
Abstract
PsbO-D158 is a highly conserved residue of the PsbO protein in photosystem II (PSII), and participates in one of the hydrogen-bonding networks connecting the manganese cluster with the lumenal surface. In order to examine the role of PsbO-D158, we mutated it to E, N or K in Thermosynechococcus vulcanus and characterized photosynthetic properties of the mutants obtained. The growth rates of these three mutants were similar to that of the wild type, whereas the oxygen-evolving activity of the three mutant cells decreased to 60–64% of the wild type. Fluorescence kinetics showed that the mutations did not affect the electron transfer from QA to QB, but slightly affected the donor side of PSII. Moreover, all of the three mutant cells were more sensitive to high light and became slower to recover from photoinhibition. In the isolated thylakoid membranes from the three mutants, the PsbU subunit was lost and the oxygen-evolving activity was reduced to a lower level compared to that in the respective cells. PSII complexes isolated from these mutants showed no oxygen-evolving activity, which was found to be due to large or complete loss of PsbO, PsbV and PsbU during the process of purification. Moreover, PSII cores purified from the three mutants contained Psb27, an assembly co-factor of PSII. These results suggest that PsbO-D158 is required for the proper binding of the three extrinsic proteins to PSII and plays an important role in maintaining the optimal oxygen-evolving activity, and its mutation caused incomplete assembly of the PSII complex.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In oxygenic photosynthesis, light-driven water oxidation is carried out by the oxygen-evolving complex (OEC) in Photosystem II (PSII). PSII is a large membrane-spanning protein complex in the thylakoid membranes of plants, algae and cyanobacteria (Shen 2015; Umena et al. 2011; Vinyard et al. 2013). In cyanobacteria, it is composed of 20 protein subunits, of which 17 are intrinsic, transmembrane subunits and 3 are membrane-extrinsic subunits. Among the transmembrane subunits, seven (D1, D2, CP47, CP43, α, β subunits of cytochrome b559, and PsbI) constitute the core of PSII and are important for the electron transfer reactions. Three extrinsic proteins, PsbO (33 kDa), PsbU (12 kDa), and PsbV (17 kDa) are located in the lumenal side of the thylakoid membrane and indispensable for maintaining the stability and maximal oxygen-evolving activity of OEC (Bricker et al. 2012; Enami et al. 2008; Ifuku 2015; Roose et al. 2016).
Among the three extrinsic proteins, PsbO is the largest one and also called manganese-stabilizing protein (MSP) on the basis of its function in maintaining the stability of the Mn4CaO5-cluster, the catalytic center of OEC. PsbO is present in all oxygenic photosynthetic organisms, and contains 240–247 residues in its mature form. PsbO plays a crucial role in photosynthetic water oxidation, and its functions have been shown to include stabilization of the manganese cluster and maintenance of the suitable ion environment for its optimal activity, based on in vitro release-reconstitution experiments (Bricker 1992; Miyao and Murata 1984). In vivo studies on the role of PsbO have been performed using psbO knock-out mutant (Al-Khaldi et al. 2000; Burnap and Sherman 1991; Burnap et al. 1992; Mayfield et al. 1987; Philbrick et al. 1991; Yi et al. 2005), which showed that deletion of this subunit in the cyanobacterium Synechocystis sp. PCC 6803 does not affect the accumulation of the PSII intrinsic core proteins, and the mutant can evolve O2 at a rate 30%-70% of the wild type (WT), but the sensitivity of PSII to photoinhibition is increased (Burnap and Sherman 1991; Burnap et al. 1992; Henmi et al. 2004; Mayes et al. 1991; Philbrick et al. 1991). Deletion of MSP also alters photo-assembly of the Mn4Ca cluster (Qian et al. 1997). In contrast to cyanobacteria, deletion of psbO from green algae and higher plants abolishes O2 evolution (Mayfield et al. 1987; Yi et al. 2005). Mutants of Arabidopsis thaliana and Chlamydomonas reinhardtii that lack the psbO gene cannot grow photoautotrophically and were not able to assemble the PSII centers correctly, suggesting that the MSP has a slightly different role between cyanobacteria and algae/plants (Mayfield et al. 1987; Yi et al. 2005). In addition, PsbO is also inferred to participate in stabilizing the PSII dimer, since PsbO in one PSII monomer interacts with the CP47 protein of the adjacent monomer (Suga et al. 2015; Umena et al. 2011).
X-ray structural analysis of cyanobacterial PSII showed that PsbO has an elongated shape and consisted of two major parts: Domain I is a cylinder composed of eight antiparallel β-strands, and domain II is a hydrophilic head domain (Umena et al. 2011; De Las Rivas and Barber 2004) (Fig. 1). Domain I is full of bulky hydrophobic amino acid residues, which may play a vital role in protecting the Mn4CaO5 cluster from attack by ions in the outside solution. The head domain is mainly composed by non-regular loops and turns located between β-strands 5 and 6, whose role appears to provide a docking site for PsbO to the lumenal surface of PSII (Umena et al. 2011; Suga et al. 2015).

Structure of PsbO in association with D1 from T. vulcanus (3WU2), showing secondary structure elements of the PsbO protein. Color used: green, D1; violet purple, PsbO. The N- and C-terminals of PsbO are depicted in yellow and cyan, respectively, and PsbO-D158 and D1-R334 are depicted in orange
The high resolution crystal structure of PSII suggested that PsbO may participate in the transport of protons generated by water oxidation (Shen 2015; Umena et al. 2011). The PsbO protein is rich in aspartate/glutamate clusters, several of which participate in the hydrogen-bond network leading from the Mn4CaO5-cluster to the bulk solution of the PSII complex, and therefore may be involved in proton transfer (Bondar and Dau 2012; Del Val and Bondar 2017; Shen 2015; Umena et al. 2011). Conformational changes of PsbO during the S-state cycle were reported based on measurements by Fourier transform infrared spectroscopy, which was proposed to reflect changes in the hydrogen-bonding networks (Offenbacher et al. 2013). Molecular dynamics simulations suggested that the water-bridged carboxylate cluster at the surface of PsbO could be important for proton transfer (Lorch et al. 2015). Multiple sequence alignment of known PsbO proteins identified 19 highly conserved residues, of which 18 are included in five regions between eukaryotic and prokaryotic cells. The most conserved DPKGR region (P149, R152, D158, R162, G167), located in the head domain of PsbO, interacts with D1, D2, CP47, CP43 and PsbU (De Las Rivas and Barber 2004; Suga et al. 2015; Umena et al. 2011). However, the exact roles of these highly conserved amino acid residues of PsbO remain unclear.
Site-directed mutagenesis has been used to study the functions of some important residues in PsbO (Burnap et al. 1994; Motoki et al. 2002; Popelkova et al. 2006, 2009; Roose et al. 2010). Among these residues, PsbO-D158 (Thermosynechococcus vulcanus numbering), homologous to PsbO-D157 in spinach and D-159 in Synechocystis sp. PCC 6803, is a highly conserved residue and participates in one of the hydrogen-bonding networks. The function of PsbO-D158 has been examined by single amino acid mutations by both in vivo mutagenesis and in vitro reconstitution approaches, and the results obtained show that this residue is involved in the interaction with PSII and important for maintaining the oxygen-evolving activity (Burnap et al. 1994; Motoki et al. 2002; Popelkova et al. 2009; Roose et al. 2010). However, contradicting results have been reported previously, since in cyanobacteria, mutations of D158 (D159) have been shown to destabilize the binding of PsbO to PSII significantly (Burnap et al. 1994; Motoki et al. 2002; Shen et al. 1995), whereas in spinach, mutations of D157 affected the oxygen-evolving activity but not the binding of PsbO to PSII based on in vitro reconstitution experiments (Popelkova et al. 2009; Roose et al. 2010). To examine the function of this highly conserved residue in more detail, we changed this residue to K, N or E, respectively, in the thermophilic cyanobacterium Thermosynechococcus vulcanus (T. vulcanus), and characterized the photosynthetic properties of the resultant mutants. Our results showed that mutation of PsbO-D158 impaired the oxygen-evolving activity and increased the vulnerability of PSII to photoinhibition. While effects of mutations on the acceptor side electron transfer were negligible and only a slight effect was observed on the donor side with altered recombination kinetics between QA− and the S-state of OEC, these mutations reduced the binding affinity of PsbO to PSII remarkably and increased the population of PSII intermediates that are involved in the PSII repair/assembly.
Materials and methods
Culture of cells
Cells of the thermophilic cyanobacterium T. vulcanus were grown in a DTN medium (Muhlenhoff and Chauvat 1996), either on 1.5% (w/v) agar plates or in liquid culture, under continuous illumination with white fluorescent lamp at a light intensity of 40 μmol photons m−2 s−1 at 45 °C. The liquid culture was continuously bubbled with air containing 3–5% (v/v) CO2.
Construction of the site-directed mutants
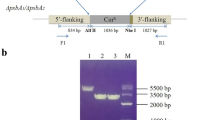
A mutagenic plasmid containing the full-length sequence of the psbO gene and an antibiotic resistance gene conferring chloramphenicol resistance (CmR) was constructed (Fig. 2a) as follows. First, a 1100 bp DNA fragment containing the psbO gene plus 5′-flanking sequences was cloned from genomic DNA of WT T. vulcanus by PCR amplification and then sub-cloned into a plasmid pUC18 between the Xba I and BamH I sites. Then, a chloramphenicol resistance gene cassette (≈ 1000 bp) was ligated to the downstream of the stop codon of psbO at the position of BamH I and Kpn I. Finally, a fragment of 800 bp of 3′-flanking sequences downstream the psbO coding region was amplified by PCR and inserted into the plasmid pUC18 at the position of EcoR I and Kpn I site downstream the chloramphenicol resistance gene cassette. For creation of the PsbO-D158E/N/K site-directed mutants, the position of D158 was modified to the corresponding residues on the plasmid by using a Site-directed Mutagenesis Kit (TransGen Biotech).

Construction and identification of the site-directed mutations in the psbO gene. a Schematic diagram of the pUC18-psbO-CmR vector. A chloramphenicol resistance gene cassette was inserted in the downstream of the psbO coding region. b Agarose gel electrophoresis of the PCR product amplified from the genome of wide type (lane 2), wide type with a Cm-resistant cassette inserted at the end of psbO gene (lane 3), D158E (lane 4), D158N (lane 5), and D158K (lane 6) cells, respectively. Lane 1 is a DNA ladder marker
The plasmids containing the site-directed mutations were transformed into T. vulcanus cells by electroporation (Kirilovsky et al. 2004; Muhlenhoff and Chauvat 1996). Single colonies were selected on Cm-containing (4 μg mL−1) DTN agar plates. The mutant strains were segregated and maintained in the presence of 6 μg mL−1 chloramphenicol, but for analytical experiments the cells were cultured in the absence of antibiotics. After transformation and segregation, fragments were amplified from the genomic DNA of the transformants with primers P1 (5′-ACCAATCGTCAGCCTTTAGCAG-3′) and P2 (5′-GGATTGGGTATAAGGGTGCTGTG-3′) by PCR, and their sequences were analyzed to identify the correct mutations. For comparison, a strain transformed with the plasmid constructed above but contained no site-directed mutation was used as the WT in this study.
Quantification of PSII
Variable chlorophyll (Chl) fluorescence was recorded with a Dual PAM 100 Chl fluorometer (Walz, Germany) to estimate the content and donor side property of PSII in the cells. Estimation of relative PSII contents was performed by detecting the charge-separating PSII centers as described previously (Chu et al. 1994; Dilbeck et al. 2013; Nixon and Diner 1992). Cells were incubated in the dark for 10 min in the presence of 300 μM 2,6-dichloro-p-benzoquinone (DCBQ) and 300 μM K3Fe(CN)6 to fully oxidize QA−. Then the cells were incubated for 1 min in the presence of 20 μM 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU) to block the electron transfer from QA to QB, followed by addition of 10 mM hydroxylamine. Chl fluorescence was recorded within 20 s after the addition of hydroxylamine by applying 30 saturating actinic flashes.
The population of PSII centers capable of donating electrons was estimated by the variable Chl fluorescence induced by a single saturating flash in the presence of 20 μM DCMU.
Measurement of oxygen evolution
Oxygen evolution of the whole cells (15 μg Chl mL−1) and thylakoid membranes (15 μg Chl mL−1) was measured by a Hansatech Clark-type oxygen electrode under saturating white light at 30 °C with 0.5 mM DCBQ as an electron acceptor. Cells grown in the logarithmic phase were harvested by centrifugation at 1500×g for 5 min at room temperature and washed once with an HN buffer (10 mM Hepes–NaOH and 30 mM NaCl, pH 7.0). The cells were suspended in a medium containing 50 mM Mes-NaOH (pH 6.5), 15 mM CaCl2, 15 mM MgCl2 and 10% glycerol (w/v). In order to determine the activity of oxygen evolution under different pH conditions, the following buffers were used: Mes-NaOH for pH 5.0, 5.5, 6.0 and 6.5; Hepes–NaOH for pH 7.0 and 7.5. Chl concentration was determined by the method of Porra et al. (Porra et al. 1989).
Measurement of fluorescence relaxation kinetics
Flash-induced Chl a fluorescence relaxation kinetics was monitored with a dual-modulated fluorescence fluorometer (FL3500, Photon Systems Instruments, Brno, Czech Republic). Cells at the logarithmic grow phase were harvested and suspended in a fresh DTN medium to a final concentration of 5 μg Chl mL−1. Measurements were performed in either the absence or presence of DCMU. Dark-adapted (5 min) cells were illuminated with a single saturating flash and then the fluorescence was monitored by a series of weak actinic flashes. The fluorescence curves were normalized according to the equation Fv = (Ft − F0)/(FM − F0), where F0 is the basic fluorescence, FM is the maximum fluorescence, and Ft is the fluorescence yield at time t.
Photoinhibition and recovery measurements
Cells grown at 45 °C under 40 μmol photons m−2 s−1 were collected by centrifugation, washed with a buffer containing 50 mM MES-NaOH (pH 6.5), 15 mM CaCl2, 15 mM MgCl2 and 10% glycerol (w/v), and suspended in the same buffer at a concentration of 15 μg Chl mL−1. The harvested cells were subjected to photoinhibition at a light intensity of 1000 μmol photons m−2 s−1 (white light) for 1 h and then followed by recovery at a normal light intensity (40 μmol photons m−2 s−1) for 1 h in either the presence or absence lincomycin at a concentration of 200 μg mL−1 as used in a previous study (Ogami et al. 2012). During the whole illumination process, the temperature was maintained at 45 °C and the oxygen-evolving activity of cells was measured at the designated time.
Isolation of PSII core complexes
PSII core complexes were isolated from T. vulcanus as described previously (Shen and Inoue 1993; Shen and Kamiya 2000). Crude PSII particles were purified by solubilization of the thylakoid membranes with lauryldimethylamine N-oxide. The crude PSII particles were further solubilized with 1.2% n-Dodecyl-β-d-maltoside (β-DDM), followed by purification of the PSII dimer and monomer with a Q Sepharose High Performance column eluted with a linear gradient of NaCl from 150 to 300 mM.
SDS-PAGE, BN-PAGE and immunoblot analyses
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was carried out as described by Ikeuchi and Inoue (Ikeuchi and Inoue 1988) with a gel containing 16–22% polyacrylamide and 7.5 M urea. Samples were solubilized with 2% (w/v) lithium dodecyl sulfate, 60 mM dithiothreitol, and 60 mM Tris-HCl (pH 8.5) for 10 min at 60 °C prior to electrophoresis. After electrophoresis, the gels were stained with Coomassie Brilliant Blue R250.
Blue native polyacrylamide gel electrophoresis (BN-PAGE) was performed as described previously (Schagger and von Jagow 1991; Kawakami et al. 2007) with minor modifications. The PSII complexes were suspended in 25 mM Bis–Tris, pH 7.0, and 20% (w/v) glycerol to a concentration of 1 mg Chl mL−1. Then β-DDM was added to a final concentration of 0.1% (w/v), followed by 10 min incubation on ice. Samples of 2 μg Chl were loaded onto a gel with 3% acrylamide for the stacking gel and 3–12% acrylamide gradient for the separation gel.
For immunoblot analysis, cells or thylakoid membranes as well as crude PSII complex containing 4 μg Chl were loaded onto each lane and separated by SDS-PAGE. After electrophoresis, polypeptides in the gels were electrophoretically transferred to a nitrocellulose membrane and proteins were detected with antibodies raised against PsbO, PsbV, PsbU and Psb27 proteins.
Results
Generation of the mutants and their photoautotrophic growth properties
The three mutants PsbO-D158E, PsbO-D158N, and PsbO-D158K were verified by PCR and DNA sequence analysis after segregation. Figure 2b shows the PCR-amplified products with the genome of the PsbO-D158E, PsbO-D158N, PsbO-D158K mutants and the control strain as templates using the P1 and P2 primes. The results clearly confirmed that the three mutants were successfully constructed. These mutations were further confirmed by DNA sequencing of each PCR-amplified fragment from the respective strains (data not shown).
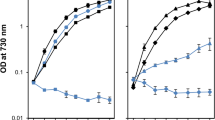
Photoautotrophic growth rates were measured at a light intensity of 40 µmol photons m−2 s−1, which showed that the growth of all of the three mutants was very similar to that of the WT strain (Fig. 3). Visible absorption spectroscopy at room temperature and 77 K fluorescence spectroscopy (not shown) showed that the pigment composition and the ratio of the two photosystems in the cells of these mutants were comparable with that of the wild type strain when the cells were cultured under standard conditions. These results suggest that substituting PsbO-D158 with E, K or N had no significant effect on the photosynthetic growth rate and pigment composition of the cells under normal growth conditions.

Photoautotrophic growth of the wild type strain (black) and PsbO-D158E (red), PsbO-D158N (blue), PsbO-D158K (green) mutants of T. vulcanus at 40 μmol photons m−2 s−1 monitored by OD730. Data are averages of three independent measurements
PSII accumulation and charge-separation activity of the mutant strains
The concentration of PSII centers capable of charge-separation was quantified by variable Chl fluorescence for the three PsbO-D158 mutants and the control strain with the acceptor side fully pre-oxidized and the donor side Mn cluster reduced and released by hydroxylamine. The hydroxylamine is a reductant that can remove the Mn4CaO5 cluster from PSII and donates electrons to YZ+ after light-induced oxidation of the P680 (Cheniae and Martin 1971). To induce the maximum amplitude of Chl fluorescence, DCMU was added to block the electron transfer from QA to QB. A trace of 30 saturating actinic flashes ensures all PSII centers to reach the high fluorescence yield state (Magyar et al. 2018). The ratio of Fv/F0 represents PSII centers that are capable of charge-separation (Chu et al. 1994; Dilbeck et al. 2013). As shown in Table 1, while the PsbO-D158E mutant was able to accumulate PSII as much as that of the WT strain, the amount of PSII centers in the D158N and D158K mutants were slightly lower than that in the WT strain.
After a single saturating flash without treatment of hydroxylamine and in the presence of DCMU, the variable Chl fluorescence Fv/F0 is proportional to the PSII centers that are able to form P680QA− and reflects the ability of electron donation of the PSII donor side (Dilbeck et al. 2013). In the PsbO-D158E, PsbO-D158N and PsbO-D158K mutants, the PSII centers forming P680QA− reached to 92%, 89%, 84% level of the WT, respectively (Table 1). This indicated that these three mutants were able to assemble the Mn cluster with a slightly lowered efficiency.
Oxygen-evolving activity of the PsbO-D158 mutants
The steady state oxygen-evolving activities of PsbO-D158E, PsbO-D158N and PsbO-D158K cells measured at pH 6.5 were reduced to 60%, 64% and 63% of the WT cells, respectively (Table 2). The lower rate of steady state O2 evolution was not proportional to the estimated concentration of PSII centers in these three mutants, suggesting that the decrease in the activity was caused by impairment of the water-splitting reaction itself instead of the reduction of the numbers of PSII centers.
In order to examine the effects of pH on the oxygen evolution rates in the mutant cells, the pH-dependence of oxygen-evolving activity of WT and the three mutant cells were determined. The results showed that both the WT and three mutant cells exhibited a maximum rate of oxygen evolution at pH 6.5 (Fig. 4). The inset in Fig. 4 showed the plots of the relative oxygen evolution rates against pH with the maximum rates at pH 6.5 as 100%. From pH 5.0–6.5, the oxygen-evolving activity of the three mutants displayed a similar rising tendency, which was slightly faster than that of WT. On the other hand, from pH 6.5–7.5, the oxygen-evolving activity of the three mutants showed a similar dependence on pH as that of the WT.

Dependence of oxygen evolution of whole cells on pH from wide type (black) and D158E (red), D158N (blue), D158K (green). The inset shows relative activities of oxygen evolution with the maximal activity observed at pH 6.5 as 100%. Data are presented as mean ± SD of seven independent measurements
The oxygen-evolving activities of isolated thylakoid membranes from the three mutants were 46%-49% of the WT value (Table 2). These results suggest that the oxygen-evolving activity was unstable and easy to lose during thylakoid membrane isolation from the mutant strains.
Kinetics of flash-induced chlorophyll a fluorescence relaxation
In order to characterize the properties of the donor and acceptor side electron transfer reactions in PSII, flash-induced fluorescence relaxation kinetics were measured from the whole cells in either the absence or presence of DCMU. Figure 5a shows the normalized decay curves of WT and the three mutant strains in the absence of DCMU, which reflects the oxidation of QA− by QB as well as by the S2-state of OEC (Vass et al. 1999). The decay curves were fitted into two exponential components and one hyperbolic component according to Vass et al. (Vass et al. 1999). The fast phase reflects QA− reoxidation by QB immediately following the flash, and the middle phase reflects QA− reoxidation by PQ molecules via an empty QB site due to the movement of PQ molecules into the QB site. The slow phase reflects QA− reoxidation via charge recombination with the S2 state. The result showed that the fast, middle and slow phases in the three mutants are similar to that in the wide type, suggesting that these mutations have no apparent effects on the electron transfer reactions of the acceptor side.

Kinetics of flash-induced chlorophyll fluorescence relaxation of whole cells from wide type (black), D158E (red), D158N (blue), D158K (green) recorded after 5 min dark adaptation in the absence (a) and presence (b) of 20 μM DCMU. Data are presented as mean ± SD of three independent measurements
It should be noted that, as shown in Fig. 5a, the decay curves of the flash-induced Chl fluorescence in the mutant and wide type cells showed a transient drop with a minimum at around 50–100 ms after the flash. This phenomenon was also observed in the thermophilic cyanobacterium T. elongatus, or in Synechocystis cells after 15 min microaerobic incubation before measurement (Deák et al. 2014), which was considered to reflect the transient changes of the redox level of the PQ pool (Deák and Vass 2008; Deák et al. 2014).
In the presence of DCMU, the fluorescence decay arises from charge recombination of QA− with the donor side due to the blockage of electron transfer to QB. This decay is much slower than that in the absence of DCMU, and the three mutants showed a slightly slower decay kinetics than that of the WT (Fig. 5b). This suggests that the charge recombination between QA− and the S2-state became slightly slower in the mutants than that in the WT, probably due to the inefficiency of S-state advancement in the three mutants compared with the WT. This implies the inhibition of S-state transition or a more stabilized S2-state in the three PsbO-D158 mutants.
Effects of PsbO-D158 mutations on photoinhibition and repair of PSII
In order to examine the photosensitivity and ability to recover from photoinhibition of the PsbO-D158 mutants, cells were exposed to high light (≈ 1000 μmol photons m−2 s−1) illumination for one hour followed by incubation under low light (≈ 40 μmol photons m−2 s−1) either in the absence or presence of lincomycin, which inhibits protein synthesis during the process of PSII repair and assembly. As shown in Fig. 6a, in the absence of lincomycin, the oxygen-evolving activity of WT cells decreased to 20% of the initial level after 40 min high light treatment (t1/2 ≈ 20 min), and the activity was kept constant during the next 20 min high light illumination. However, the oxygen-evolving activity of the three mutants decreased more rapidly to approximately 10% of the starting value after 40–60 min high light illumination (t1/2 ≈ 15 min). When the light intensity was switched to 40 μmol photons m−2 s−1, a light intensity used for cell growth, the oxygen-evolving activity of the WT cells recovered to 76% of the initial level within one hour, whereas the activity of the three mutants recovered slowly to only 30% during the one hour low light incubation.

Photosensitivity and the ability of recovery from photoinhibition in the absence (a) or presence (b) of lincomycin. Cells were exposed to high light (1000 μmol photons m−2 s−1) for 1 h, followed by recovery at normal light (40 μmol photons m−2 s−1) for another 1 h. Oxygen-evolving activity was measured at the indicated time point. Data are presented as mean ± SD of three independent measurements
Figure 6b shows the results of similar experiment as Fig. 6a in the presence of lincomycin. The oxygen-evolving activities decreased to 10% after one hour high light irradiation in both mutant and WT cells, but the activities of mutants decreased faster than that of WT strain with a t1/2 ≈ 15 min in comparison with a t1/2 ≈ 20 min for the WT cells. All strains could not recover from photoinhibition when the light intensity was reduced to the low level, indicating the requirement for de novo protein synthesis during the recovery process. These results indicate that the oxygen-evolving activities of the three PsbO-D158 mutants were more sensitive to high light treatment, and the efficiency of recovery from photoinhibition was lower than that of the WT cells.
Binding properties of the extrinsic proteins in the mutants
To further examine the effects of the site-directed mutations of PsbO-D158 on the structure and functions of PSII, thylakoid membranes and crude PSII particles were isolated from the three mutants (Shen and Inoue 1993; Shen and Kamiya 2000), and the presence and binding of the three extrinsic proteins (PsbO, PsbV and PsbU) were examined using the corresponding antibodies against these proteins. As shown in Fig. 7, the expression level of these proteins was not much affected in the cells of the three mutants in comparison with that of the WT cells, with only the level of PsbO and PsbU in the D158K mutant slightly lower than that in the WT or other mutant cells. However, in the isolated thylakoid membranes, only the PsbO and PsbV subunits were detected by Western-blotting, whereas the PsbU subunit was completely lost in the three mutant membranes. This result suggests that the mutations of PsbO-D158 disrupt or weaken the binding of PsbU to PSII remarkably, leading to its release during the isolation process of the thylakoid membranes. Moreover, the amount of PsbO and PsbV subunits were remarkably reduced or even disappeared in the crude PSII particles prepared from the three mutants (Fig. 7). These results suggest that the PsbO-D158 mutations remarkably reduced the binding affinity of the three extrinsic proteins to PSII, leading to their easy release either in the isolation of the thylakoid membranes or the crude PSII particles.

Binding properties of the extrinsic proteins in the PsbO-D158 mutants examined by Western-blotting analysis of cells, thylakoid membranes and crude PSII particles using antibodies against the PsbO, PsbU and PsbV proteins, respectively
As shown in Fig. 8a, the crude PSII particles from the D158E mutant were further purified by anion-exchange chromatography following β-DDM solubilization, from which two fractions were obtained. The protein compositions of these two fractions were analyzed by SDS-PAGE, which showed that both fractions retained the major PSII subunits including D1, D2, CP43 and CP47 but lost the three extrinsic subunits either significantly or completely (Fig. 8b). In the fraction 1, some phycobilisome subunits between PsbV and PsbU were retained. However, a new subunit with an apparent molecular weight of 11 kDa was found in both PSII fractions from the mutant (labeled ‘New’ in Fig. 8b). This subunit was identified to be Psb27 by Western-blotting analysis (Fig. 8c) which has been suggested to be a lumenal extrinsic protein involved in PSII assembly (Liu et al. 2011; Nowaczyk et al. 2006). BN-PAGE analysis showed that fraction-1 and 2 correspond to the PSII monomer and dimer, respectively (Fig. 8d), and both PSII complexes showed no oxygen-evolving activity at all. Similar results were obtained with PSII core complexes purified from the other two mutants D158N and D158K (Fig. 8e). These results are consistent with the weak binding of the extrinsic proteins in the mutants and also indicate the accumulation of PSII assembly intermediates in these mutants due to the incomplete binding of the extrinsic subunits.

Purification and analysis of PSII core complexes from the mutant cells. a Elution profile of β-DDM solubilized crude PSII particles from PsbO-D158E thylakoid membranes by a Q Sepharose High Performance column. Crude PSII particles were prepared with the two steps-lauryldimethylamine N-oxide solubilization method, and a linear gradient of 150–300 mM NaCl was used to separate the PSII monomers and dimers. The eluate was monitored at 280 nm. b SDS-PAGE analysis of PSII core complexes isolated from the PsbO-D158E mutant and WT. Lane 1: sample of fraction 1 from panel a; lane 2: sample of fraction 2 from panel a; lane 3: WT PSII dimer; lane 4: molecular weight marker. Each lane was loaded with 4 μg Chl of the samples. c Western-blotting analysis of PSII core complexes using an antibody against the Psb27 protein. Lane 1: sample of fraction 1 from panel a; lane 2: sample of fraction 2 from panel a. d BN-PAGE analysis of the PSII core complexes isolated from the PsbO-D158E mutant. Lane 1: sample of fraction 1 from panel a; lane 2: sample of fraction 2 from panel a. Each lane was loaded with 2 μg Chl of the samples. e SDS-PAGE analysis of PSII core dimer complexes isolated from WT (lane 2 and 6), PsbO-D158E (lane 3), PsbO-D158N (lane 4) and PsbO-D158K (lane 5) mutants. Lane 1 is the molecular weight marker
Discussions
D158 is a completely conserved residue in PsbO from cyanobacteria to higher plants, and its role has been studied in both cyanobacteria and higher plants. In the cyanobacterium Synechocystis sp. PCC 6803, alterations of D159 (equivalent to D158 in T. vulcanus) to N decreased the oxygen-evolving activity to 64% and 44% in the cells and isolated thylakoid membranes, respectively, in comparison with that of the WT (Burnap et al. 1994). These are very similar to the present results. The present results further showed that the decrease of the activity in the mutant cells can be ascribed to the weakening of the binding of the mutant PsbO to PSII, leading to the loss of PsbU in the isolated thylakoid membranes and almost complete loss of all the three extrinsic proteins in the purified PSII complexes. In agreement with this, the purified PSII cores were found to bind Psb27, a subunit that binds to the assembly intermediate of PSII before binding of the extrinsic proteins or with weaker binding of the extrinsic proteins (Liu et al. 2011; Nowaczyk et al. 2006). The weakening of binding of the extrinsic proteins in the D158 mutants also explains the previous results that a double mutant of Syenchocystis sp. PCC 6803 lacking PsbV and with the PsbO-D159 replaced by N could not grow photoautotrophically (Al-Khaldi et al. 2000), which is similar to the double deletion mutant lacking both PsbV and PsbO (Shen et al. 1995).
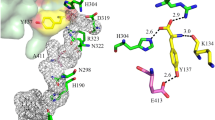
The high resolution structure of PSII showed that PsbO-D158 is located in the interface between D1-PsbO-D2, and its backbone oxygen is hydrogen-bonded directly to D1-R334 (Fig. 9). One of its carboxylate oxygen is hydrogen-bonded to PsbO-R162, which is in turn hydrogen-bonded to D2-A305 and D2-A306. The carboxylate oxygen of PsbO-D158 also interacts with PsbO-K160 and PsbO-K188 through hydrogen-bonds (Fig. 9). Mutation of the D158 residue will break these hydrogen-bonds, thereby affecting the binding of PsbO to PSII, leading to its weak binding and easy release in the mutant cells.

Structure of the environment of the PsbO-D158 residue. Yellow dashed lines indicate hydrogen-bonds. Color used: green, D1; Yellow, D2, violet purple, PsbO; magenta, CP43
The effects of mutations of PsbO-D158 on the functioning of PSII observed in the present study can be well explained by the weakened binding of this subunit in the mutant cells. Fluorescence decay kinetics showed that the electron transfer at the acceptor side was not affected by the mutations, whereas charge recombination between QA− and the donor side (S-states) was slower in the mutant than that in the WT. This can be explained by a more stable S-state intermediate present in some PSII centers that loss some of the extrinsic proteins in the mutant cells. This may be the cause for the loss of oxygen-evolving activity by around 35% caused by the mutations, since the number of PSII centers in the mutant cells did not change much as compared with the WT cells. Furthermore, the mutant cells became more sensitive to photoinhibition, and the recovery after photoinhibition was much more inefficient in the mutant cells than that in the WT cells. This is a phenomenon typical for PSII that is damaged at the donor side, since in such cases, the OEC will be unable to deliver electrons to Tyr-Z+ and P680+ rapidly or the side-path electron transfer will be affected.
The PsbO-D158 residue is also located close to a hydrogen-bond network starting from the Mn4CaO5-cluster to the bulk solution mediated by the Cl-1 binding site (Umena et al. 2011; Shen 2015). This network has been suggested to function in transporting protons generated by the water-splitting reaction toward the outside solution, and the D1-R334 residue is directly involved in this hydrogen-bond network (Service et al. 2014; Shen 2015). In order to examine the possible effects of mutations of PsbO-D158 on the proton transfer through this hydrogen-bond network, we compared the pH-dependencies of oxygen evolution between WT and mutant cells, since it is expected that if the hydrogen-bond network is affected, higher pH would be required to facilitate the proton egress and therefore the oxygen evolution would be higher at higher pH in the mutant cells than that in the WT cells. However, the pH-dependencies were not much changed in the mutant cells than that in the WT cells. This may also be due to the weak binding of the extrinsic proteins in the mutant cells, resulting in a partial loss of the proteins and therefore the opening of the Mn4CaO5-site, leading to an easy proton egress even without the hydrogen-bond network.
In spinach PSII, mutations of the corresponding PsbO-D157 decreased the oxygen-evolving activity to around 30% based on the results of in vitro reconstitution experiments, and a similar effect on the charge recombination between QA− and the donor side was observed (Popelkova et al. 2009; Roose et al. 2010). However, the binding of the mutant PsbO to PSII was not affected upon reconstitution of the mutant PsbO proteins to purified PSII cores (Roose et al. 2010). This is different from the in vivo results observed in the present and previous studies, and may be due to the non-functional binding of the mutant PsbO protein to the purified PSII core in vitro.
In conclusion, the highly conserved PsbO-D158 residue maintains the proper binding of PsbO to PSII; its mutations weaken binding of PsbO as well as PsbU and PsbV, thereby causing the decrease of the oxygen-evolving activity and increase of the sensitivity to high light. In the mutant cells, the amount of assembly intermediate of PSII that binds Psb27 was increased. Since similar results were observed with all the three mutants irrespective of the replacement of the negatively charged residue (D) by either negatively charged (E), positively charged (K) or neutral (N) residues, our results indicate that not only the chemical properties but also the volume of side chains of PsbO-D158 have an important role in maintaining the proper binding of PsbO to PSII and hence the proper assembly and functioning of PSII.
Abbreviations
- BN-PAGE:
-
Blue native polyacrylamide gel electrophoresis
- CmR :
-
Chloramphenicol-resistant gene
- Chl:
-
Chlorophyll
- DCBQ:
-
2,6-Dichloro-p-benzoquinone
- DCMU:
-
3-(3,4-Dichlorophenyl)-1,1-dimethylurea
- DDM:
-
N-Dodecyl-β-d-maltoside
- MSP:
-
Manganese-stabilizing protein
- OEC:
-
Oxygen-evolving complex
- PCR:
-
Polymerase chain reaction
- PSII:
-
Photosystem II
- WT:
-
Wild type
References
Al-Khaldi SF, Coker J, Shen JR, Burnap RL (2000) Characterization of site-directed mutants in manganese-stabilizing protein (MSP) of Synechocystis sp. PCC6803 unable to grow photoautotrophically in the absence of cytochrome c-550. Plant Mol Biol 43:33–41
Bondar AN, Dau H (2012) Extended protein/water H-bond networks in photosynthetic water oxidation. Biochim Biophys Acta 1817:1177–1190
Bricker TM (1992) Oxygen evolution in the absence of the 33-kilodalton manganese-stabilizing protein. Biochemistry 31:4623–4628
Bricker TM, Roose JL, Fagerlund RD, Frankel LK, Eaton-Rye JJ (2012) The extrinsic proteins of photosystem II. Biochim Biophys Acta 1817:121–142
Burnap RL, Sherman LA (1991) Deletion mutagenesis in Synechocystis sp. PCC6803 indicates that the Mn-stabilizing protein of photosystem II is not essential for O2 evolution. Biochemistry 30:440–446
Burnap RL, Shen JR, Jursinic PA, Inoue Y, Sherman L (1992) Oxygen yield and thermoluminescence characteristics of a cyanobacterium lacking the manganese-stabilizing protein of photosystem II. Biochemistry 31:7404–7410
Burnap RL, Qian M, Shen JR, Inoue Y, Sherman LA (1994) Role of disulfide linkage and putative intermolecular binding residues in the stability and binding of the extrinsic manganese-stabilizing protein to the photosystem II reaction center. Biochemistry 33:13712–13718
Cheniae GM, Martin IF (1971) Effects of hydroxylamine on photosystem II: I. Factors affecting the decay of O2 evolution. Plant Physiol 47:568–575
Chu HA, Nguyen AP, Debus RJ (1994) Site-directed photosystem II mutants with perturbed oxygen-evolving properties. 1. Instability or inefficient assembly of the manganese cluster in vivo. Biochemistry 33:6137–6149
De Las RJ, Barber J (2004) Analysis of the structure of the PsbO protein and its implications. Photosyn Res 81:329–343
Deák Z, Vass I (2008) Oscillating yield of flash-induced chlorophyll fluorescence decay in intact cells of Thermosynechococcus elongatus. In: Allen JF, Gantt E, Golbeck JH, Osmond B (eds) Photosynthesis, energy from the sun. Springer, Dordrecht, pp 573–576
Deák Z, Sass L, Kiss E, Vass I (2014) Characterization of wave phenomena in the relaxation of flash-induced chlorophyll fluorescence yield in cyanobacteria. Biochim Biophys Acta 1837:1522–1532
Del Val C, Bondar AN (2017) Charged groups at binding interfaces of the PsbO subunit of photosystem II: a combined bioinformatics and simulation study. Biochim Biophys Acta 1858:432–441
Dilbeck PL, Bao H, Neveu CL, Burnap RL (2013) Perturbing the water cavity surrounding the manganese cluster by mutating the residue D1-valine 185 has a strong effect on the water oxidation mechanism of photosystem II. Biochemistry 52:6824–6833
Enami I, Okumura A, Nagao R, Suzuki T, Iwai M, Shen JR (2008) Structures and functions of the extrinsic proteins of photosystem II from different species. Photosynth Res 98:349–363
Henmi T, Miyao M, Yamamoto Y (2004) Release and reactive-oxygen-mediated damage of the oxygen-evolving complex subunits of PSII during photoinhibition. Plant Cell Physiol 45:243–250
Ifuku K (2015) Localization and functional characterization of the extrinsic subunits of photosystem II: an update. Biosci Biotechnol Biochem 79:1223–1231
Ikeuchi M, Inoue Y (1988) A new photosystem II reaction center component (4.8 kDa protein) encoded by chloroplast genome. FEBS Lett 241:99–104
Kawakami K, Iwai M, Ikeuchi M, Kamiya N, Shen JR (2007) Location of PsbY in oxygen-evolving photosystem II revealed by mutagenesis and X-ray crystallography. FEBS Lett 581:4983–4987
Kirilovsky D, Roncel M, Boussac A, Wilson A, Zurita JL, Ducruet JM, Bottin H, Sugiura M, Ortega JM, Rutherford AW (2004) Cytochrome c550 in the cyanobacterium Thermosynechococcus elongatus: study of redox mutants. J Biol Chem 279:52869–52880
Liu H, Huang RY, Chen J, Gross ML, Pakrasi HB (2011) Psb27, a transiently associated protein, binds to the chlorophyll binding protein CP43 in photosystem II assembly intermediates. Proc Natl Acad Sci USA 108:18536–18541
Lorch S, Capponi S, Pieront F, Bondar AN (2015) Dynamic carboxylate/water networks on the surface of the PsbO subunit of photosystem II. J Biol Chem 119:12172–12181
Magyar M, Sipka G, Kovács L, Ughy B, Zhu Q, Han G, Špunda V, Lambrev PH, Shen JR, Garab G (2018) Rate-limiting steps in the dark-to-light transition of Photosystem II - revealed by chlorophyll-a fluorescence induction. Sci Rep 8:2755
Mayes SR, Cook KM, Self SJ, Zhang Z, Barber J (1991) Deletion of the gene encoding the photosystem II 33 kDa protein from Synechocystis sp PCC 6803 does not inactivate water-splitting but increases vulnerability to photoinhibition. Biochim Biophys Acta 1060:1–12
Mayfield SP, Bennoun P, Rochaix JD (1987) Expression of the nuclear encoded OEE1 protein is required for oxygen evolution and stability of photosystem II particles in Chlamydomonas reinhardtii. EMBO J 6:313–318
Miyao M, Murata N (1984) Role of the 33-kDa polypeptide in preserving Mn in the photosynthetic oxygen-evolution system and its replacement by chloride ions. FEBS Lett 170:350–354
Motoki A, Usui M, Shimazu T, Hirano M, Katoh S (2002) A domain of the manganese-stabilizing protein from Synechococcus elongatus involved in functional binding to photosystem II. J Biol Chem 277:14747–14756
Muhlenhoff U, Chauvat F (1996) Gene transfer and manipulation in the thermophilic cyanobacterium Synechococcus elongatus. Mol Gen Genet 252:93–100
Nixon PJ, Diner BA (1992) Aspartate 170 of the photosystem II reaction center polypeptide D1 is involved in the assembly of the oxygen-evolving manganese cluster. Biochemistry 31:942–948
Nowaczyk MM, Hebeler R, Schlodder E, Meyer HE, Warscheid B, Rogner M (2006) Psb27, a cyanobacterial lipoprotein, is involved in the repair cycle of photosystem II. Plant Cell 18:3121–3131
Offenbacher AR, Polander BC, Barry BA (2013) An intrinsically disordered photosystem II subunit, PsbO, provides a structural template and a sensor of the hydrogen-bonding network in photosynthetic water oxidation. J Biol Chem 288:29056–29068
Ogami M, Boussac A, Sugiura M (2012) Deactivation processes in PsbA1-Photosystem II and PsbA3-Photosystem II under photoinhibitory conditions in the cyanobacterium Thermosynechococcus elongatus. Biochim Biophys Acta 1817:1322–1330
Philbrick JB, Diner BA, Zilinskas BA (1991) Construction and characterization of cyanobacterial mutants lacking the manganese-stabilizing polypeptide of photosystem II. J Biol Chem 266:13370–13376
Popelkova H, Betts SD, Lydakis-Symantiris N, Im MM, Swenson E, Yocum CF (2006) Mutagenesis of basic residues R151 and R161 in manganese-stabilizing protein of Photosystem II causes inefficient binding of chloride to the oxygen-evolving complex. Biochemistry 45:3107–3115
Popelkova H, Commet A, Yocum CF (2009) Asp157 is required for the function of PsbO, the photosystem II manganese stabilizing protein. Biochemistry 48:11920–11928
Porra RJ, Thompson WA, Kriedemann PE (1989) Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim Biophys Acta 975:384–394
Qian M, Al-Khaldi SF, Putnam-Evans C, Bricker TM, Burnap RL (1997) Photoassembly of the photosystem II Mn4 cluster in site-directed mutants impaired in the binding of the manganese-stabilizing protein. Biochemistry 36:15244–15252
Roose JL, Yocum CF, Popelkova H (2010) Function of PsbO, the photosystem II manganese-stabilizing protein: probing the role of aspartic acid 157. Biochemistry 49:6042–6051
Roose JL, Frankel LK, Mummadisetti MP, Bricker TM (2016) The extrinsic proteins of photosystem II: update. Planta 243:889–908
Schagger H, von Jagow G (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem 199:223–231
Service RJ, Hillier W, Debus RJ (2014) Network of hydrogen bonds near the oxygen-evolving Mn4CaO5 cluster of photosystem II probed with FTIR difference spectroscopy. Biochemistry 53:1001–1017
Shen JR (2015) The structure of photosystem II and the mechanism of water oxidation in photosynthesis. Annu Rev Plant Biol 66:23–48
Shen JR, Inoue Y (1993) Binding and functional properties of two new extrinsic components, cytochrome c-550 and a 12 kDa protein, in cyanobacterial photosystem II. Biochemistry 32:1825–1832
Shen JR, Kamiya N (2000) Crystallization and the crystal properties of the oxygen-evolving photosystem II from Synechococcus vulcanus. Biochemistry 39:14739–14744
Shen JR, Burnap RL, Inoue Y (1995) An independent role of cytochrome c-550 in cyanobacterial photosystem II as revealed by double-deletion mutagenesis of the psbO and psbV genes in Synechocystis sp. PCC 6803. Biochemistry 34:12661–12668
Suga M, Akita F, Hirata K, Ueno G, Murakami H, Nakajima Y, Shimizu T, Yamashita K, Yamamoto M, Ago H, Shen JR (2015) Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 517:99–103
Umena Y, Kawakami K, Shen JR, Kamiya N (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473:55–60
Vass I, Kirilovsky D, Etienne AL (1999) UV-B radiation-induced donor- and acceptor-side modifications of photosystem II in the cyanobacterium Synechocystis sp. PCC 6803. Biochemistry 38:12786–12794
Vinyard DJ, Ananyev GM, Dismukes GC (2013) Photosystem II: the reaction center of oxygenic photosynthesis. Annu Rev Biochem 82:577–606
Yi X, McChargue M, Laborde S, Frankel LK, Bricker TM (2005) The manganese-stabilizing protein is required for photosystem II assembly/stability and photoautotrophy in higher plants. J Biol Chem 280:16170–16174
Acknowledgements
This work was supported by National Key R&D Program of China (2017YFA0503700); National Natural Science Foundation of China grant (31470339); A Strategic Priority Research Program of CAS (XDB17000000); a Key Research Program of Frontier Sciences, CAS, Grant (QYZDY-SSW-SMC003).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhu, Q., Yang, Y., Xiao, Y. et al. Function of PsbO-Asp158 in photosystem II: effects of mutation of this residue on the binding of PsbO and function of PSII in Thermosynechococcus vulcanus. Photosynth Res 146, 29–40 (2020). https://doi.org/10.1007/s11120-020-00715-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-020-00715-0