{\mathrm{m}\left({\mathrm{UQH}}_2/\mathrm{SQ}\right)}\sim 460\kern0.50em \mathrm{mV} \). This potential falls in the broad range of \( {E}_{\mathrm{m}\left({\mathrm{UQH}}_2/\mathrm{SQ}\right)} \) values (from 310 to 776 mV) obtained from kinetic data for the Cyt bc 1 complex in Rhodobacter sphaeroides (Hong et al. 1999; Cape et al. 2007). Similar estimates were obtained from the measurements of SQ generation at the Qo-site in the Cyt bc 1 complex (Cape et al. 2007). These estimates are in agreement with experimental evidence that in the Cyt complexes of the bc family the rate of quinol oxidation is largely determined by the first electron transfer reaction (see Crofts 2004a, b). Thus, taking into account the estimated values of \( {E}_{\mathrm{m}\left({\mathrm{PQH}}_2/\mathrm{SQ}\right)} \) and \( {E}_{\mathrm{m}\left(\mathrm{ISP}\right)} \) (Fig. 10.11), we can conclude that electron transfer from PQH2 to the ISPox is the energy-uphill process that limits the overall rate of PQH2 oxidation (see also Malnoë et al. 2011). The increase in free energy upon the first electron transfer (\( {\mathrm{PQH}}_2\overset{e^{-}}{\to }{\mathrm{ISP}}_{\mathrm{ox}} \)) is compensated by the free energy decrease in the energy-favorable reactions SQ \( \overset{e^{-}}{\to}\kern0.50em {b}_6^{\mathrm{L}}\overset{e^{-}}{\to }{b}_6^{\mathrm{H}} \).
Figure 10.12 depicts the sequence of events associated with the reaction of PQH2 oxidation according to the “proton-first-then-electron” (PT/ET) transfer mechanism favoured by Crofts et al. (2013). After the formation of the PQH2 ‐ ISPox complex (the downhill transition 1 → 2), the proton transfer from PQH2 to the ISPox occurs (the uphill transition 2 → 3). This step is accompanied by the free energy rise \( D{G}_{\mathrm{proton}}=2.303\times RT\times \left(\mathrm{p}{K}_{{\mathrm{PQH}}_2}-\mathrm{p}{K}_{{\mathrm{ISP}}_{\mathrm{ox}}}\right)>0 \) (the so-called “Brønsted barrier”). The transfer of a proton towards the ISP moiety promotes the electron transfer, resulting in the production of \( {\mathrm{ISP}}_{\mathrm{red}}^{\overline{\bullet}}{\mathrm{H}}^{+} \)and PQH• (the transition 3 → 4 → 5 over the activation barrier 4). \( {\mathrm{ISP}}_{\mathrm{red}}^{\bullet }{\mathrm{H}}^{+} \)dissociates from the Qo-center and moves towards heme f (state 5). Then, after the second PCET reaction, oxidized quinone dissociates from the Qo-center (the downhill transition 5 → 6). Further electron transfer (the downhill transition 6 → 7) will stabilize charge separation after the completion of the PQH2 oxidation cycle at the Qo-site.

Plausible energy profile of bifurcated two-electron reaction of quinol oxidation at the Qo-site (Modified Figure 11 from Tikhonov 2014)
Quantum chemical calculations for a model system consisting of TMQH2 (trimethylbenzoquinol, the “bobtail” analog of PQH2), the [Fe2S2] cluster, and surrounding them amino acid residues of the Cyt b 6 f complex (Fig. 10.13a), support the notion that the PCET from PQH2 to ISPox is the “uphill” process, which hampers the overall rate of PQH2 oxidation (Frolov and Tikhonov 2009). According to density functional theory (DFT) computations, the transfer of a hydrogen atom from the quinol molecule to the nearest nitrogen atom of His155 is the endoergonic process (ΔE ≈ 10 kcal/mol) with a rather high energy barrier (Fig. 10.13b). The rate constant for this process was calculated using the Moser–Dutton ruler for evaluation of the rate of electron tunneling between redox centers (Page et al. 1999) expanded by Crofts (2004b) for PCET reactions. The rate constant of TMQH2 oxidation in the model system was evaluated as k PCET~40 – 170 s−1 (Frolov and Tikhonov 2009). These estimates correspond to the half-times of PQH2 oxidation t 1/2 = ln 2/k PCET~4 − 17 ms. These values are in a reasonably good agreement with experimental data for electron transfer from PQH2 to \( {\mathrm{P}}_{700}^{+} \) (Sect. 10.2.5).

Panel A – Quantum chemical part of the model system, which includes trimethylbenzoquinol (TMQH2, the analogue of PQH2), the [Fe2S2] cluster, and surrounding them amino acid residues of the Cyt b 6 f complex, Cys134-Thr135-His136-Leu137-Gly138-Cys139, Cys152, Cys154-His155-Gly156-Ser157, Tyr159 (Frolov and Tikhonov 2009). TMQH2 was placed at the position of quinone inhibitor TDS in the crystal structure of the Cyt b 6 f complex from Chlamydomonas reinhardtii (PDB entry 1Q90, Stroebel et al. 2003)
Panel B – The plot of the model system energy versus the reaction coordinate R H − Nε, which simulates changes in the potential energy of the model system upon the displacement of the H atom from TMQH2 to the Nε atom of His155 (after Frolov and Tikhonov 2009)
What type of the mechanism for electron transfer in the Qo-centers, “sequential” or “concerted”, is realized in the Cyt b 6 f and bc 1 complexes? This question still remains a challenge to molecular bioenergenics. “Sequential” mechanism (\( {\mathrm{PQH}}_2\overset{-{e}^{-}}{\to}\mathrm{SQ}\overset{-{e}^{-}}{\to}\mathrm{PQ} \)) implies that the first and the second electron transfer reactions occur separately, with a certain time-delay between the steps. In this case, SQ radicals appear transiently as the intermediate species (Cape et al. 2006). A genuine “concerted” mechanism implies that the long-lived SQ species will not form if the bifurcated oxidation of PQH2 proceeds in a single step as a time-concerted transfer of two electrons (Osyczka et al. 2004, 2005). In the latter case, both steps are realized simultaneously (within picoseconds-microseconds), with no SQ intermediate available to participate in side reactions. The truly concerted mechanism could be realized when both the redox partners, the [Fe2S2] cluster and heme \( {b}_6^{\mathrm{L}} \), are oxidized. Simultaneous reduction of ISP and Cyt b L with t 1/2~250 microsecond was observed during ubiquinol oxidation in the bovine Cyt bc 1 complex (Zhu et al. 2007). Concerted oxidation of PQH2 precludes the accumulation of SQ, thereby avoiding the formation of harmful superoxide radicals (\( \mathrm{SQ}\overset{e^{-}}{\to }{\mathrm{O}}_2 \)) (Cape et al. 2006).
10.4 Regulation of the Intersystem Electron Transport
10.4.1 pH-Dependent Control of the Intersystem Electron Transport
Apart from the role of the molecular device for electron transport and proton pumping, the Cyt b 6 f complex participates in feedback regulation of electron transport. The light-induced acidification of the thylakoid lumen is the key factor of down-regulation of the intersystem electron transport (Rumberg and Siggel 1969; Tikhonov et al. 1981, 1984; Harbinson and Hedley 1989; Foyer et al. 2012; Järvi et al. 2013; Tikhonov 2013). Down-regulation of the intersystem electron transport is associated with two basic effects caused by the light-induced decrease in the intrathylakoid pHin: (i) the retardation of PQH2 oxidation, and (ii) the attenuation of PSII activity. The first mechanism of electron transport contol is realized in the stage of PQH2 oxidation at the Qo-site of the Cyt b 6 f complex (for review, see Tikhonov 2013, 2014, 2015). Acidification of the lumen impedes the oxidation of PQH2 due to the back “pressure” from the protons accumulating inside the thylakoids on the functional proton-accepting groups, thereby decreasing the rate of PQH2 oxidation (Figs. 10.5b and 10.10b). The influence of pHin on the rate of PQH2 oxidation is likely to reflect the effect of pHin on the formation of hydrogen bonds between PQH2 and the nearby proton-accepting groups in the Qo-center (the Nε atom of His155/His129 and the carboxyl group of Glu78). Note that electron transfer from Cyt f to Pc is independent of pHin (Finazzi 2002). Acidification of the lumen also acts as a signal for enhancement of excess energy dissipation in the light-harvesting antenna of PSII, i.e., non-photochemical quenching (NPQ) of Chl a excitation (Eberhard et al. 2008; Horton 2012; Ruban 2012). Both mechanisms, the slowing down of PQ turnover and generation of NPQ, cause deceleration of electron flow from PSII to PSI at excess of irradiation, protecting PSA against solar stress (for review, see Li et al. 2009, Demmig-Adams et al. 2012, Horton 2012, Rochaix 2014, Jallet et al. 2016).
10.4.2 Redox-Dependent Regulation of Electron Transport and State Transitions
Other regulatory mechanisms are associated with the light-induced activation of metabolic processes and redistribution of electrons fluxes between alternative electron transport pathways, which are controlled by the redox state of the chloroplast ETC. The light-induced activation of the CBC reactions is a striking example of pH- and redox-dependent regulation of metabolic processes in photosynthetic systems. The CBC is inactive in dark-adapted chloroplasts. The light-induced alkalization of stroma and redox-dependent activation of the CBC enzymes speed up the outflow of electrons from PSI to the CBC (Buchanan 1980; Edwards and Walker 1983; Andersson 2008; Michelet et al. 2013; Balsera et al. 2016). In addition to acceleration of LEF (PSII → PSI → CBC) due to activation of the CBC reactions, there are redox-dependent mechanisms of redistribution of electron fluxes between the LEF and CEF1 pathways. The partitioning of electron fluxes on the donor side of PSI is controlled by competition between the LEF and CEF1 pathways for reducing equivalents (Joliot and Joliot 2005; Breyton et al. 2006; Johnson 2011). Relatively slow consumption of NADPH in the CBC, which is peculiar to dark-adapted chloroplasts, would divert electrons from LEF to CEF1, thereby supporting generation of ΔpH and ATP synthesis (Kramer et al. 2004). In the meantime, with the light-induced activation of the CBC reactions, the contribution of CEF1 will decrease in favor of LEF.
The mechanism of electron transport control associated with the redistribution of light energy between the light-harvesting complexes of PSI and PSII is triggered by the “check-point device” operating at the level of the Cyt b 6 f complex. This regulatory mechanism relates to the so-called “state transitions” phenomenon. It is well established fact that the redox state of the PQ pool serves as a signal about the “traffic jam” on the pathway between PSII and PSI (see Bennett 1977; Allen 1992; Haldrup et al. 2001; Lemeille and Rochaix 2010; Rochaix 2014; Puthiyaveetil et al. 2016). The over-reduction of the PQ pool triggers a protein kinase that catalyses phosphorylation of the loosely bound light-harvesting complexes of PSII. This process is induced by PQH2 binding to the Qo-site of the Cyt b 6 f complex (Vener et al. 1997; Zito et al. 1999). In plants, there are three forms of LHCII trimers, which differ with respect to their association with PSII: “strong”, “moderate”, and “loose” binding to PSII (Galka et al. 2012; Puthiyaveetil et al. 2016). The loosely bound form of phosphorylated LHCII dissociates from the PSII-LHCII supercomplex and migrates to PSI, thereby increasing the light harvesting capacity of PSI (see cartoon in Fig. 10.14). Such a structural remodelling of PSA, termed “State 1 → State 2” transition, enhances PSI activity at expense of PSII, promoting the reoxidation of the PQH2 pool. Redistribution of energy in favour of PSI is a reversible process: after a decrease in the concentration of PQH2, LHCII becomes dephosphorylated due to chloroplast phosphatases. Dephosphorylation of LHCII allows its return to the grana domains enriched with PSII (“State 2 → State 1” transition), increasing the light absorption in PSII. Remodelling of PSA associated with state transitions provides short-term responses (in the minute range) of photosynthetic organisms to variations of light intensity. Thus, reversible transitions “State 1 ↔ State 2” provide the optimal balance of absorbed light energy between PSI and PSII upon fluctuations of the environment light (Tikkanen and Aro 2012, Tikkanen et al. 2012).

A schematis representation of state transitions triggered by the PQH2 binding to the Cyt b 6 f complex (see explanations in text) (Modified Figure 15 from Tikhonov 2014)
A molecular device, which receives the signal from the PQH2 pool and triggers state transitions, is inherent to the Cyt b 6 f complex. Activation of the LHCII kinase is initiated by PQH2 binding to the Qo-site. A strong correlation between the PQH2 occupancy at the Qo-center and the LHCII kinase activity of chloroplasts has been found in (Vener et al. 1997, Zito et al. 1999). In C. reinhardtii, activation of the LHCII kinase is mediated through the protein kinase Stt7, which becomes phosphorylated in State 2 conditions. Protein kinase Stt7 acts in catalytic amounts (Stt7/LHCII ~ 1/200) (Lemeille et al. 2009). Stt7 has an ortholog STN7 in Arabidopsis and other land plants (Bellafiore et al. 2005). The knock out of STN7 inhibits phosphorylation of LHCII and state transitions. A putative mechanism of kinase activation due to PQH2-induced conformational changes in the Cyt b 6 f complex has been suggested in (Hasan et al. 2013a). However, a complete chain of events leading to state transitions is still unclear. The Stt7/STN7 kinase is the redox-regulated enzyme, which contains the catalytic and regulatory domains (Bellafiore et al. 2005; Lemeille et al. 2009). The catalytic domain is facing the stroma, the regulatory domain is exposed to the thylakoid lumen. These domains are separated by a transmembrane helix with two Cys residues essential for the enzyme activity. The redox state of Cys residues may be regulated by Fd and thioredoxin through the membrane-bound thiol oxidoreductases (Lennartz et al. 2001; Motohashi and Hisabori 2006; Dietz and Pfannschmidt 2011). The over-reduced electron acceptors on the acceptor side of PSI will cause the reduction of Cys residues, thereby promoting dephosphorylation of the kinase and eventually inducing the “State 2 → State 1” transition, upon which PSI activity will lessen.
10.5 Concluding Remarks
Summing up a brief overview of structural and functional properties of the Cyt b 6 f complex, one can conclude that the overall rate of quinol oxidation is determined by PQH2 turnover at the Qo-site. The rates of PQ reduction to PQH2 in PSII and its diffusion to the Cyt b 6 f complex do not limit the overall rate of electron transfer between PSII and PSI. The rate-limiting step in the intersystem electron transport is associated with PQH2 oxidation at the Qo-site in the Cyt b 6 f complex. It is the first step of the bifurcated reaction of PQH2 oxidation (electron transfer from PQH2 to ISPox) that restricts the rate of PQH2 oxidation. The rate of this process is controlled by the intrathylakoid pH, values of which determine the protonation/deprotonation events in the Qo-center of the Cyt b 6 f complex. The feedback control of PQH2 oxidation is governed by the intrathylakoid pHin. The acidification of the lumen causes deceleration of PQH2 oxidation, thus impeding the intersystem electron transport. The Cyt b 6 f complex stands at the crossroad of alternative pathways of electron transport, providing flexibility and adaptability of the photosynthetic apparatus by means of two regulatory mechanisms: (i) redistribution of electron fluxes between alternative pathways (LEF and CEF1), and (ii) “state transitions” associated with the redistribution of solar energy between PSI and PSII.
Notes
- 1.
Here and below, the general terms QH2 and Q are used to denote the quinol and quinone species, regardless of their origin (plastoquinone, PQ, or ubiquinone, UQ). SQ designates the redox states of semiquinone species, either plastosemiquinone (in the b 6 f complex) or ubisemiquinone (in the bc 1 complex), regardless of their protonation state.
- 2.
It is likely that this species will appear in the form of the anion-radical \( {\mathrm{PQ}}^{\underset{\_}{\bullet }} \), because the pK values of semiquinones are usually fall in the range below the stromal pH establishes under the normal physiological conditions (pHout~7 − 8).
Abbreviations
- CBC:
-
Calvin-Benson cycle
- CEF1:
-
cyclic electron flow around photosystem I
- ETC:
-
electron transport chain
- Fd:
-
ferredoxin
- FNR:
-
ferredoxin-NADP-oxidoreductase
- ISP:
-
iron-sulfur protein
- LEF:
-
linear electron flow
- LHCI:
-
light-harvesting complex I
- LHCII:
-
light-harvesting complex II
- NDH:
-
NDH(P)H dehydrogenase complex
- NPQ:
-
non-photochemical quenching
- P680 :
-
special chlorophyll pair in PSII
- P700 :
-
special chlorophyll pair in PSI
- Pc:
-
plastocyanin
- PCET:
-
proton-coupled electron transfer
- PQ and PQH2 :
-
plastoquinone and plastoquinol, respectively
- PSA:
-
photosynthetic apparatus
- PSI and PSII:
-
photosystem I and photosystem II respectively
- Q, SQ and QH2 :
-
general notations for three accessible redox states of quinone species – quinone (Q), semiquinone (SQ) and quinol (QH2)
- TMQH2 :
-
trimethylbenzoquinol
- UQ and UQH2 :
-
ubiquinone and ubiquinol, respectively
- WOC:
-
water-oxidizing complex
References
Albertsson PÅ (2001) A quantitative model of the domain structure of the photosynthetic membrane. Trends Plant Sci 6:349–354
Allen JF (1992) Protein phosphorylation in regulation of photosynthesis. Biochim Biophys Acta 1098:275–335
Allen JF (2003) Cyclic, pseudocyclic and noncyclic photophosphorylation: new links in the chain. Trends Plant Sci 8:15–19
Alric J, Pierre Y, Picot D et al (2005) Spectral and redox characterization of the heme c i of the cytochrome b 6 f complex. Proc Natl Acad Sci U S A 102:15860–15865
Anderson JM (1982) Distribution of the cytochromes of spinach chloroplasts between the appressed membranes of grana stacks and stroma-exposed thylakoid regions. FEBS Lett 138:62–66
Anderson JM, Chow WS, Goodchild DJ (1988) Thylakoid membrane organisation in sun/shade acclimation. Funct Plant Biol 15:11–26
Andersson I (2008) Catalysis and regulation in Rubisco. J Exp Bot 59:1555–1568
Balsera M, Schürmann P, Buchanan BB (2016) Redox regulation in chloroplasts. In: Kirchhoff H (ed) Chloroplasts. Current research and future trends. Caister Academic Press, Norfolk, pp 187–207
Bellafiore S, Barneche F, Peltier G et al (2005) State transitions and light adaptation require chloroplast thylakoid protein kinase STN7. Nature 433:892–895
Bendall DS, Manasse RS (1995) Cyclic photophosphorylation and electron transport. Biochim Biophys Acta 1229:23–38
Bennett J (1977) Phosphorylation of chloroplast membrane polypeptides. Nature 269:344–346
Benz JP, Lintala M, Soll J et al (2010) A new concept for ferredoxin-NADP(H) oxidoreductase binding to plant thylakoids. Trends Plant Sci 15:608–613
Berry EA, Guergova-Kuras M, Huang L et al (2000) Structure and function of cytochrome bc complexes. Annu Rev Biochem 69:1005–1075
Blankenship RE (2002) Molecular mechanisms of photosynthesis. Blackwell Science, Oxford
Bleier L, Dröse S (2013) Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim Biophys Acta 1827:1320–1231
Brandt U (1996) Bifurcated ubihydroquinone oxidation in the cytochrome bc 1 complex by proton-gated charge transfer. FEBS Lett 387:1–6
Brandt U, Okun JG (1997) Role of deprotonation events in ubihydroquinone: cytochrome c oxidoreductase from bovine heart and yeast mitochondria. Biochemistry 36:11234–11240
Brettel K (1997) Electron transfer and arrangement of the redox cofactors in photosystem I. Biochim Biophys Acta 1318:322–373
Breyton C (2000) Conformational changes in the cytochrome b 6 f complex induced by inhibitor binding. J Biol Chem 275:13195–13201
Breyton C, Nandha B, Johnson G et al (2006) Redox modulation of cyclic electron flow around photosystem I in C3 plants. Biochemistry 45:13465–13475
Buchanan BB (1980) Role of light in the regulation of chloroplast enzymes. Annu Rev Plant Physiol 31:341–374
Burrows PA, Sazanov LA, Svab Z et al (1998) Identification of a functional respiratory complex in chloroplasts through analysis of tobacco mutants containing disrupted plastid ndh genes. EMBO J 17:868–876
Cape JL, Bowman MK, Kramer DM (2006) Understanding the cytochrome bc complexes by what they don’t do. The Q-cycle at 30. Trends Plant Sci 11:46–55
Cape JL, Bowman MK, Kramer DM (2007) A semiquinone intermediate generated at the Qo site of the cytochrome bc 1 complex: importance for the Q-cycle and superoxide production. Proc Natl Acad Sci U S A 104:7887–7892
Cardona T, Sedoud A, Cox N et al (2012) Charge separation in Photosystem II: a comparative and evolutionary overview. Biochim Biophys Acta 1817:26–43
Chobot SE, Zhang H, Moser CC et al (2008) Breaking the Q-cycle: finding new ways to study Qo through thermodynamic manipulations. J Bioenerg Biomembr 40:501–507
Chovancová E, Pavelka A, Beneš P et al (2012) CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PloS Comput Biol 8:e1002708
Clark WM (1960) Oxidation-reduction potentials of organic systems. Williams and Wilkins, Baltimore
Cramer WA, Zhang H, Yan J et al (2006) Transmembrane traffic in the cytochrome b 6 f complex. Annu Rev Biochem 75:769–790
Cramer WA, Hasan SS, Yamashita E (2011) The Q cycle of cytochrome bc complexes: A structure perspective. Biochim Biophys Acta 1807:788–802
Crofts AR (2004a) The cytochrome bc 1 complex: function in the context of structure. Annu Rev Physiol 66:689–733
Crofts AR (2004b) Proton-coupled electron transfer at the Qo-site of the bc 1 complex controls the rate of ubihydroquinone oxidation. Biochim Biophys Acta 1655:77–92
Crofts AR, Guergova-Kuras M, Huang L-S et al (1999a) Mechanism of ubiquinol oxidation by the bc 1 complex: role of the iron sulfur protein and its mobility. Biochemistry 38:15791–15806
Crofts AR, Hong S, Zhang Z et al (1999b) Physicochemical aspects of the movement of the Rieske iron sulfur protein during quinol oxidation by the bc 1 complex from mitochondria and photosynthetic bacteria. Biochemistry 38:15827–15839
Crofts AR, Guergova-Kuras M, Kuras R et al (2000) Proton-coupled electron transfer at the Qo-site: what type of mechanism can account for the high activation barrier? Biochim Biophys Acta 1459:456–466
Crofts AR, Lhee S, Crofts SB et al (2006) Proton pumping in the bc 1 complex: a new gating mechanism that prevents short circuits. Biochim Biophys Acta 1757:1019–1034
Crofts AR, Hong S, Wilson C et al (2013) The mechanism of ubihydroquinone oxidation at the Qo-site of the cytochrome bc 1 complex. Biochim Biophys Acta 1827:1362–1377
Cruz JA, Avenson TJ, Kanazawa A et al (2007) Plasticity in light reactions of photosynthesis for energy production and photoprotection. J Exp Bot 56:395–406
DalCorso G, Pesaresi P, Masiero S et al (2008) A complex containing PGRL1 and PGR5 is involved in the switch between linear and cyclic electron flow in Arabidopsis. Cell 132:273–285
Darrouzet E, Moser CC, Dutton PL et al (2001) Large scale domain movement in cytochrome bc 1: a new device for electron transfer in proteins. Trends Biochem Sci 26:445–451
de Vitry C, Ouyang Y, Finazzi G et al (2004) The chloroplast Rieske iron-sulfur protein: at the crossroad of electron transport and signal transduction. J Biol Chem 279:44621–44627
Dekker JP, Boekema EJ (2005) Supermolecular organization of the thylakoid membrane proteins in green plants. Biochim Biophys Acta 1706:12–39
Demmig-Adams B, Cohu CM, Muller O et al (2012) Modulation of photosynthetic energy conversion efficiency in nature: from seconds to seasons. Photosynth Res 113:75–88
Dietz K-J, Pfannschmidt T (2011) Novel regulators in photosynthetic redox control of plant metabolism and gene expression. Plant Physiol 155:1477–1485
Eberhard S, Finazzi G, Wollman FA (2008) The dynamics of photosynthesis. Annu Rev Genet 42:463–515
Edwards GE, Walker DA (1983) C3, C4: mechanisms, and cellular and environmental regulation of photosynthesis. Blackwell, Oxford
Endo T, Shikanai T, Sato F et al (1998) NAD(P)H dehydrogenase-dependent, antimycin A-sensitive electron donation to plastoquinone in tobacco chloroplasts. Plant Cell Physiol 39:1226–1231
Esser L, Gong X, Yang S et al (2006) Surface-modulated motion switch: capture and release of iron-sulfur protein in the cytochrome bc 1 complex. Proc Natl Acad Sci U S A 103:13045–13050
Finazzi G (2002) Redox-coupled proton pumping activity in cytochrome b 6 f, as evidenced by the pH dependence of electron transfer in whole cells of Chlamydomonas reinhardtii. Biochemistry 41:7475–7482
Foyer CH, Neukermans J, Queval G et al (2012) Photosynthetic control of electron transport and the regulation of gene expression. J Exp Bot 63:1637–1661
Frolov AE, Tikhonov AN (2009) The oxidation of plastoquinol by a cytochrome b 6 f complex: a density functional theory study. Russian J Phys Chem A 83:506–508
Fromme P, Jordan P, Krauss N (2001) Structure of photosystem I. Biochim Biophys Acta 1507:5–31
Galka P, Santabarbara S, Khuong TTH et al (2012) Functional analyses of the plant photosystem I-light-harvesting complex II supercomplex reveal that light-harvesting complex II loosely bound to Photosystem II is a very efficient antenna for Photosystem I in state II. Plant Cell 24:2963–2978
Gill SS, Tuteja N (2010) Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol Biochem 48:909–930
Haehnel W (1973) Electron transport between plastoquinone and chlorophyll aI. Biochim Biophys Acta 305:618–631
Haehnel W (1976a) The reduction kinetics of chlorophyll a1 as an indicator for proton uptake between the light reactions in chloroplasts. Biochim Biophys Acta 440:506–521
Haehnel W (1976b) The ratio of the two light reactions and their coupling in chloroplasts. Biochim Biophys Acta 423:499–509
Haehnel W (1982) On the functional organization of electron transport from plastoquinone to photosystem I. Biochim Biophys Acta 682:245–247
Haehnel W (1984) Photosynthetic electron transport in higher plants. Annu Rev Plant Physiol 35:659–693
Haldrup A, Jensen PE, Lunde C, Scheller HV (2001) Balance of power: a view of the mechanism of photosynthetic state transitions. Trends Plant Sci 6:301–305
Harbinson J, Hedley CL (1989) The kinetics of P-700+ reduction in leaves: a novel in situ probe of thylakoid functioning. Plant Cell and Environ 12:357–369
Hasan SS, Yamashita E, Cramer WA (2013a) Transmembrane signaling and assembly of the cytochrome b 6 f-lipidic charge transfer complex. Biochim Biophys Acta 1827:1295–1308
Hasan SS, Stofleth JT, Yamashita E et al (2013b) Lipid-induced conformational changes within the cytochrome b 6 f complex in oxygenic photosynthesis. Biochemistry 52:2649–2654
Hasan SS, Yamashita E, Baniulis D et al (2013c) Quinone-dependent proton transfer pathways in the photosynthetic cytochrome b 6 f complex. Proc Natl Acad Sci 110:4297–4302
Heimann S, Ponamarev MV, Cramer WA (2000) Movement of the Rieske iron-sulfur protein in the p-side bulk aqueous phase: effect of lumenal viscosity on redox reactions of the cytochrome b 6 f complex. Biochemistry 39:2692–2699
Hertle AP, Blunder T, Wunder T et al (2013) PGRL1 is the elusive ferredoxin-plastoquinone reductase in photosynthetic cyclic electron flow. Mol Cell 49:511–523
Hong SJ, Ugulava N, Guergova-Kuras M et al (1999) The energy landscape for ubihydroquinone oxidation at the Qo-site of the bc 1 complex in Rhodobacter sphaeroides. J Biol Chem 274:33931–33944
Hope AB (2000) Electron transfers amongst cytochrome f, plastocyanin and photosystem I: kinetics and mechanisms. Biochim Biophys Acta 1456:5–26
Hope AB, Valent P, Matthews DB (1994) Effects of pH on the kinetics of redox reactions in and around the cytochrome bf complex in an isolated system. Photosynth Res 42:111–120
Horton P (2012) Optimization of light harvesting and photoprotection: molecular mechanisms and physiological consequences. Phil Trans R Soc B 367:3455–3465
Hsueh K-L, Westler WM, Markley JL (2010) NMR investigations of the Rieske protein from Thermus thermophilus support a coupled proton and electron transfer mechanism. J Am Chem Soc 132:7908–7918
Iwai M, Takizawa K, Tokutsu R et al (2010) Isolation of the elusive supercomplex that drives cyclic electron flow in photosynthesis. Nature 464:1210–1213
Iwaki M, Yakovlev G, Hirst J et al (2005) Direct observation of redox-linked histidine protonation changes in the iron–sulfur protein of the cytochrome bc 1 complex by ATR-FTIR spectroscopy. Biochemistry 44:4230–4237
Iwata S, Lee JW, Okada K et al (1998) Complete structure of the 11-subunit bovine mitochondrial cytochrome bc 1 complex. Science 281:64–71
Izrailev S, Crofts AR, Berry EA et al (1999) Steered molecular dynamics simulation of the Rieske subunit motion in the cytochrome bc 1 complex. Biophys J 77:1753–1768
Jallet D, Cantrell M, Peers G (2016) New players for photoprotection and light acclimation. In: Kirchhoff H (ed) Chloroplasts. Current research and future trends. Caister Academic Press, Norfolk, pp 133–159
Järvi S, Gollan PJ, Aro E-M (2013) Understanding the roles of the thylakoid lumen in photosynthetic regulation. Front Plant Sci. https://doi.org/10.3389/fpls.2013.00434
Joët T, Cournac L, Horvath EM et al (2001) Increased sensitivity of photosynthesis to antimycin A induced by inactivation of the chloroplast ndhB gene. Evidence for a participation of the NADH-dehydrogenase complex to cyclic electron flow around photosystem I. Plant Physiol 125:1919–1929
Johnson GN (2011) Physiology of PSI cyclic electron transport in higher plants. Biochim Biophys Acta 1807:906–911
Joliot P, Joliot A (2005) Quantification of cyclic and linear flows in plants. Proc Natl Acad Sci U S A 102:4913–4918
Joliot P, Joliot A (2006) Cyclic electron flow in C3 plants. Biochim Biophys Acta 1757:362–368
Jordan P, Fromme P, Witt HT et al (2001) Three-dimensional structure of photosystem I at 2.5 Å resolution. Nature 411:909–917
Junge W, Nelson N (2015) ATP synthase. Annu Rev Biochem 83:631–657
Kim H, Xia D, Yu CA et al (1998) Inhibitor binding changes domain mobility in the iron–sulphur protein of the mitochondrial bc 1 complex from bovine heart. Proc Natl Acad Sci U S A 95:8026–8033
Kirchhoff H (2008) Significance of protein crowding, order and mobility for photosynthetic membrane functions. Biochem Soc Trans 36:967–970
Kirchhoff H (2013) Architectural switches in plant thylakoid membranes. Photosynth Res 116:481–487
Kirchhoff H (2014) Diffusion of molecules and macromolecules in thylakoid membranes. Biochim Biophys Acta 1837:495–502
Kono M, Terashima I (2014) Long-term and short-term responses of the photosynthetic electron transport to fluctuating light. J Photochem Photobiol B 137:89–99
Kono M, Noguchi K, Terashima I (2014) Roles of cyclic electron flow around PSI (CEF-PSI) and O2-dependent alternative pathways in regulation of the photosynthetic electron flow in short-term fluctuating light in Arabidopsis thaliana. Plant Cell Physiol 55:990–1004
Kramer DM, Sacksteder CA, Cruz JA (1999) How acidic is the lumen? Photosynth Res 60:151–163
Kramer DM, Avenson TJ, Edwards GE (2004) Dynamic flexibility in the light reactions of photosynthesis governed by both electron and proton transfer reactions. Trends Plant Sci 9:349–357
Kubicki A, Funk E, Westhoff P et al (1996) Differential expression of plastome-encoded ndh genes in mesophyll and bundle-sheath chloroplasts of the C4 plant Sorghum bicolor indicates that the complex I-homologous NAD(P)H-plastoquinone oxidoreductase is involved in cyclic electron transport. Planta 199:276–281
Kubota K, Sakurai I, Katayama K et al (2009) Purification and characterization of photosystem I complex from Synechocystis sp. PCC 6803 by expressing histidine-tagged subunits. Biochim Biophys Acta 1797:98–105
Kurisu G, Zhang H, Smith JL et al (2003) Structure of the cytochrome b 6 f complex of oxygenic photosynthesis: tuning the cavity. Science 302:1009–1014
Laisk A, Eichelmann H, Oja V et al (2005) Control of cytochrome b 6 f at low and high light intensity and cyclic electron transport in leaves. Biochim Biophys Acta 1708:79–90
Lavergne J, Joliot P (1991) Restricted diffusion in photosynthetic membranes. Trends Biochem Sci 16:129–134
Lemeille S, Rochaix J-D (2010) State transitions at the crossroad of thylakoid signaling pathways. Photosynth Res 106:33–46
Lemeille S, Willig A, Depège-Fargeix N et al (2009) Analysis of the chloroplasts protein kinase Stt7 during state transition. PLoS Biol 7 e1000045, 664–673
Lennartz K, Plucken H, Seidler A et al (2001) HCF164 encodes a thioredoxin-like protein involved in the biogenesis of the cytochrome b 6 f complex in Arabidopsis. Plant Cell 13:2539–2551
Lhee S, Kolling DRJ, Nair SK et al (2010) Modifications of protein environment of the [2Fe-2S] cluster of the bc 1 complex: effects on biophysical properties of the Rieske iron-sulfur protein and on the kinetics of the complex. J Biol Chem 285:9233–9248
Li Z, Wakao S, Fischer BB et al (2009) Sensing and responding to excess light. Annu Rev Plant Biol 60:239–260
Lichtenthaler HK, Babani F (2004) Light adaptation and senescence of the photosynthetic apparatus. Changes in pigment composition, chlorophyll fluorescence parameters and photosynthetic activity. In: Papageorgiou GC, Govindjee (eds) Chlorophyll a fluorescence: a signature of photosynthesis, Advances in photosynthesis and respiration, vol 19. Springer, Dordrecht, pp 713–736
Lin I-J, Chen Y, Fee JA, Song J et al (2006) Rieske protein from Thermus thermophilus: 15N NMR titration study demonstrates the role of iron-ligated histidines in the pH dependence of the reduction potential. J Am Chem Soc 128:10672–10673
Link TA (1997) The role of the “Rieske” iron sulfur protein in the hydroquinone oxidation (Qp) site of the cytochrome bc 1 complex: the “proton-gated affinity change” mechanism. FEBS Lett 412:257–264
Malnoë A, Wollman F-A, de Vitry C et al (2011) Photosynthetic growth despite a broken Q-cycle. Nature Communications 2:301. https://doi.org/10.1038/ncomms1299/www.nature.com/naturecommunications
Mamedov M, Govindjee, Nadtochenko V et al (2015) Primary electron transfer processes in photosynthetic reaction centers from oxygenic organisms. Photosynth Res 125:51–63
Michelet L, Zaffagnini M, Morisse S et al (2013) Redox regulation of the Calvin–Benson cycle: something old, something new. Front Plant Sci 4:Article 470. https://doi.org/10.3389/fpls.2013.00470
Mitchell P (1966) Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol Rev 41:445–502
Mitchell P (1976) Possible molecular mechanisms of the protonmotive function of cytochrome systems. J Theor Biol 62:327–367
Motohashi K, Hisabori T (2006) HCF164 receives reducing equivalents from stromal thioredoxin across the thylakoid membrane and mediates reduction of target proteins in the thylakoid lumen. J Biol Chem 281:35039–35047
Müh F, Glöckner C, Hellmich J et al (2012) Light-induced quinone reduction in photosystem II. Biochim Biophys Acta 1817:44–65
Mulkidjanian AY (2005) Ubiquinol oxidation in the cytochrome bc 1 complex: reaction mechanism and prevention of short-circuiting. Biochim Biophys Acta 1709:5–34
Munekage Y, Hojo M, Meurer J et al (2002) PGR5 is involved in cyclic electron flow around photosystem I and is essential for photoprotection in Arabidopsis. Cell 110:361–371
Munekage Y, Hashimoto M, Miyake C et al (2004) Cyclic electron flow around photosystem I is essential for photosynthesis. Nature 429:579–582
Munekage Y, Genty B, Peltier G (2008) Effect of PGR5 impairment on photosynthesis and growth in Arabidopsis thaliana. Plant Cell Physiol 49:1688–1698
Nelson N, Yocum CF (2006) Structure and function of photosystems I and II. Annu Rev Plant Biol 57:521–565
Osyczka A, Moser CC, Daldal F et al (2004) Reversible redox energy coupling in electron transfer chains. Science 427:607–612
Osyczka A, Moser CC, Dutton L (2005) Fixing the Q cycle. Trends Biochem Sci 30:176–182
Page CC, Moser CC, Chen X et al (1999) Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 402:47–52
Peng LW, Shimizu H, Shikanai T (2008) The chloroplast NAD(P)H dehydrogenase complex interacts with Photosystem I in Arabidopsis. J Biol Chem 283:34873–34879
Peng LW, Fukao Y, Fujiwara M et al (2009) Efficient operation of NAD(P)H dehydrogenase requires supercomplex formation with Photosystem I via minor LHCI in Arabidopsis. Plant Cell 21:3623–3640
Pesaresi P, Hertle A, Pribil M et al (2010) Optimizing photosynthesis under fluctuating light. The role of the Arabidopsis STN7 kinase. Plant Signal Behav 5:21–25
Pierre Y, Breyton C, Kramer D, Popot J-L (1995) Purification and characterization of the cytochrome b 6 f complex from Chlamydomonas reinhardtii. J Biol Chem 270:29342–29349
Ponamarev MV, Cramer WA (1998) Perturbation of the internal water chain in cytochrome f of oxygenic photosynthesis: loss of concerted reduction of cytochromes f and b. Biochemistry 37:17199–17208
Postila PA, Kaszuba K, Sarewicz M et al (2013) Key role of water in proton transfer at the Qo-site of the cytochrome bc 1 complex predicted by atomistic molecular dynamics simulations. Biochim Biophys Acta 1827:761–768
Puthiyaveetil S, Kirchhoff H, Hohner R (2016) Structural and functional dynamics of the thylakoid membrane system. In: Kirchhoff H (ed) Chloroplasts. Current research and future trends. Caister Academic Press, Norfolk, pp 59–87
Rich PR (2004) The quinone chemistry of bc complexes. Biochim Biophys Acta 1658:165–171
Roberts AG, Bowman MK, Kramer DM (2002) Certain metal ions are inhibitors of cytochrome b 6 f complex ‘Rieske’ iron-sulfur protein domain movements. Biochemistry 41:4070–4079
Rochaix J-D (2014) Regulation and dynamics of the light-harvesting system. Annu Rev Plant Biol 65:287–309
Ruban A (2012) The photosynthetic membrane: molecular mechanisms and biophysics of light harvesting. Wiley-Blackwell, Oxford
Rumberg B, Siggel U (1969) pH changes in the inner phase of the thylakoids during photosynthesis. Naturwissenschaften 56:130–132
Sainz G, Carrell CJ, Ponamarev MV et al (2000) Interruption of the internal water chain of cytochrome f impairs photosynthetic function. Biochemistry 39:9164–9173
Samoilova RI, Kolling D, Uzawa T et al (2002) The interaction of the Rieske iron sulfur protein with occupants of the Qo-site of the bc 1 complex, probed by 1D and 2D electron spin echo envelope modulation. J Biol Chem 277:4605–4608
Schöttler MA, Tôth SZ, Boulouis A, Kahlau S (2015) Photosynthetic complex stoichiometry dynamics in higher plants: biogenesis, function, and turnover of ATP synthase and the cytochrome b 6 f complex. J Exp Bot 66:2373–2400
Seelert H, Poetsch A, Dencher NA et al (2000) Structural biology. Proton-powered turbine of a plant motor. Nature 405:418–419
Shelaev IV, Gostev FE, Mamedov MD et al (2010) Femtosecond primary charge separation in photosystem I. Biochim Biophys Acta 1797:1410–1420
Shikanai T (2007) Cyclic electron transport around photosystem I: Genetic approaches. Plant Biol 58:199–217
Shikanai T (2016) Chloroplast NDH: a different enzyme with a structure similar to that of respiratory NADH dehydrogenase. Biochim Biophys Acta 1857:1015–1022
Shikanai T, Endo T, Hashimoto et al (1998) Directed disruption of the tobacco ndhB gene impairs cyclic electron flow around photosystem I. Proc Natl Acad Sci U S A 95:9705–9709
Siggel U, Renger G, Stiehl HH et al (1972) Evidence for electronic and ionic interaction between electron transport chains in chloroplasts. Biochim Biophys Acta:328–335
Snyder CH, Gutierrez-Cirlos EB, Trumpower BL (2000) Evidence for a concerted mechanism of ubiquinol oxidation by the cytochrome bc 1 complex. J Biol Chem 275:13535–13541
Soriano GM, Ponamarev MV, Tae G-S et al (1996) Effect of the interdomain basic region of cytochrome f on its redox reactions in vivo. Biochemistry 35:14590–14598
Soriano GM, Guo L-W, de Vitry C et al (2002) Electron transfer from the Rieske iron-sulfur protein (ISP) to cytochrome f in vitro. Is a guided trajectory of the ISP necessary for competent docking? J Biol Chem 277:41865–41871
Staehelin LA (2003) Chloroplast structure: from chlorophyll granules to supra-molecular architecture of thylakoid membranes. Photosynth Res 76:185–196
Stiehl HH, Witt HT (1969) Quantitative treatment of the function of plastoquinone in photosynthesis. Z Naturforsch 24b:1588–1598
Stirbet A (2013) Excitonic connectivity between photosystem II units: what is it, and how to measure it. Photosynth Res 116:189–214
Strand DD, Fisher N, Kramer DM (2016) Distinct energetics and regulatory functions of the two major cyclic electron flow pathways in chloroplasts. In: Kirchhoff H (ed) Chloroplasts. Current research and future trends. Caister Academic Press, Norfolk, pp 89–100
Stroebel D, Choquet Y, Popot J-L et al (2003) An atypical heam in the cytochrome b 6 f complex. Nature 426:413–418
Suorsa M, Järvi S, Grieco M et al (2012) PROTON GRADIENT REGULATION5 is essential for proper acclimation of Arabidopsis photosystem I to naturally and artificially fluctuating light conditions. Plant Cell 24:2934–2948
Tikhonov AN (2013) pH-dependent regulation of electron transport and ATP synthesis in chloroplasts. Photosynth Res 116:511–534
Tikhonov AN (2014) The cytochrome b 6 f complex at the crossroad of photosynthetic electron transport pathways. Plant Physiol Biochem 81:163–183
Tikhonov AN (2015) Induction events and short-term regulation of electron transport in chloroplasts: an overview. Photosynth Res 125:65–94
Tikhonov AN (2016) Modeling electron and proton transport in chloroplasts. In: Kirchhoff H (ed) Chloroplasts. Current Research and Future Trends. Caister Academic Press, Norfolk, pp 101–134
Tikhonov AN, Ruuge EK (1979) Electron paramagnetic resonance study of electron transport in photosynthetic systems. VIII. The interplay between two photosystems and kinetics of P700 redox transients under various conditions of flash excitation. Molec Biol (Moscow) 13:1085–1097
Tikhonov AN, Vershubskii AV (2017) Connectivity between electron transport complexes and modulation of photosystem II activity in chloroplasts. Photosynth Res 133:103–114
Tikhonov AN, Khomutov GB, Ruuge EK (1980) Electron spin resonance study of electron transport in photosynthetic systems. IX. Temperature dependence of the kinetics of P700 redox transients in bean chloroplasts induced by flashes with different duration. Molec Biol (Moscow) 14:157–172
Tikhonov AN, Khomutov GB, Ruuge EK et al (1981) Electron transport control in chloroplasts. Effects of photosynthetic control monitored by the intrathylakoid pH. Biochim Biophys Acta 637:321–333
Tikhonov AN, Timoshin AA, Blumenfeld LA (1983) The kinetics of electron transport translocation of protons and photophosphorylation in chloroplasts and their relationship with the thermo-induced structural changes in the thylakoid membrane. Molec Biol (Moscow) 17:1236–1248
Tikhonov AN, Khomutov GB, Ruuge EK (1984) Electron transport control in chloroplasts. Effects of magnesium ions on the electron flow between two photosystems. Photobiochem Photobiophys 8:261–269
Tikkanen M, Aro E-M (2012) Thylakoid protein phosphorylation in dynamic regulation of photosystem II in higher plants. Biochim Biophys Acta 1817:232–238
Tikkanen M, Grieco M, Nurmi M et al (2012) Regulation of the photosynthetic apparatus under fluctuating growth light. Philos Trans R Soc Lond B 367:3486–3493
Tremmel IG, Kirchhoff H, Weis E et al (2003) Dependence of plastoquinol diffusion on the shape, size, and density of integral thylakoid proteins. Biochim Biophis Acta 1603:97–109
Turina P, Petersen J, Gräber P (2016) Thermodynamics of proton transport coupled ATP synthesis. Biochim Biophys Acta 1857:653–664
Vener AV, van Kan PJ, Rich PR et al (1997) Plastoquinol at the quinol oxidation site of reduced cytochrome bf mediates signal transduction between light and protein phosphorylation: Thylakoid protein kinase deactivation by a single-turnover flash. Proc Natl Acad Sci U S A 94:1585–1590
Victoria D, Burton R, Crofts AR (2013) Role of the –PEWY-glutamate in catalysis at the Qo-site of the Cyt bc 1 complex. Biochim Biophys Acta 1827:365–386
Witt HT (1979) Energy conversion in the functional membrane of photosynthesis. Analysis by light pulse and electric pulse methods. The central role of the electric field. Biochim Biophys Acta 505:355–427
Wood PM (1988) The potential diagram for oxygen at pH 7. Biochem J 253:287–289
Xia D, C-A Y, Kim H et al (1997) Crystal structure of the cytochrome bc 1 complex from bovine heart mitochondria. Science 277:60–66
Xia D, Esser L, Tang W-K et al (2013) Structural analysis of cytochrome bc 1 complexes: Implications to the mechanism of function. Biochim Biophys Acta 1827:1278–1294
Yamashita E, Zhang H, Cramer WA (2007) Structure of the cytochrome b 6 f complex: Quinone analogue inhibitors as ligands of heme c n . J Mol Biol 370:39–52
Yamori W, Shikanai T (2016) Physiological functions of cyclic electron transport around photosystem I in sustaining photosynthesis and plant growth. Annu Rev Plant Biol 67:81–106
Yan J, Cramer WA (2003) Functional insensitivity of the cytochrome b 6 f complex to structure changes in the hinge region of the Rieske iron–sulfur protein. J Biol Chem 278:20926–20933
Zhang H, Carrell CJ, Huang D et al (1996) Characterization and crystallization of the lumen side domain of the chloroplast Rieske iron-sulfur protein. J Biol Chem 271:31360–31366
Zhang Z, Huang L, Shulmeister VM et al (1998) Electron transfer by domain movement in cytochrome bc 1. Nature 392:677–684
Zhang H, Whitelegge JP, Cramer WA (2001) Ferredoxin: NADP+ oxidoreductase is a subunit of the chloroplast cytochrome b 6 f complex. J Biol Chem 276:38159–38165
Zhu J, Egawa T, Yeh S-R et al (2007) Simultaneous reduction of iron-sulfur protein and cytochrome b L during ubiquinol oxidation in cytochrome bc 1 complex. Proc Natl Acad Sci U S A 104:4864–4869
Zito F, Finazzi G, Joliot P et al (1998) Glu78, from the conserved PEWY sequence of subunit IV, has a key function in the cytochrome b 6 f turnover. Biochemistry 37:10395–10403
Zito F, Finazzi G, Delosme R et al (1999) The Qo site of cytochrome b 6 f complexes controls the activation of the LHCII kinase. EMBO J 18:2961–2969
Zu Y, Manon M-J, Couture MM-J et al (2003) Reduction potentials of Rieske clusters: Importance of the coupling between oxidation state and histidine protonation state. Biochemistry 42:12400–12408
Acknowledgments
This work was supported in part by the Russian Foundation for Basic Research (RFBR Project 15-04-03790a).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Tikhonov, A.N. (2018). The Cytochrome b 6 f Complex: Biophysical Aspects of Its Functioning in Chloroplasts. In: Harris, J., Boekema, E. (eds) Membrane Protein Complexes: Structure and Function. Subcellular Biochemistry, vol 87. Springer, Singapore. https://doi.org/10.1007/978-981-10-7757-9_10
Download citation
DOI: https://doi.org/10.1007/978-981-10-7757-9_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-7756-2
Online ISBN: 978-981-10-7757-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)