
Abstract
In photosynthetic reaction centers (RCs) of purple bacteria, conserved histidine residues [His L173 and His M202 in Rhodobacter (Rba.) sphaeroides] are known to serve as fifth axial ligands to the central Mg atom of the bacteriochlorophyll (BChl) molecules (PA and PB, respectively) that constitute the homodimer (BChl/BChl) primary electron donor P. In a number of previous studies, it has been found that replacing these residues with leucine, which cannot serve as a ligand to the Mg ion of BChl, leads to the assembly of heterodimer RCs with P represented by the BChl/BPheo pair. Here, we show that a homodimer P is assembled in Rba. sphaeroides RCs if the mutation H(M202)L is combined with the mutation of isoleucine to histidine at position M206 located in the immediate vicinity of PB. The resulting mutant H(M202)L/I(M206)H RCs are characterized using pigment analysis, redox titration, and a number of spectroscopic methods. It is shown that, compared to wild-type RCs, the double mutation causes significant changes in the absorption spectrum of the P homodimer and the electronic structure of the radical cation P+, but has only minor effect on the pigment composition, the P/P+ midpoint potential, and the initial electron-transfer reaction. The results are discussed in terms of the nature of the axial ligand to the Mg of PB in mutant H(M202)L/I(M206)H RCs and the possibility of His M202 participation in the previously proposed through-bond route for electron transfer from the excited state P* to the monomeric BChl BA in wild-type RCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In plant cells, algae, and phototrophic microorganisms, the harvested light energy is converted into the energy of chemical bonds as a result of photosynthesis. A key initial step of this process is the light-driven charge separation that occurs in a specialized cofactor–protein complex—the photosynthetic reaction center (RC). The RC of the purple bacterium Rhodobacter (Rba.) sphaeroides consists of three protein subunits and ten cofactors integrated in the protein matrix and arranged in two membrane-spanning branches, A and B, around an axis of quasi twofold symmetry (Blankenship 2014). Both branches have a common cofactor, a dimer (P) of two strongly interacting bacteriochlorophyll (BChl) molecules (PA and PB) that serves as a primary electron donor, and each of the two branches includes a monomeric BChl molecule (BA or BB), a bacteriopheophytin (BPheo) molecule (HA or HB), and a quinone molecule (QA or QB). The RC structure also contains a non-heme iron atom located between QA and QB, and a carotenoid molecule that is asymmetrically placed near BB. In the wild type (Wt) RC, only the A cofactor branch is active in photoinduced charge separation. The charge separation process is initiated by excitation of P to its lowest singlet excited state (P*), which transfers an electron to the monomeric BChl BA, forming the initial short-lived state P+BA− within ~ 3 ps at 293 K. Within ~ 1 ps, the electron is then transferred from BA− to the BPheo HA to produce the radical pair P+HA−. A subsequent electron transfer to the primary quinone acceptor QA forms the state P+QA− in about 200 ps with essentially 100% yield (Woodbury and Allen 1995; Parson 2008).
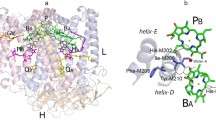
It is well known that the protein environment of P has a significant effect on its composition and spectral and redox properties (Bylina and Youvan 1988; Kirmaier et al. 1991; Lin et al. 1994; DiMagno et al. 1998; Spiedel et al. 2002; Vasilieva et al. 2012). In particular, important interactions of the P dimer with surrounding amino acid residues are two coordination bonds formed between the central Mg ions of BChls PA and PB and His L173 and His M202, respectively (Fig. 1). Based on site-directed mutagenesis studies performed with purple bacteria RCs from Rba. capsulatus (Bylina and Youvan 1988), Blastochloris viridis (Ponomarenko et al. 2009), and Rba. sphaeroides (McDowell et al. 1991), it has been found that the replacement of His L173 or His M202 with Leu not acting as a ligand for the Mg ion led to the assembly of the so-called heterodimeric RCs, in which the special pair is represented by the BChl/BPheo pair. The absorption spectrum and redox properties of the heterodimeric special pair are drastically different from those of P in the Wt RC, and the rate of primary charge separation in the heterodimeric complexes is significantly reduced (Kirmaier et al. 1988; McDowell et al. 1991). Previously, conservation of the BChl/BChl homodimer in His-substituted RCs was demonstrated in Rba. sphaeroides single mutants, in which His M202 was changed for Gly, Ser, Cys, or Asn (Goldsmith et al. 1996; Nabedryk et al. 2000), as well as in the single H(L173)G and double H(M202)G/H(L173)G mutants (Goldsmith et al. 1996).

View of the geometry and environment of the primary donor dimer P BChls (PA and PB) and the monomeric BChl BA, as determined in the crystal structure of the Wt Rba. sphaeroides RC (Vasilieva et al. 2012; PDB: 3V3Y). In the double mutant H(M202)L/I(M206)H studied in this work, His at position M202 and Ile at position M206 are replaced with Leu and His, respectively. Dashed lines show distances in angstroms
Our recent measurements have shown that the homodimer P appears to be retained also in the mutant Rba. sphaeroides RC with the double substitution H(L173)L/I(L177)H, as indicated by the P/P+ midpoint potential value, the results of the pigment analysis, and the presence of the long-wavelength Qy band of P in the absorption spectrum measured at 90 K (Vasilieva et al. 2012). In this mutant, the replacement of His L173 with Leu was combined with the replacement of Ile with His at position L177, shifted by one turn of the protein alpha-helix from His L173. The double substitution was accompanied by an unusually large (46 nm) blue shift of the Qy absorption band of P. Here, we show that a heterodimer primary electron donor also fails to assemble in the mutant containing a symmetric double amino acid substitution H(M202)L/I(M206)H on the other side of the Rba. sphaeroides RC. Instead, the homodimeric structure of P is retained in the H(M202)L/I(M206)H mutant, although its properties are somewhat different from those of P in the Wt. The study of this mutant is of significant interest from the point of view of obtaining further insight into the role of the nearest amino acid environment in the organization and structural stability of P. In addition, this mutation offers a potential possibility to address a hypothesis of the possible involvement of His M202 in the initial charge separation event as an integral element of a through-bond route for electron transfer from P* to the BA molecule (Yakovlev et al. 2002, 2005). Based on the RC crystal structure (Ermler et al. 1994; Potter et al. 2005), it has been proposed that a chain of polar atoms N–Mg(PB)–N–C–N(HisM202)–HOH(water A)–O=(BA) could provide a direct electron-transfer link between the primary reactants (Yakovlev et al. 2002, 2005). While the influence of the removal of the bound water A on the rate of primary electron transfer is known from the time-resolved absorption measurements (Potter et al. 2005; Yakovlev et al. 2005; Gibasiewicz et al. 2016), as far as we know, no transient absorption data are available for the initial electron-transfer reaction in mutant RCs which do not have His M202, but contain the P homodimer.
Here, we present a detailed characterization of isolated H(M202)L/I(M206)H mutant Rba. sphaeroides RCs using pigment analysis, steady-state visible/near-IR absorption spectroscopy, mid-IR light-induced Fourier transform infrared (FTIR) spectroscopy, electron paramagnetic resonance (EPR) spectroscopy, and femtosecond transient absorption spectroscopy.
Materials and methods
Site-directed mutagenesis and reaction center purification
Mutations were introduced using PCR oligonucleotides as described previously (Khatypov et al. 2005). Nucleotide changes were confirmed by DNA sequencing. Altered pufM genes were re-cloned into the broad-host-range vector, a derivative of pRK415, containing a 4.2 kb EcoRI–HindIII restriction fragment that included the pufLMX genes. The resulting plasmids were transferred into the Rba. sphaeroides strain DD13 through conjugative crossing to give transconjugant strains with RC-only phenotypes (Jones et al. 1992). The control strain comprised DD13 complemented with a pRK415-derivative plasmid containing a Wt copy of the pufLMX genes. Details of the growth of the mutant bacterial strains were described earlier (Khatypov et al. 2005). RCs were isolated according to (Fufina et al., 2015) and resuspended in 20 mM Tris–HCl, pH 8.0/0.1% LDAO/180 mM NaCl buffer. For FTIR and transient absorption spectroscopy measurements, RCs were dissolved in a 20 mM Tris–HCl (pH 8.0) buffer with 0.1% Triton X-100 as a detergent.
Pigment composition analysis and redox potentials determination
Pigment extraction and analysis of pigment composition of RCs were performed as described in detail previously (Vasilieva et al. 2012). P/P+ midpoint potentials were determined by chemical titrations using potassium ferricyanide and sodium ascorbate as described previously (Vasilieva et al. 2012).
Steady-state electronic absorption spectroscopy
Room-temperature electronic absorption spectra of RCs were recorded with a Shimadzu UV1800 spectrophotometer. Absorption spectra at 100 K were measured with a Shimadzu UV-1601PC spectrophotometer using an optical liquid nitrogen cryostat of the local design and a cuvette with an optical pathlength of ~ 2 mm. For low-temperature measurements, glycerol was added to the sample to a final concentration of 60% (v/v). Sodium ascorbate was added to a final concentration of 1 mM in order to keep the primary donor in the reduced state.
FTIR spectroscopy
Light-induced difference FTIR spectra were recorded as described previously (Zabelin et al. 2009) on Bruker IFS66v/s Fourier transform infrared spectrometer equipped with an MCT (D313/6) detector and a KBr beamsplitter. The spectral resolution was 4 cm−1. Concentrated suspension of RCs (5 μl, ~ 0.25 mM) solubilized in 20 mM Tris–HCl buffer (pH 8.0) containing 0.1% Triton X-100 was applied to a CaF2 plate, partially dehydrated using an argon gas jet, and covered with another CaF2 plate. Light-induced difference (light minus dark) P+Q−/PQ FTIR spectra were recorded at room temperature under steady-state continuous illumination of the samples (720–1100 nm, ~ 2 mW/cm2). Illumination cycles were repeated several times to achieve an acceptable signal-to-noise ratio.
EPR spectroscopy
EPR spectra were recorded with an EMX6 spectrometer (Bruker, Germany) at room temperature under the following spectrometer settings: mw power, 2 mW, modulation amplitude, 0.4 mT. The dark spectrum was subtracted from the spectrum under illumination (λ > 640 nm). To improve the precision of parameter determination, the difference spectrum was fitted with a Gaussian function.
Transient electronic absorption spectroscopy
Femtosecond transient absorption spectra were measured essentially as described previously (Zabelin et al. 2019a), with the exceptions that the pump spectrum was centered at about 870 nm for Wt and at 840 nm for the double mutant, and the relative polarization between the pump and probe pulses was set to the magic angle (54.7°).
The transient absorption datasets were analyzed globally (van Stokkum et al. 2004) using the Glotaran software (Snellenburg et al. 2012). The program includes mathematical description of spectral dispersion of the maximum of the instrument response function (IRF). The half width at half maximum of the IRF was estimated to be ~ 35–40 fs for both investigated spectral regions.
Results
Figure 2 shows the normalized electronic ground-state absorption spectra of Wt (dotted line) and mutant I(M206)H/H(M202)L RCs (solid line), measured at room temperature (main panel) and 100 K (inset). The spectrum of Wt RCs corresponds well to the spectrum published for Rba. sphaeroides RCs (McDowell et al. 1991). It can be seen that introduction of the double mutation I(M206)H/H(M202)L leads to significant changes in the spectral properties of the RC. At room temperature, the long-wavelength band at 866 nm in the spectrum of Wt RCs, which is attributed to the low-energy Qy exciton transition of the homodimer primary electron donor P, in the mutant is strongly shifted to the blue, and is discernible only as a shoulder on the long-wavelength side of the band at about 800 nm. The intensity of this latter band, which is assigned mainly to the Qy transitions of the two monomeric BChls (BA and BB) with possible contribution of the high-energy exciton transition of the P dimer, is slightly decreased in the spectrum of the mutant compared to Wt. In the region of the Qx optical transitions, a band peaking at 599 nm in the spectrum of Wt includes contributions from all BChl molecules (PA, PA, BA, and BB). In the spectrum of mutant RCs, maximum of this band shifts to 596 nm. At 100 K, the band with longest wavelength in the spectrum of mutant RCs is resolved as a separate peak at 857 nm, the intensity of which is significantly smaller than that of the low-energy exciton transition of P at 888 nm in the spectrum of Wt. This 31-nm blue absorption shift corresponds to a 50-meV increase in the P/P* energy difference.

Electronic ground-state absorption spectra of Rba. sphaeroides RCs from the Wt (dashed line) and the mutant I(M206)H/H(M202)L (solid line). Spectra were measured at room temperature (main panel) and 100 K (inset) and normalized at the maximum of the BPheo Qy band near 760 nm
The pigment composition in the Wt and mutant RCs was determined by performing analysis of acetone/methanol (7:2) extracts. Similar BChl/BPheo ratio of 1.9 ± 0.1 was found for both RCs.
The P/P+ midpoint potential was determined to be approximately 485 mV for the Wt RC and 470 mV for the RC H(M202)L/I(M206)H (estimated error ± 10 mV), with the value for the Wt being in agreement with the previous data (Moss et al. 1991; Williams et al. 1992).
Figures 3 and 4 present the mid-IR (4500–1200 cm−1) P+Q−/PQ FTIR difference spectra corresponding to the photo-oxidation of the primary electron donor and photo-reduction of quinone acceptors (QA and QB) in isolated RCs of Wt (a) and of the double mutant H(M202)L/I(M206)H (b). The negative and positive features in the spectra mainly reflect the disappearance of the neutral forms of P and the appearance of their radical ions, respectively. We note that the frequency range between 3700 and 3100 cm−1 is saturated due to the strong background absorption of water and is excluded from Fig. 3. The amide I frequency region of the spectra (around ~ 1650 cm−1) (Fig. 4) is also distorted due to the water absorption and is not analyzed in this paper.

Light-minus-dark FTIR difference spectra, reflecting photo-oxidation of the primary electron donor and reduction of quinone electron acceptors in Rba. sphaeroides Wt RCs (a) and H(M202)L/I(M206)H RCs (b), measured at room temperature in the region of 4500–1200 cm−1. The spectra are normalized by the amplitude of the differential signal in the region of 1749–1747/1739 cm−1. The region at ~ 3700–3100 cm−1 is saturated due to the strong absorption of the sample and water and is excluded from the figure

Expanded comparison of the low-frequency region (1800–1200 cm−1) of the FTIR P+Q−/PQ spectra for Rba. sphaeroides Wt RCs (a) и H(M202)L/I(M206)H RCs (b), replotted from Fig. 3
The P+Q−/PQ FTIR difference spectrum of the Wt RC (Figs. 3a, 4a) is similar to the spectra of previously well-characterized native Rba. sphaeroides RCs (Breton et al. 1992; Johnson et al. 2002; Zabelin et al. 2009). In this spectrum, the broad band centered at ~ 2680 cm−1 has been assigned to a specific electronic transition that corresponds to the hole transfer between the two halves of the oxidized P dimer and characterize the electronic coupling of PA and PB BChls in the P+ state (Breton et al. 1992; Reimers and Hush 2003). The spectrum includes also three vibrational (phase-phonon) bands at ~ 1550, 1477 ,and ~ 1290 cm−1 due to deformations of the porphyrin macrocycles of BChls PA and PB (Reimers and Hush 2003). The phase-phonon modes are formally symmetry forbidden in monomeric BChl, but they intensify due to the coupling with the hole transfer electronic transition in the dimer (Reimers and Hush 2003). Notably, all these IR bands are spectroscopic indicators of the dimeric structure of P+ (Nabedryk et al. 1992, 1993, 2000). (Although the P+Q−/PQ FTIR spectra are expected to contain contributions from quinone modes, the intense P and P+ bands dominate over the signals due to Q and Q−as suggested by Nabedryk et al. 1993). From comparison of the spectra in Figs. 3 and 4, it is clear that similar IR features are also present in the FTIR spectrum of the H(M202)L/I(M206)H mutant. However, upon mutation (Fig. 3b), the broad positive band significantly decreases in intensity and its maximum upshifts to ~ 2740 cm−1 (the FTIR spectra were normalized according to Malferrari et al. 2015 on the basis of the differential signals at 1749–1747/1739 cm−1 attributed to the 133-ester C=O mode of BChl Nabedryk et al. 1993). The phase-phonon peaks in the spectrum of the mutant show maxima at 1544, 1484, and 1317 cm−1 and decrease in intensity compared to Wt.
In the 1760–1650 cm−1 range, the FTIR difference spectra (Fig. 4) show differential signals associated mainly with high-frequency shifts of the ester and keto carbonyl modes of the primary donor upon its photo-oxidation (Nabedryk et al. 1993). In the Wt RC, a positive peak at 1749 cm−1 and a negative peak at 1739 cm−1 are assigned to at least one of the two 133-ester C=O groups of the primary donor in the P+ and P states, respectively (Fig. 4a) (Nabedryk et al. 1993; Johnson et al. 2002). Figure 4b shows that a differential signal of similar shape is observed for the H(M202)L/I(M206)H RC at 1747( +)/1739( −) cm−1, indicating that the local environment of the ester carbonyls in the double mutant remains close to that in Wt in both the neutral and the cation state of P.
The FTIR spectrum of the double mutant is noticeably perturbed compared to the Wt spectrum in the frequency region of 131-keto carbonyl modes (Fig. 4). The negative peak at 1684 cm−1, accounting for both PA and PB contributions in Wt, is downshifted to 1680 cm−1 in the mutant. Positive signals observed in the spectrum of Wt at ~ 1713 and 1704 cm−1 and assigned to the free 131-keto-C=O groups of PA+ and PB+, respectively (Nabedryk et al. 1993) shift to 1714 and 1697 cm−1, respectively, in the spectrum of the mutant. Along with the splitting of the PA+ and PB+ peaks, a reversion in the relative amplitude of these peaks is observed in the mutant as compared to Wt.
Light-induced EPR signal of the H(M202)L/I(M206)H mutant primary donor in isolated RCs is characterized by the g-factor of 2.0026 ± 0.0001 and linewidth ΔB = 1.18 ± 0.01 mT. The somewhat high microwave power used to record the spectra had little effect on the linewidth, as confirmed by the light-induced P+ signal of the Wt RCs taken under the same conditions demonstrating the well-known ΔB = 0.95 ± 0.01 mT value (data not shown).
The dynamics and mechanism of charge separation in Wt and the double H(M202)L/I(M206)H mutant were studied using femtosecond time-resolved difference absorption spectroscopy at room temperature. The experimentally measured entire sets of transient absorption spectra for Wt and H(M202)L/I(M206)H (not shown) were analyzed globally using a sequential, irreversible kinetic model (1 → 2 → 3 → …) and a parallel kinetic model with independent decays (van Stokkum et al. 2004). Two exponential kinetic components and a non-decaying component satisfactorily fit the data.
The evolution-associated difference spectra (EADS) and the corresponding time constants resulting from applying the sequential model to Wt and the double mutant are shown in Figs. 5a, b and 6a, b, respectively. It can be seen that the EADS for the two RCs show clear similarities but also exhibit some differences, in particular, reflecting differences between the ground-state absorption spectra of the RCs (Fig. 2). The EADS obtained for Wt are in agreement with the published data (see, for example, Sun et al. 2016).

Evolution-associated difference spectra (EADS, a, b) and decay-associated difference spectra (DADS, c, d) obtained from global analysis of the transient absorption data in the 500–720 nm (a, c) and 740–1000 nm (b, d) regions for Rba. sphaeroides Wt RCs

Evolution-associated difference spectra (EADS, a, b) and decay-associated difference spectra (DADS, c, d) obtained from global analysis of the transient absorption data in the 500–720 nm (a, c) and 740–1000 nm (b, d) regions for H(M202)H/I(M206)H mutant RCs
The EADS associated with the lifetime of 3.4 ps for Wt and 4.2 ps for mutant RCs show spectral features typical for the excited state P* (Figs. 5a, b, 6a, b, black lines). Both spectra demonstrate broad negative band in the Qy region which includes contribution from the P bleaching at ~ 865 nm for Wt and ~ 840 nm for mutant RCs along with stimulated emission from P* at ~ 900 nm (panels b). The negative band at ~ 600 nm is present in the Qx spectral regions on the background of the weakly structured absorption of P* in the range 500–720 nm (panels a). The P* state evolves into the charge-separated state P+HA− characterized by 197 ps and 217 ps EADS for the Wt and mutant RCs, respectively, demonstrating the negative band at ~ 600 nm, bleaching of the Qx absorption band of the BPheo HA at 545 nm, and appearance of the absorption bands of the radical anion HA− at ~ 670 and 960 nm. The EADS of the mutant RCs also contains the BPheo radical anion band at 915 nm, which is not seen in the Wt spectrum due to overlap with the P bleaching at ~ 865 nm. Bleaching of the P absorption band is observed at 838 nm in the Qy region of the mutant spectrum (Fig. 6b, red line) in accordance with the different spectral position of the long-wavelength band of P in the ground-state absorption spectra (Fig. 2). Both spectra also contain electrochromic shift in the region of ~ 800 nm (Figs. 5a, b, 6a, b, red lines). The state P+HA− further evolves into the non-decaying state P+QA− with a time constant of ~ 200 ps in both RCs. The almost equal amplitudes of the P bleaching at ~ 865 nm in the 197 ps EADS and the non-decaying EADS (Fig. 5b) correspond to essentially 100% yield of the P+HA− → P+QA− electron transfer in Wt. A slightly reduced amplitude of the P bleaching at ~ 840 nm in the non-decaying P+QA− EADS as compared to that in the 217 ps EADS suggests that some ground-state recovery of P and a decrease in the P+QA− yield (by about 10%) are observed in the double mutant (Fig. 6b). Interestingly, there is a pronounced decrease in the amplitude of the negative signal at 545 nm relative to that at ~ 600 nm in the 217 ps EADS of the double mutant (Fig. 6a), while the amplitudes of these features are close to each other in the 197 ps EADS of Wt (Fig. 5a). In principle, this might reflect a smaller yield of P+HA− in the double mutant than in Wt (see, however, below).
Some additional information about the possible effects of the double mutation on the yields of electron-transfer reactions involved in charge separation can be extracted from the decay-associated difference spectra (DADS) obtained from global analysis using a parallel kinetic model (Figs. 5c, d, 6c, d). The DADS corresponding to 3.4 ps in Wt and 4.2 ps in double mutant contain positive bands at 545, 760, and 810 nm and a negative band at ~ 670 representing the formation of the P+HA− state (Figs. 5c, d, 6c, d, red lines). The broad negative band at ~ 900 nm shows the decay of stimulated emission of P*. Both spectra instead of a bleaching demonstrate band shifts centered at ~ 600 nm (i.e., there is no ground-state recovery of P) suggesting a practically 100% yield of P+HA− in both Wt and the double mutant. Therefore, it is likely that the aforementioned difference in the amplitudes of negative signals at 545 nm and ~ 600 nm in the 217 ps EADS of the double mutant is due to another reason, for example, a change in the absorption properties of HA. In the Wt RCs, 197 ps DADS reflect the spectral changes associated with the electron-transfer reaction P+HA− → P+QA including the decay of the 545 nm Qx ground-state bleaching and radical anion-positive absorption bands at 670, 914, and 965 nm. It is noteworthy that in addition to the features associated with electron transfer from HA− to QA, the mutant DADS with 217 ps lifetime include a negative signal at 840 nm indicating a ground-state recovery of P. This implies that, unlike Wt RCs, in H(M202)L/I(M206)H RCs, recombination of P+HA− to the ground state effectively competes with forward electron transfer from HA− to QA resulting in a lower quantum yield of the P+QA− state.
Summarizing, the transient absorption measurements show that (i) the sequence of electron-transfer reactions P* → P+HA− → P+QA− is preserved in the double mutant, (ii) the double mutation does not significantly affect the P* lifetime, and (iii) the yield of the P+QA− state is slightly decreased in the double mutant.
Discussion
Homodimer structure of the primary electron donor in the H(M202)L/I(M206)H mutant RC
Both absorption spectra and results of pigment analysis come to an agreement that in the mutant RC the special pair is the BChl homodimer. The long-wavelength Qy band of P is clearly visible in the 100 K absorption spectrum of the mutant RCs (Fig. 2, inset), although it shifts to the blue and decreases in intensity compared to Wt, apparently due to some changes in the structure and/or interactions of the homodimer, caused by the double amino acid substitution. The spectral position of the Qy band of P depends on various factors such as formation of charge transfer states (Parson and Warshel 1987), conformation of acetyl carbonyl groups (Parson and Warshel 1987; Thompson et al. 1991), formation of hydrogen bonds (Thompson et al. 1991), electrostatic interactions (Thompson et al. 1991; Johnson et al. 2003), and small changes (~ 0.1 Å) in spacing between the two BChls of the dimer (Gudowska-Nowak et al. 1990; Thompson et al. 1991; Parson and Warshel 1987). A similar BChl/BPheo ratio of 1.9 ± 0.1 determined here for both Wt and double mutant RCs suggests that these RCs have the same pigment composition (i.e., four BChls and two BPheos per RC) and that the BChl homodimer is most likely retained in the mutant RC.
In the absence of crystallographic data, valuable information on the nature and electronic structure of the primary electron donor in the double mutant H(M202)L/I(M206)H can be obtained by comparing its light-induced FTIR difference spectrum for the primary donor oxidation with the P+Q−/PQ FTIR spectrum of Wt that contains the homodimer P (Figs. 3, 4). Previously, it has been shown that the hole transfer electronic transition localized in the FTIR spectrum of Wt RCs at ~ 2680 cm−1 (Fig. 3a), as well as vibrational (phase-phonon) bands at ~ 1550, 1477 ,and ~ 1290 cm−1 (Fig. 4a), is not observed in the FTIR difference spectrum of isolated BChl radical cation (Nabedryk et al. 1993, 2000), thus being markers of the dimeric structure of the oxidized primary donor (Breton et al. 1992). In addition, these IR features are not observed in the spectra of heterodimer mutants in which His M202 (or His L173) is replaced by Leu (Nabedryk et al. 1992). The presence of the P+ electronic band at ~ 2740 cm−1 (Fig. 3b) and associated phase-phonon bands at 1544, 1484, and 1317 cm−1 in the FTIR spectrum of H(M202)L/I(M206)H (Fig. 4b) demonstrate that the double mutant contains a homodimer with the charge on P+ distributed between the two coupled BChl molecules.
However, a decrease in the intensity (and change in position) of the electronic and phase-phonon bands in the spectrum of H(M202)L/I(M206)H compared to Wt (Figs. 3, 4) suggest that the electronic structure of P+ is different in these RCs. Apparently, the double mutation is accompanied by a decrease in the extent of electronic coupling between the two BChls of P+ and/or a more asymmetric charge distribution over these BChls (Breton et al. 1992; Reimers and Hush 2003). The fact that the relative amplitude of the 131-keto C = O peaks of PA+ and PB+ in the double mutant is reversed compared to Wt (Fig. 4) can be interpreted as indicating a stronger charge localization on PA+ in the mutant than in Wt (Malferrari et al. 2015). These data, as well as small shifts of the 131-keto carbonyl frequencies in the double mutant with respect to those in Wt (ranging from ca. + 1 to − 7 cm−1; Fig. 4) probably reflect small mutation-induced rearrangement of P BChl macrocycles and/or of their substituents (Nabedryk et al. 2000; Spiedel et al. 2002).
The linewidth of the H(M202)L/I(M206)H mutant EPR signal (1.18 mT) exceeds considerably that of the Wt RCs and is close to the 1.22–1.26 mT value obtained for the heterodimer primary donor of the H(M202)L mutant (Huber et al. 1990; Rautter et al. 1995). It indicates a highly asymmetrical spin density distribution in the double mutant and is in agreement with the weakly coupled dimer of the primary donor molecule.
Possible candidates for the axial ligand to PB in the double mutant H(M202)L/I(M206) H RC
As outlined in Introduction, the replacement of His M202, the fifth axial ligand to Mg of the PB BChl in the homodimer P of the wild-type strain of Rba. sphaeroides, with Leu leads to the formation of the BChl/BPheo heterodimer (McDowell et al. 1991). In this context, the preservation of the overall BChl/BChl homodimer structure of the primary electron donor when a combination of substitutions H(M202)L and I(M206)H is introduced into the mutant RC is rather surprising and suggests the presence of a ligand to PB. Apparently, there are three potential candidates for the role of the fifth ligand to the Mg ion of PB in the H(M202)L/I(M206)H double mutant RC.
One possibility is that the Mg of the PB BChl in the double mutant is coordinated by the new His side chain introduced at position M206. According to Camara-Artigas et al. (2002), the crystal structure of the H(M202)L heterodimer RC from Rba. sphaeroides showed that, compared to Wt RCs, His to Leu substitution resulted in small shifts of residues M196 and M206, a rotation of the side chain of Ile M206, and a loss of the bound water molecule (water A in Fig. 1). Based on this structure (PDB: 1KBY), modeling of His at M206 using PyMOL (DeLano 2002) does not suggest that the side chain of His M206 could be in a suitable position to serve as an axial ligand to the Mg atom at the center of the PB macrocycle (not shown). In fact, when the His M206 residue is directed toward P, its imidazole group is positioned at an acute angle to the macrocycle plane of PB. It is most likely that a scenario with a coordinating interaction between His M206 and the Mg of PB could be realized only if the double mutation leads to rather strong (or even gross) changes in the position/conformation of the P dimer and/or in the surrounding protein. Our data do not seem to provide evidence for such alterations. Although the electronic absorption spectrum (Fig. 2) and the P+Q−/PQ FTIR spectrum (Figs. 3, 4) of H(M202)L/I(M206)H RCs show significant differences with the corresponding spectra of Wt RCs, these results cannot be used as indisputable indication of the specific large-scale structural changes in the double mutant RC. Indeed, blue shifts of different sizes were previously described for the Qy absorption band of P in purple bacteria RCs resulting from various perturbations of the P environment, such as changes caused by the type and concentration of the solubilizing detergent used (Wang et al. 1994; Müh et al. 1996, 1997), the hydration state of the RC (Malferrari et al. 2015; Zabelin et al. 2019b), as well as by various point mutations (see, for example, Bylina and Youvan 1988; Kirmaier et al. 1991; DiMagno et al. 1998). Regarding IR data, our recent measurements have shown that the P+Q−/PQ FTIR spectrum of the L(M196)H mutant Rba. sphaeroides RCs (Zabelin et al. 2019a) demonstrate features similar to those observed here for the H(M202)L/I(M206)H RCs, while the crystal structure of the L(M196)H RC did not reveal global alterations in the conformation of the P dimer (Fufina et al. 2015). It was shown that mutation of His L168 to Phe led to a significant blue shift of the Qy band of P and was accompanied by a strong intensity decrease and a downshift of the broad electronic transition at 2600 cm−1 along with some decrease in intensities of the phase-phonon bands, whilst only relatively small changes were found in the crystal structure of the H(L168)F mutant RC (Spiedel et al. 2002). These changes include a small shift in the position of the PA BChl relative to PB, as well as a change in the conformation of the acetyl carbonyl group of PA (Spiedel et al. 2002). It is worth noting that the unexpected assembly of the P homodimer observed in the H(M202)L/I(M206)H mutant is not unprecedented. It has been reported (but apparently not described in detail) that the homodimer P assembled in Rba. sphaeroides double mutant in which the H(M202)L mutation was combined with the distant L(M214)H mutation located near the BPheo HA molecule (see Heller et al. 1995). As already mentioned above, Vasilieva et al. (2012) has proposed that the homodimer P is retained in the structure of the mutant Rba. sphaeroides RC containing symmetric double substitution H(L173)L/I(L177)H. Properties of the RC H(L173)L/I(L177)H were largely similar to those of the RC H(M202)L/I(M206)H, and the blue shift of the Qy band of P was even more pronounced (46 nm at 90 K). Similar changes in the P environment caused by symmetric double mutations suggest analogous mechanism for replacing the natural axial ligands of PA and PB with alternative ligands.
Another possibility is that the Mg ion of PB in the double mutant is coordinated by the carbonyl oxygen of the 31-acetyl C=O group of PA BChl if this group is undergoing out-of-plane rotation, as it has been discussed for the H(M202)G homodimer mutant in which His at M202 is replaced with non-coordinating glycine (Goldsmith et al. 1996). In the Wt RC, the His L168 residue has been shown to donate a strong hydrogen bond to the 31-acetyl carbonyl of PA (Murchison et al. 1993; Spiedel et al. 2002). Therefore, it seems reasonable to expect that the rotation of the PA acetyl carbonyl group would lead to removal (or strong weakening) of this hydrogen-bonding interaction and, as a result, to a large change in the redox potential of P. In fact, the removal of this hydrogen bond in the H(L168)F mutant led to a decrease in the P/P+ mid-point redox potential in purified RCs by 80–95 mV (Murchison et al. 1993; Lin et al. 1994). The results of our redox titration measurements did not show a significant difference in the P/P+ midpoint potential between the H(M202)L/I(M206)H mutant and Wt. In this regard, it is interesting to note that using FT-Raman spectroscopy, Goldsmith et al. (1996) showed that the acetyl carbonyl groups of both BChls of the P homodimer in the H(M202)G mutant remained in a conformation similar to that in Wt.
Finally, it is possible that coordination of Mg in PB of the H(M202)L/I(M206)H double mutant is provided by a water molecule incorporated into the mutant structure. Although the model substitution of Ile M206 with His in the crystal structure of the H(M202)L heterodimer RC (Camara-Artigas et al. 2002) using PyMOL (DeLano 2002) did not suggest the presence of a cavity to accommodate a water molecule (not shown), a cavity might be created in the H(M202)L/I(M206)H RC as a result of structural alterations caused by the double mutation. It is conceivable that due to the close proximity of His M206, the Leu M202 side chain in the double mutant adopts geometry when it is no longer located over the center of the PB macrocycle [as is observed in the RC crystal structure of the H(M202)L heterodimer Camara-Artigas et al. 2002], making room for a new water molecule. The ability of water molecule to serve as an adventitious axial ligand to the Mg atom of the PB BChl has previously been proposed for the homodimer mutants in which His M202 was replaced with a number of amino acid residues (such as Gly, Ser, Cys, or Asn) (Goldsmith et al. 1996; Nabedryk et al. 2000).
Primary charge separation in H(M202)L/I(M206)H mutant RCs
The results presented show that the combination of amino acid substitutions H(M202)L/I(M206)H leads to a significant shift of the low-energy component of the Qy optical transition of P to higher energies (50 meV at 100 K) and noticeably affects the electronic structure of the radical cation P+, but have a relatively small influence on the oxidation potential of P. Despite the changes in the spectroscopic and electronic properties of P and P+, the primary photochemistry in the double mutant RC is basically similar, although not identical, to that observed in the Wt RC (Figs. 5, 6). Specifically, the rate of the initial electron transfer from P* to HA (most likely via BA) to form the P+HA− radical pair in the mutant is essentially unchanged compared to Wt. This suggests that the mutation is not accompanied by a significant change (at least an increase) in the inherent rate of P* deactivation to the ground state via internal conversion. Notably, a small but distinct decrease in the yield of the P+QA− state is observed for the mutant, while (in agreement with the published data) this state is formed with a nearly 100% yield in Wt. The reason for this difference remains to be understood. Most likely, unlike the situation in Wt, deactivation of the P+HA− state to the ground state in the mutant effectively competes with the forward electron transfer from HA− to QA. At present, we can only speculate that the introduced amino acid substitutions modify the energetics of the P+BA− and P+HA− states, leading to enhanced charge recombination of P+HA− to the ground state via the thermally activated short-lived P+BA− state. The inherent time constants for the decay of P+HA− and P+BA− to the ground state in Wt RCs were estimated to be ~ 20 ns (Chidsey et al. 1984; Budil et al. 1987; Ogrodnik et al. 1988) and ~ 1 ns (Kirmaier et al. 1991, 1995; Shkuropatov and Shuvalov 1993), respectively. The observed decrease in the P/P+ midpoint potential of the double mutant by about 15 mV compared to Wt should lead to uniform changes in the free energy of all charge-separated states in the RC, including P+BA− and P+HA−. The relative increase in the P/P* energy difference by 50 meV in the double mutant is expected to increase the driving force for the P* → P+BA− electron-transfer reaction. However, it cannot be excluded that the double mutation might affect the redox potentials of BA and/or HA, making the free energy levels of P+HA− and P+BA− closer to each other. In this case, the short-lived P+BA− could be accessible for thermal repopulation from the relaxed state P+HA−, and, as a result, deactivation of P+HA− to the ground state would be more efficient in the double mutant than in Wt.
With regard to the mechanism of charge separation in purple bacteria RCs, an interesting hypothesis exists that the initial electron-transfer reaction occurs via a set of connected atoms, which provide through-bond electron transfer between the primary reactants, P and BA (Yakovlev et al. 2002, 2005). The key elements of the putative through-bond electron-transfer route are water A and His M202 (Yakovlev et al. 2002, 2005). According to the RC crystal structure (see, for example, Fig. 1), water A is within hydrogen-bond distance from both oxygen of the 131-keto carbonyl of BA and nitrogen of the imidazole of His M202 that coordinates the Mg ion of PB. It has been shown that steric exclusion of water A from the structure of the membrane-bound G(M203)L mutant RCs of Rba. sphaeroides has a strong effect on the rate of primary electron transfer at room temperature, increasing P* lifetime in the mutant to 20–40 ps compared to ~ 5 ps in the Wt (Potter et al. 2005; Gibasiewicz et al. 2016). A slowing down of the initial electron-transfer reaction was also observed for purified G(M203)L RCs at 90 K (Yakovlev et al. 2005). These data are consistent with a through-bond electron tunneling from P* to BA; however, alternative explanations are also possible, such as the effect of water A on the BA/BA− mid-point potential (for further discussion see Potter et al. 2005; Yakovlev et al. 2005). Based on theoretical calculations, it was proposed that the reversible rotation of His M202 and the associated displacement of the water A protons toward Gly M203 and the 131-carbonyl of BA are coupled to the P* → P+BA− electron-transfer reaction in the Rba. sphaeroides RC (Eisenmayer et al. 2013).
The results of our measurements are not conclusive for answering the question of whether the proposed through-bond connection is important for the initial electron transfer in the RC. However, the fact that the rate and yield of the initial electron transfer are practically not affected by the H(M202)L/I(M206)H double mutation strongly suggests that the presence of His M202 is not critical for the primary charge separation process. It is possible that if the putative through-bond electron-transfer route functions in Wt, its loss due the removal of His M202 in the double mutant is effectively compensated by competitive (through space) electron tunneling routes through the atoms of the closest approach between P* and BA. It can also be assumed that in the absence of His M202, the functional through-bond connection in the double mutant RC might be completed by an alternative ligand (see above). The absence of a significant effect of the double mutation on the initial electron transfer may indicate that water A remains in the structure of the H(M202)L/I(M206)H RC, while it is absent in the crystal structure of the H(M202)L heterodimer RC (Camara-Artigas et al. 2002).
Conclusions
Unlike a single mutation H(M202)L, replacing His M202 with Leu does not prevent the assembly of the BChl homodimer in the H(M202)L/I(M206)H double mutant. Apparently, other structural elements of the double mutant RC (e.g., His M206) or an incorporated water molecule may serve as the fifth axial ligand to Mg of PB, while retaining the overall BChl/BChl homodimeric structure and the basic functional properties of the primary electron donor. The fact that the double mutation has only a minor effect on the electron-transfer reaction P* → (P+BA−) → P+HA− suggests that water A is preserved in the structure of the double mutant RC, and that His M202 is not essential in controlling the optimal rate and yield of the initial charge separation reaction in Wt RCs.
Abbreviations
- RC:
-
Reaction center
- Wt:
-
Wild type
- BChl:
-
Bacteriochlorophyll
- BPheo:
-
Bacteriopheophytin
- P:
-
Dimer of BChls in the RC
- PA and PB :
-
BChls constituting P
- BA and BB :
-
Monomeric BChls in the active and inactive cofactors branch, respectively
- HA and HB :
-
BPheos in the active and inactive cofactors branch, respectively
- QA :
-
Primary quinone acceptor
- QB :
-
Secondary quinone acceptor
- Rba. :
-
Rhodobacter
- FTIR:
-
Fourier transform infrared
- EPR:
-
Electron paramagnetic resonance
References
Blankenship RE (2014) Molecular mechanisms of photosynthesis. Wiley Blackwell, Chichester
Breton J, Nabedryk E, Parson WW (1992) A new infrared electronic transition of the oxidized primary electron donor in bacterial reaction centers: a way to assess resonance interactions between the bacteriochlorophylls. Biochemistry 31:7503–7510. https://doi.org/10.1021/bi00148a010
Budil DE, Kolaczkowski SV, Norris JR (1987) The temperature dependence of electron back-transfer from the primary radical pair of bacterial photosynthesis. In: Biggins G (ed) Progress in photosynthesis research, vol 1. Martinus Nijhoff Publishers, Dordrecht, pp 25–28
Bylina EJ, Youvan DC (1988) Directed mutations affecting spectroscopic and electron transfer properties of the primary donor in the photosynthetic reaction center. Proc Natl Acad Sci USA 85:7226–7230. https://doi.org/10.1073/pnas.85.19.7226
Camara-Artigas A, Magee C, Goetsch A, Allen JP (2002) The structure of the heterodimer reaction center from Rhodobacter sphaeroides at 2.55 Å resolution. Photosynth Res 74:87–93. https://doi.org/10.1023/A:1020882402389
Chidsey CED, Kirmaier C, Holten D, Boxer SG (1984) Magnetic field dependence of radical-pair decay kinetics and molecular triplet quantum yield in quinone-depleted reaction centers. Biochim Biophys Acta 766:424–437. https://doi.org/10.1016/0005-2728(84)90258-5
DeLano WL (2002) The PyMOL molecular graphics system. https://www.pymol.org
DiMagno TJ, Laible PD, Reddy NR, Small GJ, Norris JR, Schiffer M, Hanson DK (1998) Protein–chromophore interactions: spectral shifts report the consequences of mutations in the bacterial photosynthetic reaction center. Spectrochim Acta A 54:1247–1267. https://doi.org/10.1016/S1386-1425(98)00074-2
Eisenmayer TJ, Lasave JA, Monti A, de Groot HJM, Buda F (2013) Proton displacements coupled to primary electron transfer in the Rhodobacter sphaeroides reaction center. J Phys Chem B 117:11162–11168. https://doi.org/10.1021/jp401195t
Ermler U, Fritzsch G, Buchanan SK, Michel H (1994) Structure of the photosynthetic reaction centre from Rhodobacter sphaeroides at 2.65 Å resolution: cofactors and protein–cofactor interactions. Structure 2:925–936. https://doi.org/10.1016/s0969-2126(94)00094-8
Fufina TY, Vasilieva LG, Gabdulkhakov AG, Shuvalov VA (2015) The L(M196)H mutation in Rhodobacter sphaeroides reaction center results in new electrostatic interactions. Photosynth Res 125:23–29. https://doi.org/10.1007/s11120-014-0062-0
Gibasiewicz K, Białek R, Pajzderska M, Karolczak J, Burdzinski G, Jones MR, Brettel K (2016) Weak temperature dependence of P+HA− recombination in mutant Rhodobacter sphaeroides reaction centers. Photosynth Res 128:243–258. https://doi.org/10.1007/s11120-016-0239-9
Goldsmith JO, King B, Boxer SG (1996) Mg coordination by amino acid side chains is not required for assembly and function of the special pair in bacterial photosynthetic reaction centers. Biochemistry 35:2421–2428. https://doi.org/10.1021/bi9523365
Gudowska-Nowak E, Newton MD, Fajer J (1990) Conformational and environmental effects on bacteriochlorophyll optical spectra: correlations of calculated spectra with structural results. J Phys Chem 94:5795–5801. https://doi.org/10.1021/j100378a036
Heller BA, Holten D, Kirmaier C (1995) Characterization of bacterial reaction centers having mutations of aromatic residues in the binding site of the bacteriopheophytin intermediary electron carrier. Biochemistry 34:5294–5302. https://doi.org/10.1021/bi00015a045
Huber M, Lous EJ, Isaacson RA, Feher G, Gaul D, Schenck CC (1990) EPR and ENDOR studies of the oxidized donor in reaction centers of Rhodobacter sphaeroides strain R-26 and two heterodimer mutants in which histidine M202 or Ll73 was replaced by leucine. In: Michel-Beyerle ME (ed) Reaction centers of photosynthetic bacteria. Springer, Berlin, pp 219–228
Johnson ET, Müh F, Nabedryk E, Williams JC, Allen JP, Lubitz W, Breton J, Parson WW (2002) Electronic and vibronic coupling of the special pair of bacteriochlorophylls in photosynthetic reaction centers from wild-type and mutant strains of Rhodobacter sphaeroides. J Phys Chem B 106:11859–11869. https://doi.org/10.1021/jp021024q
Johnson ET, Nagarajan V, Zazubovich V, Riley K, Small GJ, Parson WW (2003) Effects of ionizable residues on the absorption spectrum and initial electron-transfer kinetics in the photosynthetic reaction center of Rhodobacter sphaeroides. Biochemistry 42:13673–13683. https://doi.org/10.1021/bi035366d
Jones MR, Visschers RW, van Grondelle R, Hunter CN (1992) Construction and characterization of a mutant of Rhodobacter sphaeroides with the reaction center as the sole pigment–protein complex. Biochemistry 31:4458–4465. https://doi.org/10.1021/bi00133a011
Khatypov RA, Vasilieva LG, Fufina TY, Bolgarina TI, Shuvalov VA (2005) Substitution of isoleucine L177 by histidine affects the pigment composition and properties of the reaction center of the purple bacterium Rhodobacter sphaeroides. Biochemistry (Mosc) 70:1256–1261. https://doi.org/10.1007/s10541-005-0256-3
Kirmaier C, Holten D, Bylina EJ, Youvan DC (1988) Electron transfer in a genetically modified bacterial reaction center containing a heterodimer. Proc Natl Acad Sci USA 85:7562–7566. https://doi.org/10.1073/pnas.85.20.7562
Kirmaier C, Gaul D, DeBey R, Holten D, Schenck CC (1991) Charge separation in a reaction center incorporating bacteriochlorophyll for photoactive bacteriopheophytin. Science 251:922–927. https://doi.org/10.1126/science.2000491
Kirmaier C, Laporte L, Schenck CC, Holten D (1995) The nature and dynamics of the charge-separated intermediate in reaction centers in which bacteriochlorophyll replaces the photoactive bacteriopheophytin 2 The rates and yields of charge separation and recombination. J Phys Chem 99:8910–8917. https://doi.org/10.1021/j100021a068
Lin X, Murchison HA, Nagarajan V, Parson WW, Allen JP, Williams JC (1994) Specific alteration of the oxidation potential of the electron donor in reaction centers from Rhodobacter sphaeroides. Proc Natl Acad Sci USA 91:10265–10269. https://doi.org/10.1073/pnas.91.22.10265
Malferrari M, Turina P, Francia F, Mezzetti A, Leibl W, Venturoli G (2015) Dehydration affects the electronic structure of the primary electron donor in bacterial photosynthetic reaction centers: evidence from visible–NIR and light-induced difference FTIR spectroscopy. Photochem Photobiol Sci 14:238–251. https://doi.org/10.1039/c4pp00245h
McDowell LM, Gaul D, Kirmaier C, Holten D, Schenck CC (1991) Investigation into the source of electron transfer asymmetry in bacterial reaction centers. Biochemistry 30:8315–8322. https://doi.org/10.1021/bi00098a006
Moss DA, Leonhard M, Bauscher M, Mäntele W (1991) Electrochemical redox titration of cofactors in the reaction center from Rhodobacter sphaeroides. FEBS Lett 283:33–36. https://doi.org/10.1016/0014-5793(91)80547-G
Müh F, Rautter J, Lubitz W (1996) Effects of zwitterionic detergents on the primary donor of bacterial reaction centers. Ber Bunsenges Phys Chem 100:1974–1977. https://doi.org/10.1002/bbpc.19961001208
Müh F, Rautter J, Lubitz W (1997) Two distinct conformations of the primary electron donor in reaction centers from Rhodobacter sphaeroides revealed by ENDOR/TRIPLE-spectroscopy. Biochemistry 36:4155–4162. https://doi.org/10.1021/bi962859s
Nabedryk E, Robles JSJ, Goldman E, Youvan DC, Breton J (1992) Probing the primary donor environment in the histidine M200-leucine and histidine L173-leucine heterodimer mutants of Rhodobacter capsulatus by light-induced Fourier transform infrared difference spectroscopy. Biochemistry 31:10852–10858. https://doi.org/10.1021/bi00159a028
Nabedryk E, Schulz C, Müh F, Lubitz W, Breton J (2000) Heterodimeric versus homodimeric structure of the primary electron donor in Rhodobacter sphaeroides reaction centers genetically modified at position M202. Photochem Photobiol 71:582–588. https://doi.org/10.1562/0031-8655(2000)071%3C0582:hvhsot%3E2.0.co;2
Naberdyk E, Allen JP, Taguchi AKW, Williams JC, Woodbury NW, Breton J (1993) Fourier transform infrared study of the primary electron donor in chromatophores of Rhodobacter sphaeroides with reaction centers genetically modified at residues M160 and L131. Biochemistry 32:13879–13885. https://doi.org/10.1021/bi00213a017
Ogrodnik A, Volk M, Letterer R, Feick R, Michel-Beyerle ME (1988) Determination of free energies in reaction centers of Rb. sphaeroides. Biochim Biophys Acta 936:361–371. https://doi.org/10.1016/0005-2728(88)90012-6
Parson WW (2008) Functional patterns of reaction centers in anoxygenic photosynthetic bacteria. In: Renger G (ed) Primary processes of photosynthesis, Part 2 principles and apparatus. RSC Publishing, Cambridge, pp 57–109. https://doi.org/10.1039/9781847558169-00057
Parson WW, Warshel A (1987) Spectroscopic properties of photosynthetic reaction centers. 2. Application of the theory to Rhodopseudomonas viridis. J Am Chem Soc 109:6152–6163. https://doi.org/10.1021/ja00254a040
Ponomarenko NS, Li L, Marino AR, Tereshko V, Ostafin A, Popova JA, Bylina EJ, Ismagilov RF, Norris JRJ (2009) Structural and spectropotentiometric analysis of Blastochloris viridis heterodimer mutant reaction center. Biochim Biophys Acta Biomembr 1788:1827–1831. https://doi.org/10.1016/j.bbamem.2009.06.006
Potter JA, Fyfe PK, Frolov D, Wakeham MC, van Grondelle R, Robert B, Jones MR (2005) Strong effects of an individual water molecule on the rate of light-driven charge separation in the Rhodobacter sphaeroides reaction center. J Biol Chem 280:27155–27164. https://doi.org/10.1074/jbc.M501961200
Rautter J, Lendzian F, Schulz C, Fetsch A, Kuhn M, Lin X, Williams JC, Allen JP, Lubitz W (1995) ENDOR studies of the primary donor cation radical in mutant reaction centers of Rhodobacter sphaeroides with altered hydrogen-bond interactions. Biochemistry 34:8130–8143. https://doi.org/10.1021/bi00025a020
Reimers JR, Hush NS (2003) Modeling the bacterial photosynthetic reaction center. VII. Full simulation of the intervalence hole-transfer absorption spectrum of the special-pair radical cation. J Chem Phys 119:3262–3277. https://doi.org/10.1063/1.1589742
Shkuropatov AYa, Shuvalov VA (1993) Electron transfer in pheophytin a-modified reaction centers from Rhodobacter sphaeroides (R-26). FEBS Lett 322:168–172. https://doi.org/10.1016/0014-5793(93)81561-d
Snellenburg JJ, Laptenok SP, Seger R, Mullen KM, van Stokkum IHM (2012) Glotaran: a Java-based graphical user interface for the R package TIMP. J Stat Soft 49:1–22. https://doi.org/10.18637/jss.v049.i03
Spiedel D, Roszak AW, McKendrick K, McAuley KE, Fyfe PK, Nabedryk E, Breton J, Robert B, Cogdell RJ, Isaacs NW, Jones MR (2002) Tuning of the optical and electrochemical properties of the primary donor bacteriochlorophylls in the reaction centre from Rhodobacter sphaeroides: spectroscopy and structure. Biochim Biophys Acta 1554:75–93. https://doi.org/10.1016/s0005-2728(02)00215-3
Sun C, Carey A-M, Gao B-R, Wraight CA, Woodbury NW, Lin S (2016) Ultrafast electron transfer kinetics in the LM dimer of bacterial photosynthetic reaction center from Rhodobacter sphaeroides. J Phys Chem B 120:5395–5404. https://doi.org/10.1021/acs.jpcb.6b05082
Thompson MA, Zerner MC, Fajer J (1991) A theoretical examination of the electronic structure and excited states of the bacteriochlorophyll b dimer from Rhodopseudomonas viridis. J Phys Chem 95:5693–5700. https://doi.org/10.1021/j100167a058
van Stokkum IHM, Larsen DS, van Grondelle R (2004) Global and target analysis of time-resolved spectra. Biochim Biophys Acta 1657:82–104. https://doi.org/10.1016/j.bbabio.2004.04.011
Vasilieva LG, Fufina TY, Gabdulkhakov AG, Leonova MM, Khatypov RA, Shuvalov VA (2012) The site-directed mutation I(L177)H in Rhodobacter sphaeroides reaction center affects coordination of PA and BB bacteriochlorophylls. Biochim Biophys Acta 1817:1407–1417. https://doi.org/10.1016/j.bbabio.2012.02.008
Wang S, Lin S, Lin X, Woodbury NW, Allen JP (1994) Comparative study of reaction centers from purple photosynthetic bacteria: isolation and optical spectroscopy. Photosynth Res 42:203–215. https://doi.org/10.1007/BF00018263
Williams JC, Alden RG, Murchison HA, Peloquin JM, Woodbury NW, Allen JP (1992) Effect of mutations near the bacteriochlorophylls in reaction centers from Rhodobacter sphaeroides. Biochemistry 31:11029–11037. https://doi.org/10.1021/bi00160a012
Woodbury NW, Allen JP (1995) The pathway, kinetics and thermodynamics of electron transfer in wild type and mutant reaction centers of purple nonsulfur bacteria. In: Blankenship RE, Madigan MT, Bauer CE (eds) Anoxygenic photosynthetic bacteria. Kluwer Academic Publishers, Dordrecht, pp 527–557. https://doi.org/10.1007/0-306-47954-0_24
Yakovlev AG, Shkuropatov AYa, Shuvalov VA (2002) Nuclear wave packet motion between P* and P+BA− potential surfaces with a subsequent electron transfer to HA in bacterial reaction centers at 90 K. Electron transfer pathway. Biochemistry 41:14019–14027. https://doi.org/10.1021/bi020250n
Yakovlev AG, Jones MR, Potter JA, Fyfe PK, Vasilieva LG, Shkuropatov AYa, Shuvalov VA (2005) Primary charge separation between P* and BA: electron-transfer pathways in native and mutant GM203L bacterial reaction centers. Chem Phys 319:297–307. https://doi.org/10.1016/j.chemphys.2005.08.018
Zabelin AA, Fufina TY, Vasilieva LG, Shkuropatova VA, Zvereva MG, Shkuropatov AY, Shuvalov VA (2009) Mutant reaction centers of Rhodobacter sphaeroides I(L177)H with strongly bound bacteriochlorophyll a: structural properties and pigment–protein interactions. Biochemistry (Mosc) 74:68–74. https://doi.org/10.1134/S0006297909010106
Zabelin AA, Fufina TYu, Khristin AM, Khatypov RA, Shkuropatova VA, Shuvalov VA, Vasilieva LG, Shkuropatov AYa (2019a) Effect of leucine M196 substitution by histidine on electronic structure of the primary electron donor and electron transfer in reaction centers from Rhodobacter sphaeroides. Biochemistry (Mosc) 84:520–528. https://doi.org/10.1134/S0006297919050067
Zabelin AA, Shkuropatova VA, Shuvalov VA, Shkuropatov AYa (2019b) Spectral and photochemical properties of Rhodobacter sphaeroides R-26 reaction center films in vacuum. Biochemistry (Mosc) 84:1107–1115. https://doi.org/10.1134/S000629791909013X
Acknowledgements
This work was performed under State Task No. AAAA-A17030110140-5 and partially financially supported by the Russian Foundation for Basic Research (Grant No. 17-00-00207 KOMFI).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by AMK, AAZ, and TYuF. The first draft of the manuscript was written by AYaS and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript. AMK, AAZ, and TYuF are equally contributed to the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Khristin, A.M., Zabelin, A.A., Fufina, T.Y. et al. Mutation H(M202)L does not lead to the formation of a heterodimer of the primary electron donor in reaction centers of Rhodobacter sphaeroides when combined with mutation I(M206)H. Photosynth Res 146, 109–121 (2020). https://doi.org/10.1007/s11120-020-00728-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-020-00728-9