
Abstract
The absolute amount of plastocyanin (PC), ferredoxin-NADP+-oxidoreductase (FNR), hydrogenase (HYDA1), and ferredoxin 5 (FDX5) were quantified in aerobic and anaerobic Chlamydomonas reinhardtii whole cells using purified (recombinant) proteins as internal standards in a mass spectrometric approach. Quantified protein amounts were related to the estimated amount of PSI. The ratios of PC to FNR to HYDA1 to FDX5 in aerobic cells were determined to be 1.4:1.2:0.003:0. In anaerobic cells, the ratios changed to 1.1:1.3:0.019:0.027 (PC:FNR:HYDA1:FDX5). Employing sodium dithionite and methyl viologen as electron donors, the specific activity of hydrogenase in whole cells was calculated to be 382 ± 96.5 μmolH2 min−1 mg−1. Importantly, these data reveal an about 70-fold lower abundance of HYDA1 compared to FNR. Despite this great disproportion between both proteins, which might further enhance the competition for electrons, the alga is capable of hydrogen production under anaerobic conditions, thus pointing to an efficient channeling mechanism of electrons from FDX1 to the HYDA1.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Due to its versatility and its plant- and animal-like characteristics, the unicellular green alga Chlamydomonas reinhardtii is an important model organism for the elucidation of eukaryotic photosynthesis, chloroplast biology, flagellar biosynthesis and function, and reactions to abiotic stress. In natural environments, the soil-thriving eukaryote often experiences periods of anoxia, when during the night cellular respiration consumes the photosynthetically generated oxygen. C. reinhardtii has an intricate anaerobic metabolism and reacts to hypoxic and anaerobic conditions with a strong up-regulation of various genes and pathways. One of the key processes in response to anaerobiosis is hydrogen production. In nature, it is released for a fleeting period after dark incubation of the alga and its re-exposure to light and serves as a valve system to protect the electron transport chain (ETC) from over-reduction (Melis and Happe 2001). Since the development of the two-stage method for sustained hydrogen production by Melis et al. (2000), there has been an increased interest towards hydrogen as a direct product of photosynthesis and therefore a renewable and environmentally safe energy source.
In green algae, the reduction of protons to hydrogen is mediated by an FeFe-type hydrogenase, which contains a catalytic site (known as the H-cluster) consisting of a [4Fe–4S] center linked to a [2Fe–2S] sub-cluster (Peters et al. 1998; Mulder et al. 2010). While the [4Fe–4S] domain is synthesized and inserted independently by a generalized host-cell system, the synthesis and insertion of the 2Fe sub-cluster is performed by a maturation machinery (the maturation enzymes HYDE, HYDF, and HYDG) (Mulder et al. 2009, 2010). The enzyme is matured exclusively in the chloroplast, both with the [4Fe–4S] as well as with the [2Fe–2S] sub-cluster, and thus the chloroplast assembly system cannot be replaced by the cytosolic one (Sawyer et al. 2017). C. reinhardtii possesses two hydrogenase isoforms: HYDA1 (Happe and Kaminski 2002) and HYDA2 (Forestier et al. 2003). The main isoform, which accounts for approximately 75% of the H2 photoproduction (Meuser et al. 2012) is HYDA1, which is encoded in the nucleus and localized in the chloroplast. The hydrogenase of eukaryotic microalgae exhibits high specific activity of above 700 μmolH2 min−1 mg−1 (Roessler and Lien 1984b; Happe and Naber 1993; Florin et al. 2001; Girbal et al. 2005; Kamp et al. 2008) and is inactivated by oxygen (Ghirardi et al. 1997; Rupprecht et al. 2006).
Hydrogen production is coupled to the oxidation of ferredoxin (FDX1) (Winkler et al. 2009), which is reduced either by electrons from the photosynthetic ETC or by the fermentative pyruvate-ferredoxin oxidoreductase (Hemschemeier and Happe 2011). Electrons fed into the ETC can originate either from the light-induced water oxidation at photosystem II (PSII) [main electron source for H+-reduction (Volgusheva et al. 2013; Steinbeck et al. 2015)] or from degradation of starch, glucose, or acetate and subsequent reduction of the plastoquinone pool by the type-II NADH dehydrogenase (NDH-2) (Mus et al. 2005). FDX1 is a branch point in electron flow from photosystem I (PSI) to many competing metabolic pathways such as CO2 fixation, hydrogen production, nitrite, sulfite, and thioredoxin reduction, glutamate synthesis, and cyclic electron flow (CEF) around PSI. In vitro experiments showed that with 85% of the electrons directed to the NADPH production under aerobic conditions, the enzyme ferredoxin-NADP+-oxidoreductase (FNR) is the main downstream electron acceptor (Yacoby et al. 2011; Eilenberg et al. 2016). One reason for this is the spatial localization of the FNR at PSI (Andersen et al. 1992; Iwai et al. 2010), which allows the direct interaction of FNR with the PSI-reduced FDX1 at the expense of other competing reductive reactions (Yacoby et al. 2011; Eilenberg et al. 2016; Mosebach et al. 2017). In vivo, however, in the initial 120 s during a shift from dark anoxia to light, the PGRL1-dependent CEF is the main competitor of the hydrogenase for reduced ferredoxin, followed later by the Calvin cycle (Godaux et al. 2015). The electron flow towards hydrogenase and/or the PSI-CEF were shown to be crucial for the reactivation of the Calvin cycle and thus photosynthesis and growth of the cells after exposure to anoxia (Godaux et al. 2015).
Another protein that is strongly up-regulated by anoxia is FDX5 (Jacobs et al. 2009). In C. reinhardtii, thirteen ferredoxin genes have been identified (Yang et al. 2015); six of them have been studied in detail (FDX1-6) [for a review (Sawyer and Winkler 2017)]. They are differentially regulated in response to changing environmental conditions and nutrient supply, which together with their redox properties suggest specialized function in specific metabolic pathways (Terauchi et al. 2009). With more than 95% of the total FDX-transcript pool, the FDX1 (encoded by petf) is the most abundant isoform. FDX5 was shown to function in the desaturation of fatty acids and maintenance of the thylakoid membrane composition and therefore to have a crucial role in dark metabolism (Yang et al. 2015). A possible specificity of FDX5 for the hydrogenase could be disproven (Jacobs et al. 2009). FDX5 is furthermore not able to photoreduce NADP+ via the FNR (Jacobs et al. 2009). The fdx5 gene is induced not only in oxygen but also in copper deficiency, as well as in the presence of nickel, concomitant with the protein level (Terauchi et al. 2009; Mus et al. 2007; Jacobs et al. 2009), since the expression of fdx5 is controlled by transcriptional factor CRR1, which regulates the expression of target genes as response to oxygen and copper deficiency (Lambertz et al. 2010). Furthermore, sulfur starvation induces fdx5 expression independently from anoxia treatment (Zalutskaya et al. 2017).
Determination of the stoichiometry of key photosynthetic proteins will give important insights into the partitioning of photosynthetic electrons and will contribute to the understanding of the corresponding pathways and their adjustment under environmental changes. The most accurate method for absolute quantification (AQ) of proteins is utilization of the stable isotope dilution (Ong and Mann 2005).
This study aimed at the quantification of the absolute amount of PC, FNR, and the anaerobically induced/activated FDX5 and HYDA1 in aerobic and anaerobic cells. Our strategy was to produce and purify the proteins of interest and to introduce them at known concentration to whole cells extracts using differential isotope labeling for absolute mass spectrometric quantification.
Results and discussion
The study aimed at the mass spectrometry-based AQ of key proteins of the ETC–PC, FNR, and the anaerobically induced HYDA1 and FDX5.
Preparation of the internal standards
A straightforward way to perform mass spectrometry-based AQ is the use of synthetic, stable isotope labeled peptides as internal standards, introduced at a known concentration to the sample of interest during digestion [the AQUA method (Gerber et al. 2003; Kirkpatrick et al. 2005)]. They are chemically identical to their native proteolytic counterparts with respect to chromatographic co-elution, ionization efficiency, and relative distribution of fragment ions, but differ in their mass-to-charge (m/z) ratio and are easily detected and distinguished in a tandem mass spectrometry measurement.
However, while the AQUA method results in an accurate quantification when the sample is trypsin-digested in-solution, in-gel digestion leads to destruction of the quantitative integrity of the sample due to losses of individual peptides during peptide recovering from the polyacrylamide gel (Havlis and Shevchenko 2004). Furthermore, variations in the efficiency of the tryptic digestion are not accounted for. Therefore, to increase the accuracy of the AQ approach we expressed and purified recombinant Chlamydomonas PC, HYDA1, and FDX5 and isolated native FNR and added them as internal standards to isotopically labeled whole cell extracts before gel separation and tryptic digestion (Fig. 1). Thus, the internal standards are processed together with the native proteins.

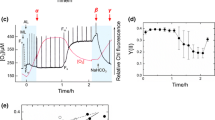
Schematic overview of the procedure for absolute protein quantification of HYDA1, FNR, PC, and FDX5 via stable 15N isotopic labeling. a Heterologous expression of HYDA1, PC, and FDX5, isolation of the native FNR, and their purification. The His-tagged HYDA1 and the Strep-tagged FDX5 were purified via affinity chromatography, whereas for purification of PC and FNR anion exchange chromatography (AEC) was employed. Protein concentration was determined using UV/Vis spectrometry, except for the hydrogenase, whose concentration was determined by BCA assay. b 15N-labeled C. reinhardtii cells were incubated either aerobically or anaerobically for 4 h, while the in vitro H2 production of the culture was measured. c SDS-PAGE gel of the purified recombinant HYDA1 and protein ladder. d Three 15N-labeled aerobic and three anaerobic samples from the same culture (technical replicates) were mixed with the purified HYDA1, PC, FDX5, and FNR and separated via SDS-PAGE. Three bands per sample were cut, digested with trypsin, and analyzed by mass spectrometry. Quantification was performed by qTrace (Terashima et al. 2010) after identification by OMSSA (Geer et al. 2004) and X! Tandem (Craig and Beavis 2004)
All proteins besides HYDA1 were expressed and purified according to already established protocols described in "Materials and methods" (Fig. 1а). After induction with IPTG, the hydrogenase was expressed under anaerobic conditions created by aeration with nitrogen and purified semi-anaerobically with buffers that have been purged with argon to remove the oxygen. The presence of oxygen leads not only to the irreversible inactivation of the enzyme, but also to its fast degradation. For this reason, the 15N-labeled whole cell extracts (Fig. 1b) were kept in the presence of PMSF as well as plant-specific protease inhibitors.
Furthermore, the purified proteins were analyzed with respect to their tryptic digestibility, chromatographic behavior of the resulting peptides, ionization efficiency, and quality of fragmentation spectra. The analysis of tryptically digested proteins resulted in the unambiguous identification of some of the in silico predicted tryptic peptides (Table 1).
An important aspect of the AQ strategy is the determination of the exact protein concentration. FDX5, HYDA1, and PC were expressed heterologously in E. coli in the presence of copper (PC) or iron (FXD5 and HYDA1) (see "Materials and methods"). The concentration of the purified apoproteins FDX5, PC, and the native FNR was determined spectrophotometrically, which accounts only for the concentration of the proteins of interest and eliminate the influence of possible contamination. The concentration of the hydrogenase was determined by the indiscriminative BCA assay. However, the protein extract exhibits only one strong band on the SDS-PAGE gel corresponding to the HYDA1 (Fig. 1c) and has therefore very good purity.
In-gel quantification of PC, FNR, FDX5, and HYDA1 in aerobic and anaerobic whole cells
For the AQ experiment, three 15N-labeled, independent biological replicates were measured, each with three technical repeats, employing the same batch of purified proteins (see "Materials and methods"). 10 µg chlorophyll per sample was supplemented with the purified proteins before gel separation. Corresponding light and heavy peptides yield distinct isotopic envelopes and were detected for all four proteins of interest (Fig. 2).

LC–MS/MS analysis of the 15N-labeled anaerobic whole cells mixed with the purified 14N HYDA1, FNR, PC, and FDX5. A spectrum for a peptide belonging to a FNR, b PC, c FDX5, and d HYDA1 is shown
Quantified protein amounts (Table 2) were related to the number of chlorophyll molecules associated with PSI in the thylakoid membrane (see "Calculation," "Materials and methods"). To do this, we determined the ratio of PSI/PSII spectroscopically, which resulted in a value of 1.3 for strain CC-400. The number of chlorophylls per PSI has been reported to be in the range of 220–320 Chl molecules for cells being in state I or state II, respectively (Nawrocki et al. 2016). Thus, absolute amounts of quantified HYDA1, FDX5, FNR, and PC are presented as a molar ratio to 249 chlorophylls for aerobic cells (considering 440 Chl molecules in total and taking into account the 1:1.3 ratio of PSII/PSI) and 319 chlorophylls (about 70 Chl molecules more than in state I) for anaerobic cells (Fig. 3).

Protein-to-PSI ratio. Purified 14N-labeled PC, FNR, HYDA1, and FDX5 were added to 15N-labeled, aerobic (AR, gray), and anaerobic (AN, black) whole cell extracts at a known concentration and separated by SDS-PAGE. After band excision and digestion with trypsin, the peptides were analyzed by mass spectrometry. Quantified protein amounts were related to the chlorophyll molecules associated to PSI (249 for aerobic and 319 for anaerobic conditions). Values are mean ± SD of at least three independent biological, with three technical replicates. The ratios of PC:FNR:HYDA1:FDX5 in aerobic cells were 1.4:1.2:0.003:0 and in anaerobic cells 1.1:1.3:0.019:0.027
For PC, a ratio of 1.4/249 Chl molecules under aerobic conditions was measured, which decreased to 1.1 molecules/319 Chl in anaerobic cells (Fig. 3). Plastocyanin concentration for different organisms has been reported in several publications. For spinach chloroplasts, Graan and Ort (1984) reported slightly more than two molecules PC for each PSI reaction center, whereas Burkey et al. (1996) mentioned a molar ratio of 3 to 4 PC/PSI, not only in spinach, but also in barley. In C. reinhardtii and spinach a PC-to-P700 ratio of 2 to 2.5 molecules has been documented [Katoh 1977, cited in (Lien and Pietro 1979)]. Plastocyanin was shown furthermore to react with the PsaF as the sole interaction partner from the PSI protein complex in a 1:1 stoichiometry (Hippler et al. 1997). In conclusion, our determined ratios indicate a slightly lower PC-to-PSI ratio compared to the ratios seen before.
For tobacco leaves, a PC/PSI molar ratio of 4–5 within leaves with high CO2 assimilation rates was reported (Schottler et al. 2004), which decreases to values below one in leaves with reduced carbohydrate export and thus reduced carbon fixation. The authors postulated that the electron flux is adjusted to the carbon assimilation rate via downregulation of the PC/Cyt b6f-to-PSI ratio as one way to protect PSI from over-reduction and formation of reactive oxygen species (ROS). In C. reinhardtii, the enzymes of the Calvin–Benson cycle do not change in amount under anaerobic conditions induced by argon bubbling (Terashima et al. 2010) contrary to sulfur deficiency-induced anaerobiosis. Nevertheless, when oxygen is removed by argon, cellular respiration is restricted, which leads to an excess of reducing equivalents. Thus, the diminished PC ratio is in accordance with the observation of Schoettler et al. (2004), also considering that cells face limitations not only to O2, but also to CO2, when flushed with argon. In addition, limited oxygen availability triggers the expression of several genes controlled by the transcriptional factor copper response regulator (CRR1), which responds also to copper (Cu) deficiency (Quinn et al. 2000, 2002; Eriksson et al. 2004). Since plastocyanin is a copper-binding protein, its reduced amount under anaerobic condition might reflect disturbed Cu homeostasis. Notably, under anoxic conditions, the induction of cytochrome c6 is observed (Quinn et al. 2000), possibly adjusting for the loss of PC.
Böhme (1978) reported a FDX1-to-FNR-to-PC ratio of 5:3:4 per P700 in spinach chloroplasts quantified by quantitative immunoelectrophoresis. Here, we report a FNR-to-PSI ratio of 1.2 with almost no change under anoxia (Fig. 3) and thus almost a 1:1 stoichiometry to PC under both conditions, here supporting the previous finding (Böhme 1978).
FDX5 has been reported to interact with two fatty acid desaturases (CrΔ4FAD and CrΔFAD6) (Yang et al. 2015). It is involved in maintenance of the proper fatty acid desaturation state and thus thylakoid membrane composition in dark oxic conditions (Yang et al. 2015). The protein assures the normal activity of many cellular processes that are dependent on a normal membrane composition; thus, its loss leads to seriously compromised photosynthesis and respiration and is lethal for the cells in the dark with little impact on daytime growth. Many studies, including this one, report a strong up-regulation of the protein under hypoxic or anaerobic conditions (Jacobs et al. 2009; Lambertz et al. 2010; Mus et al. 2007). However, anaerobiosis is not the only condition that triggers an increase in the levels of the redox carrier. fdx5 gene expression is controlled by transcriptional factor CRR1, which regulates the expression of target genes as response not only to oxygen, but also to copper deficiency (Lambertz et al. 2010). Zalutskaya et al. (2017) reported further factors that influence the expression of fdx5 gene under sulfur deprivation: (1) it is induced by sulfur starvation independently of anoxia treatment; (2) it is negatively regulated by nitric oxide in a truncated hemoglobin 1 (THB1)-dependent pathway, (3) the Ser/Thr kinase SNRK2.1 is necessary for full activation of the gene, and (4) its expression does not depend on CRR1 in sulfur-deficient aerobic cells. In this study, FDX5 was identified and quantified only in anaerobic cells at a ratio of 0.027 to PSI (Fig. 3) and thus a 0.028:1 stoichiometry to PC.
The hydrogenase HYDA1 was identified and quantified in the aerobic samples at a ratio of 0.003 and in the anaerobic sample at a ratio of 0.019 per PSI Chl (Fig. 3). These results indicate that for competition of electrons from FDX1, HYDA1 is about 70-fold less abundant than its competitor FNR under anaerobic conditions. In aerobic cells, the high standard deviation of the ratio (± 0.003) indicates that expression of the protein is variable in the different biological samples. The presence of the hydrogenase in aerobic cells might seem unexpected. For many years now, it has been accepted that the irreversible inactivation of the enzyme by oxygen is one of the main obstacles for a large-scale hydrogen photoproduction, since hydrogen production is a transient phenomenon and ceases within minutes after the onset of light (Ghirardi et al. 1997; Ghirardi 2015; Rupprecht et al. 2006).
The mechanism of the O2-dependent inactivation of the enzyme involves the initial attack of O2 on the vacant position of the distal (to the cubane domain) Fe within the 2Fe sub-cluster (Stripp et al. 2009; Lambertz et al. 2011). According to the X-ray absorption measurements, performed in the latter studies, formation of ROS [probably OOH radical or H2O2 (Bruska et al. 2011; Finkelmann et al. 2014; Hong and Pachter 2012)] follows, which diffuses to the [4Fe–4S] sub-cluster and destroys it. Another study, however, reports a different mechanism of degradation of the H-cluster: O2 interacts and destroys the 2Fe sub-cluster through a series of reactions, which depend on the initial enzyme reduction/oxidation state, thus leaving an inactive [4Fe–4S]-containing form of the hydrogenase. The latter can be reactivated in vitro by the 2Fe sub-cluster maturation machinery or is eventually destructed after prolonged exposure to high O2 concentrations (Swanson et al. 2015). Kubas et al. (2017) reported that differences in the proton/electron availability, required for completely reducing the O2 to water, may account for some of this variability described in the above studies. According to the authors, four electron/proton transfer steps are needed to reduce O2 to water, the third step being the limiting one; thus, water production is competing with formation of ROS.
Furthermore, recent studies indicate that the fast drop of hydrogen production after illumination is due to the competition for reducing equivalents with other metabolic pathways rather than to irreversible O2-dependent degradation of the hydrogenase (Godaux et al. 2015; Liran et al. 2016).
It has been reported that in the initial period of anaerobiosis nearly all of hydrogenase activity is attributed to the activation of a constitutive polypeptide precursor, followed by de novo protein synthesis (Roessler and Lien 1984a), which is in line with the quantification of the protein in aerobic cells (Fig. 3). Furthermore, no H2 production could be detected at time t0 of the in vitro experiment (Fig. 4), thus indicating that hydrogenase is activated when the cells are shifted to anaerobiosis.

In vitro hydrogenase activity of the 15N-labeled wild-type CC-400. Cells grown to the logarithmic (2 × 106 cells mL−1) growth phase were concentrated to 30 × 106 cells mL−1 (Chl concentration of 100 mg L−1) and bubbled with N2 for 4 h. Every hour the hydrogenase activity was measured in vitro. Shown is the average of three biological replicates ± SD
Absolute quantification of the hydrogenase and concomitant measurement of the H2 production in vitro allowed for the determination of the HYDA1 specific activity. Herein we determined an amount of HYDA1 of 0.065 ± 0.02 µmol protein g−1 Chl in anoxic cells (Table 2). This value is comparable to the estimated amount of HYDA1 at 0.036 µmol protein g−1 Chl (C. reinhardtii CC-124) based on in vitro produced hydrogen and calculated according to a specific activity 1000 μmolH2 min−1 mg−1 (Yacoby et al. 2012) of purified hydrogenase (Eilenberg et al. 2016). Several works reported different specific activities of the enzyme. Roessler and Lien (1984b) measured a specific activity of 1800 μmolH2 min−1 mg−1 for purified hydrogenase, isolated from C. reinhardtii 137C (+) and saturated with ferredoxin as electron mediator, whereas Happe and Naber (1993) reported a specific activity for H2 evolution of 935 μmolH2 min−1 mg−1 for purified hydrogenase, isolated from the same strain. More recent study reveals a HYDA1 specific activity of ~ 741 μmolH2 min−1 mg−1 (Kamp et al. 2008). In an assay with sodium dithionite as electron donor and methyl viologen as electron mediator, the specific activity of the hydrogenase in C. reinhardtii whole cells was calculated to be 382 ± 96.5 μmolH2 min−1 mg−1, representing the first determination of the specific enzyme activity of hydrogenase, based on the absolute amount of HYDA1 in C. reinhardtii, as determined in this study. Despite the low amounts of HYDA1 in comparison to FNR, the ability to drive hydrogen production points to an efficient channeling of electrons from FDX1 or FDX2 [another electron donor to the HYDA1, (Peden et al. 2013)] towards hydrogenase in C. reinhardtii.
Materials and methods
Strains and growth conditions
The cell wall-less wild type of C. reinhardtii CC-400 (cw15 mt+) was maintained on TAP medium (Harris 1989), pH 7.0, solidified with 1.2% (w/v) agar at 25 °C and a constant, photosynthetically active illumination of 60 µmol photons m−2 s−1. It was cultured in standard TAP medium at 25 °C under continuous light of 60 µmol photons m−2 s−1 on a rotary shaker (120 rpm). For metabolic 15N labeling, the cells were cultivated on 15N TAP plates (transferred at least 4 times) and inoculated in liquid 15N TAP medium.
For the construction of the expression strains, E. coli TOP10 (Invitrogen GmbH, Darmstadt, Germany) and BL21 (DE3) (Novagen, Merck KGaA, Darmstadt, Germany) were employed. They were grown on LB plates (25 g L−1 LB, solidified with 1.5% (w/v) agar) and cultured in liquid LB medium with added ampicillin (100 µg mL−1) at 37 °C on a rotary shaker (120 rpm) overnight.
Construction of the pET15b_HydA1 expression plasmid and expression strains
The hyda1-containing plasmid pPCR Script_hyd1 was obtained from Sloning BioTechnology GmbH (Puchheim, Germany). The hyda1 sequence was codon-optimized. After PCR amplification with Phusion High-Fidelity DNA polymerase (ThermoFisher Scientific) using the oligonucleotids 5′-GCAATTCCATATGGC-TGAATTAGC-3′ and 5′-CCGCTCGAGAGATCTTTAC-3′), the hyda1 gene was cloned into the expression plasmid pET15b (Novagen, Merck KGaA, Darmstadt, Germany) at the NdeI and XhoI restriction sites. For heterologous synthesis of the HYDA1, E. coli strain BL21 DE3 was transformed with the expression vector. Colony PCR was performed with the T7 primer 5′-TAATACGACTC-ACTATAGGG-3′ and M13 primer 5′-CAGGAAACAGCTATGACC-3′. Before protein expression, the plasmid was isolated from the expression strain and sequenced (Eurofins MWG Operon, Ebersberg, Deutschland).
For FDX5 expression, plasmid pJJ18 was kindly provided by Prof. Happe, University of Bochum (Jacobs et al. 2009).
For information on the pET17b vector-expressing plastocyanin, see Kuhlgert et al. (2012).
Heterologous expression and purification of HYDA1
For the expression of HYDA1, 1 L LB/ampicillin (100 mg L−1) medium was inoculated with the preculture and grown at 37 °C to an OD600 nm of 0.5. Fe(III)EDTA was added to a final concentration of 191 µM together with 1 mM isopropyl-β-d-thiogalactoside (IPTG) to induce the expression of the protein, which took place under anaerobic conditions (aeration with nitrogen) at room temperature and 150 rpm for 3 h. Cells were harvested by centrifugation and lysed (sonication for 10 min, pulse 40 s, pause 40 s, 20% amplitude) in the presence of 0.1 mM phenylmethylsulfonyl fluoride and 1x Halt™ Protease & Phosphatase Inhibitor Single-Use Cocktail (ThermoFisher Scientific). The lysis buffer was furthermore purged with argon to remove the oxygen.
Four mL of the lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 8 M urea) was added to the pellet after centrifugation for 20 min, at 40,000×g and 7 °C. Following a sonication for 3 min 20 s (amplitude 15%, pulse duration 40 s, pause 40 s), the cell debris was removed by centrifugation (30 min, 5000×g, 4 °C). The protein was purified using affinity chromatography with Ni–NTA agarose (QIAGEN). HYDA1 was eluted with 2 mL of the elution buffer pH 5.9 and 2 mL of the elution buffer pH 4.5 (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazol, 8 M urea). Protein concentration was measured by BCA assay (Thermo Fisher Scientific) as described by the manufacturer.
Heterologous expression and purification of PC and FDX5
Plastocyanin was expressed and purified as described in Hulsker et al. (2007) and Kuhlgert et al. (2012). Expression medium was M9 minimal medium. Instead of French press, cells were lysed by sonication (200 s, pulse 10 s, pause 40 s, 15% amplitude). The buffer for all chromatographic steps was 20 mM Tricine, 10 mM KCl, pH 7.8. The protein was eluted with a gradient of 10–400 mM KCl. The anion exchange chromatography (AEC) was performed only once, followed by size exclusion chromatography (HiLoad 16/60 Superdex 75, GE Healthcare). The concentration of the PC was determined spectrophotometrically using the ε597 nm = 4.7 mM−1 cm−1.
For the expression of FDX5, the preculture was inoculated in 1 L LB medium, supplemented with ampicillin (100 µg mL−1) and was grown aerobically at 37 °C and 180 rpm to an OD600 nm of 0.5. The expression was induced by addition of anhydrotetracycline to a final concentration of 0.1 µg mL−1. Fe(III)EDTA was added to a final concentration of 191 µM at the moment of the induction. The cells were incubated under the same conditions for 4 h, followed by cell harvest and lysis (sonication for 4 min, 15% amplitude, pulse 40 s, pause 1 min). The Strep-Tag II-FDX5 fusion proteins were purified on a Strep-Tactin Superflow Plus (QIAGEN, Hilden Germany) column according to the manufacturer’s instructions. FDX5 concentration was determined using ε419 nm = 9.7 mM−1 cm−1.
Purification of the native FNR from C. reinhardtii culture
The purification of the native FNR was done in kind cooperation with M. Twachtmann and G. Hanke (Universität Osnabrück, Germany) and is based on the protocol for recombinant FNR isolation presented in Onda et al. (2000). C. reinhardtii WT strain 21gr cells were harvested from a 15 L culture at 5000×g for 5 min and the pellet was resuspended in 750 mL 20 mM Tricine, pH 7.8, 10 mM KCl. Cells were lysed via three consecutive freeze–thaw cycles (at − 20, − 80 °C, in liquid nitrogen). The last step was followed by defrosting of the sample at 4 °C overnight. NaCl was added to the supernatant after centrifugation at 15,000×g for 15 min at a final concentration of 200 mM NaCl, which was then loaded on a DEAE Sepharose column (Whatman, Maidstone, UK). The next purification steps are performed as described in Onda et al. (2000), employing Q-Sepharose as anion exchanger instead of DEAE-Toyopearl. Prior to affinity chromatography purification, the combined FNR-containing fractions were dialyzed overnight. FNR concentration was measured by the absorbance peak at 460 nm deriving from the bound flavin molecule. Extinction of a 1 mM solution at 460 nm is approximately 10 AU.
In vitro hydrogen production
Chlamydomonas reinhardtii culture, grown to a concentration of around 2 × 106 cells mL−1, was harvested (1000×g, 5 min) and resuspended in fresh TAP medium at a final concentration of 30 × 106 cells/mL (corresponds to a chlorophyll concentration of 100 mg L−1). Concentrated cells were incubated in a 15-mL gastight vial, sealed with rubber septa (Sigma-Aldrich) at room temperature, under continuous one-side illumination at 60 µE m−2 s−1, and sparged with argon for 4 h. Every hour, 500 µl of the culture were taken anaerobically and mixed with an oxygen-free pre-mix containing 60 mM (K)PO4, pH 6.8, 1% (w/v) Triton X-100, 10 mM methyl viologen and 0.2 mM Na2S2O4 as electron donor. The in vitro assay mixture was then vortexed vigorously in order to break the cells and incubated at 37 °C and 180 rpm for 30 min. Subsequently, the gas composition of the headspace of the reaction vessel was analyzed by gas chromatography (GC-2010, Shimadzu).
Preparation of C. reinhardtii whole cell samples
One liter C. reinhardtii culture was grown to a cell concentration of around 2 × 106 cells mL−1. 200 mL out of it was used to perform in vitro hydrogen production assay. The rest was split into two 1-L flasks and incubated at 60 µE m−2 s−1 for 4 h either anaerobically (sparging with argon, AN sample) or aerobically (aeration with filtered air, AR sample) (Fig. 1). Afterwards, cells were harvested by centrifugation for 5 min at 2000×g at 4 °C and resuspended in 5 mM HEPES, pH 7.5, 10 mM EDTA, 0.1 mM PMSF, and plant-specific protease inhibitors (Serva Electrophoresis GmbH, Protease-Inhibitor Mix P), aliquoted, and kept at − 80 °C.
Determination of PSI/PSII ratio
Time-resolved absorption changes were measured using a JTS10 spectrophotometer (BioLogic), equipped with a dye laser emitting at 640 nm, pumped by the second harmonic of a Minilite II Nd:YAG laser (continuum). For the measurements, cells were harvested and resuspended to a chlorophyll concentration of 20 µg/mL in a Hepes-KOH buffer adjusted to pH 7.2, containing 10% (w/v) Ficoll to avoid cell sedimentation. Prior to measurements, samples were dark-incubated for at least 20 min on a shaker set to ~ 120 rpm.
PSI/PSII ratio was determined as described previously (Joliot and Delosme 1974) by evaluating the amplitude of the photosystem associated initial phase of the increase of the trans-thylakoid electric field, detected as an absorption change at 520 nm, and corrected with signals measured at 546 nm for non-electric field-specific contributions, following a saturating single-turnover flash. Measurements done in the presence of the photosystem II inhibitors 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU; 40 µM) and hydroxylamine (HA; 1 mM) yield an amplitude corresponding to the relative amount of active PSI, while measuring in the absence of inhibitors yields an amplitude corresponding to the relative amount of active PSI and PSII together.
Protein separation and sample preparation for LC–MS/MS
Three AR and three AN sample from the same culture (technical replicates) were thawed right before use and kept on ice. Chlorophyll concentration was measured employing 80% acetone as a solvent as described in Porra et al. (1989) and volume corresponding to 10 µg total chlorophyll was taken from each sample and supplemented with the recombinant proteins (all replicates were measured with the same batch of purified proteins) and loading buffer. The samples were then denatured at 65 °C for 15 min and loaded on a discontinuous 13% (w/v) SDS-PAGE gel according to Laemmli (1970), followed by Coomassie Brilliant Blue staining overnight as described previously (Candiano et al. 2004). Besides the protein ladder, 3 µg of the recombinant proteins was loaded on separate lanes, which served as running marker.
Three bands per sample [hydrogenase (band width 1 cm), FNR (band width 0.5 cm), and plastocyanin + ferredoxin 5 (band width 1 cm)] were excised and in-gel digested with trypsin as described in Hippler et al. (2001) and Terashima et al. (2010) with acetonitrile as the organic phase. The HYDA1-containing bands were furthermore reduced (10 mM DTT for 30 min at 37 °C) and alkylated (55 mM iodoacetamide for 20 min at room temperature in the dark).
Calculation
Absolute quantification of the proteins of interest was performed in whole cell extracts with total chlorophyll concentration of 10 µg. Since the Chl a-to-Chl b ratio in whole cells is approx. 2.7, the weighted mean of the chlorophyll molar mass is 897 g mol−1, and thus 10 µg Chl sample contains 1.11483 × 10−8 mol Chl. The amount of added purified proteins was roughly estimated, so that the amount of the (recombinant) 14N and (native) 15N proteins is similar, e.g., in a ratio of 1.43:1 reaction center for PC and FNR, 0.1:1 for HYDA1 and 0.3:1 for FDX5 (PC: 0.348 µg, FNR: 1.18 µg, HYDA1: 0.11 µg, FDX5: 0.09 µg).
After mass spectrometric analysis, based on the calculated 14N/15N ratio the amount (in µg and mol) of the corresponding proteins in the samples was determined. Quantified protein amounts (Table 2) were related to the number of chlorophyll molecules associated with PSI in the thylakoid membrane, which has been reported to be in the range of 220–320 Chl molecules for cells being in state I or state II, respectively (Nawrocki et al. 2016). Furthermore, we determined the ratio of PSI/PSII spectroscopically, which resulted in a value of 1.3 for strain CC-400. Thus, based on the approximately 440 Chl molecules for PSI and PSII in total, the calculated amount of Chl in the sample (1.11483 × 10−8 mol), and the PSI/PSII ratio of 1.3, absolute amounts of quantified proteins were calculated as a molar ratio to 249 chlorophylls [PSI Chl = (440/2.3) × 1.3] for aerobic cells and 319 chlorophylls (about 70 Chl molecules more than in state I) for anaerobic cells.
Protein analysis via LC–MS/MS and data analysis
Peptides were separated using the Ultimate 3000 Nanoflow HPLC system (Dionex). The mobile phases were composed of 2% (v/v) acetonitrile and 0.1% (v/v) formic acid in ultrapure water (A) and 80% acetonitrile and 0.1% formic acid in ultrapure water (B). The sample (1 µl) was loaded on a trap column (C18 5UM × 0.3 × 5MM; 100-Å pore size; Thermo Scientific) and desalted for 4 min using eluent A at a flow rate of 20 µl min−1. Then chromatographic separation was carried out on an RP18 capillary column (PepMap C18 3UM 75UM × 150MM, 100-Å pore size; Thermo Scientific). Peptides were eluted at a flow rate of 300 nL min−1 with the following gradient: (1) for the first biological replicate: 0–35% B (0–90 min), 35–100% B (90–97 min), and 100% B (97–107 min) and (2) for the second and third biological replicate: 0–40% B (0–35 min), 40–60% B (35–42 min), 60–100% B (42–45 min), and 100% B (45–55 min). The column was re-equilibrated with 100% A for 12 min (1st biological replicate) and for 20 min (2nd–3rd biological replicates). The LC system was coupled via a nanospray source to an LTQ Orbitrap XL mass spectrometer (Thermo Finnigan). MS full scans (mass-to-charge ratio 375:1600) were acquired by Fourier transform-MS in the Orbitrap at a resolution of 60,000 (full width at half maximum) with internal lock mass calibration on mass-to-charge ratio 445:12003. The twelve most intense ions were fragmented in the linear ion trap by collision-induced dissociation (35% normalized collision energy). Automatic gain control was enabled with target values of 5e6 and 2e5 (1st biological replicate) or 5e6 and 3e4 (2nd–3rd biological replicate) for MS full scans and MS/MS, respectively.
Each sample was analyzed twice, with 500-ms (one microscan) and 100-ms (one microscan) maximum ion trap fill time for MS/MS. Dynamic exclusion was enabled with an exclusion duration of 90 s (1st biological replicate) or 30 s (2nd–3rd biological replicate), repeat count of one, list size of 500, and exclusion mass width of 10 ppm. Unassigned charge states and charged state 1 were rejected. Peak lists were generated from raw files using msconvert 3.0.5047, developed by the ProteoWizard project (Chambers et al. 2012). For the identification of peptides, multistage activation spectra were matched against the JGI4.3 Augustus 10.2 protein sequence database using OMSSA 2.1.9 (Geer et al. 2004) and X! Tandem [version 2013.09.01; (Craig and Beavis 2004)]. The maximum number of missed cleavages allowed was two. Mass accuracy was set to 20 ppm for MS1 precursor ions and 0.8D for product ions. Carbamidomethylation of Cys was set as a fixed modification for the HYDA1-containing samples. Protein quantification was performed using qTrace (Terashima et al. 2010). Peptide ratios were calculated using all MS1 scans that contained both sister peptides, i.e., the light (14N) and heavy (15N) peptide. The peptide ratio was calculated by dividing the sum of all light by the sum of all heavy amounts. Considered were only protein ratios with at least two spectral counts and a relative standard deviation ≤ 0.5.
MS data have been deposited to ProteomeXchange (Deutsch et al. 2017) via the PRIDE partner repository (Vizcaino et al. 2016) with the dataset identifier PXD008313.
References
Andersen B, Scheller HV, Moller BL (1992) The PSI-E subunit of photosystem I binds ferredoxin:NADP + oxidoreductase. FEBS Lett 311(2):169–173
Böhme H (1978) Quantitative determination of ferredoxin, ferredoxin-NADP + reductase and plastocyanin in spinach chloroplasts. Eur J Biochem 83:137–141
Bruska MK, Stiebritz MT, Reiher M (2011) Regioselectivity of H cluster oxidation. J Am Chem Soc 133(50):20588–20603. https://doi.org/10.1021/ja209165r
Burkey KO, Gizlice Z, Carter TE Jr (1996) Genetic variation in soybean photosynthetic electron transport capacity is related to plastocyanin concentration in the chloroplast. Photosynth Res 49(2):141–149. https://doi.org/10.1007/BF00117664
Candiano G, Bruschi M, Musante L, Santucci L, Ghiggeri GM, Carnemolla B, Orecchia P, Zardi L, Righetti PG (2004) Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25(9):1327–1333. https://doi.org/10.1002/elps.200305844
Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 30(10):918–920. https://doi.org/10.1038/nbt.2377
Craig R, Beavis RC (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20(9):1466–1467. https://doi.org/10.1093/bioinformatics/bth092
Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, Moritz RL, Carver JJ, Wang M, Ishihama Y, Bandeira N, Hermjakob H, Vizcaino JA (2017) The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 45(D1):D1100–D1106. https://doi.org/10.1093/nar/gkw936
Eilenberg H, Weiner I, Ben-Zvi O, Pundak C, Marmari A, Liran O, Wecker MS, Milrad Y, Yacoby I (2016) The dual effect of a ferredoxin-hydrogenase fusion protein in vivo: successful divergence of the photosynthetic electron flux towards hydrogen production and elevated oxygen tolerance. Biotechnol Biofuels 9(1):182. https://doi.org/10.1186/s13068-016-0601-3
Eriksson M, Moseley JL, Tottey S, Del Campo JA, Quinn J, Kim Y, Merchant S (2004) Genetic dissection of nutritional copper signaling in chlamydomonas distinguishes regulatory and target genes. Genetics 168(2):795–807. https://doi.org/10.1534/genetics.104.030460
Finkelmann AR, Stiebritz MT, Reiher M (2014) Activation barriers of oxygen transformation at the active site of [FeFe] hydrogenases. Inorg Chem 53(22):11890–11902. https://doi.org/10.1021/ic501049z
Florin L, Tsokoglou A, Happe T (2001) A novel type of iron hydrogenase in the green alga Scenedesmus obliquus is linked to the photosynthetic electron transport chain. J Biol Chem 276(9):6125–6132. https://doi.org/10.1074/jbc.M008470200
Forestier M, King P, Zhang L, Posewitz M, Schwarzer S, Happe T, Ghirardi ML, Seibert M (2003) Expression of two [Fe]-hydrogenases in Chlamydomonas reinhardtii under anaerobic conditions. Eur J Biochem 270(13):2750–2758
Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH (2004) Open mass spectrometry search algorithm. J Proteome Res 3(5):958–964. https://doi.org/10.1021/pr0499491
Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci USA 100(12):6940–6945. https://doi.org/10.1073/pnas.0832254100
Ghirardi ML(2015) Implementation of photobiological H2 production: the O2 sensitivity of hydrogenases. Photosynth Res 125(3):383–393. https://doi.org/10.1007/s11120-015-0158-1
Ghirardi ML, Togasaki RK, Seibert M (1997) Oxygen sensitivity of algal H2- production. Appl Biochem Biotechnol 63–65:141–151. https://doi.org/10.1007/BF02920420
Girbal L, von Abendroth G, Winkler M, Benton PM, Meynial-Salles I, Croux C, Peters JW, Happe T, Soucaille P (2005) Homologous and heterologous overexpression in Clostridium acetobutylicum and characterization of purified clostridial and algal Fe-only hydrogenases with high specific activities. Appl Environ Microbiol 71(5):2777–2781. https://doi.org/10.1128/AEM.71.5.2777-2781.2005
Godaux D, Bailleul B, Berne N, Cardol P (2015) Induction of photosynthetic carbon fixation in anoxia relies on hydrogenase activity and proton-gradient regulation-like1-mediated cyclic electron flow in Chlamydomonas reinhardtii. Plant Physiol 168(2):648–658. https://doi.org/10.1104/pp.15.00105
Graan T, Ort DR (1984) Quantitation of the rapid electron donors to P700, the functional plastoquinone pool, and the ratio of the photosystems in spinach chloroplasts. J Biol Chem 259(22):14003–14010
Happe T, Kaminski A (2002) Differential regulation of the Fe-hydrogenase during anaerobic adaptation in the green alga Chlamydomonas reinhardtii. Eur J Biochem 269(3):1022–1032
Happe T, Naber JD (1993) Isolation, characterization and N-terminal amino acid sequence of hydrogenase from the green alga Chlamydomonas reinhardtii. Eur J Biochem 214(2):475–481
Harris EH (1989) The Chlamydomonas sourcebook: a comprehensive guide to biology and laboratory use. Academic Press, San Diego
Havlis J, Shevchenko A (2004) Absolute quantification of proteins in solutions and in polyacrylamide gels by mass spectrometry. Anal Chem 76(11):3029–3036. https://doi.org/10.1021/ac035286f
Hemschemeier A, Happe T (2011) Alternative photosynthetic electron transport pathways during anaerobiosis in the green alga Chlamydomonas reinhardtii. Biochim Biophys Acta 1807(8):919–926. https://doi.org/10.1016/j.bbabio.2011.02.010
Hippler M, Drepper F, Farah J, Rochaix JD (1997) Fast electron transfer from cytochrome c6 and plastocyanin to photosystem I of Chlamydomonas reinhardtii requires PsaF. Biochemistry 36(21):6343–6349. https://doi.org/10.1021/bi970082c
Hippler M, Klein J, Fink A, Allinger T, Hoerth P (2001) Towards functional proteomics of membrane protein complexes: analysis of thylakoid membranes from Chlamydomonas reinhardtii. Plant J 28(5):595–606
Hong G, Pachter R (2012) Inhibition of biocatalysis in [Fe-Fe] hydrogenase by oxygen: molecular dynamics and density functional theory calculations. ACS Chem Biol 7(7):1268–1275. https://doi.org/10.1021/cb3001149
Hulsker R, Mery A, Thomassen EA, Ranieri A, Sola M, Verbeet MP, Kohzuma T, Ubbink M (2007) Protonation of a histidine copper ligand in fern plastocyanin. J Am Chem Soc 129(14):4423–4429. https://doi.org/10.1021/ja0690464
Iwai M, Takizawa K, Tokutsu R, Okamuro A, Takahashi Y, Minagawa J (2010) Isolation of the elusive supercomplex that drives cyclic electron flow in photosynthesis. Nature 464(7292):1210–1213. https://doi.org/10.1038/nature08885
Jacobs J, Pudollek S, Hemschemeier A, Happe T (2009) A novel, anaerobically induced ferredoxin in Chlamydomonas reinhardtii. FEBS Lett 583(2):325–329. https://doi.org/10.1016/j.febslet.2008.12.018
Joliot P, Delosme R (1974) Flash-induced 519 nm absorption change in green algae. Biochim Biophys Acta 357(2):267–284
Kamp C, Silakov A, Winkler M, Reijerse EJ, Lubitz W, Happe T (2008) Isolation and first EPR characterization of the [FeFe]-hydrogenases from green algae. Biochim Biophys Acta 1777(5):410–416. https://doi.org/10.1016/j.bbabio.2008.02.002
Katoh S (1977) Encyclopedia of plant physiology,” new series. In: Trebst A, Avron M (eds) Photosynthesis I, Vol. 5, Springer, Berlin, pp 247–252
Kirkpatrick DS, Gerber SA, Gygi SP (2005) The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods 35(3):265–273. https://doi.org/10.1016/j.ymeth.2004.08.018
Kubas A, Orain C, De Sancho D, Saujet L, Sensi M, Gauquelin C, Meynial-Salles I, Soucaille P, Bottin H, Baffert C, Fourmond V, Best RB, Blumberger J, Leger C (2017) Mechanism of O2 diffusion and reduction in FeFe hydrogenases. Nat Chem 9(1):88–95. https://doi.org/10.1038/nchem.2592
Kuhlgert S, Drepper F, Fufezan C, Sommer F, Hippler M (2012) Residues PsaB Asp612 and PsaB Glu613 of photosystem I confer pH-dependent binding of plastocyanin and cytochrome c(6). Biochemistry 51(37):7297–7303. https://doi.org/10.1021/bi300898j
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Lambertz C, Hemschemeier A, Happe T (2010) Anaerobic expression of the ferredoxin-encoding FDX5 gene of Chlamydomonas reinhardtii is regulated by the Crr1 transcription factor. Eukaryot Cell 9(11):1747–1754. https://doi.org/10.1128/EC.00127-10
Lambertz C, Leidel N, Havelius KG, Noth J, Chernev P, Winkler M, Happe T, Haumann M (2011) O2 reactions at the six-iron active site (H-cluster) in [FeFe]-hydrogenase. J Biol Chem 286(47):40614–40623. https://doi.org/10.1074/jbc.M111.283648
Lien S, Pietro AS (1979) Interaction of plastocyanin and P700 in PSI reaction centre particles from Chlamydomonas reinhardtii and spinach. Arch Biochem Biophys 194(No.1):128–137
Liran O, Semyatich R, Milrad Y, Eilenberg H, Weiner I, Yacoby I (2016) Microoxic Niches within the thylakoid stroma of air-grown Chlamydomonas reinhardtii protect [FeFe]-hydrogenase and support hydrogen production under fully aerobic environment. Plant Physiol 172(1):264–271. https://doi.org/10.1104/pp.16.01063
Melis A, Happe T (2001) Hydrogen production. Green algae as a source of energy. Plant Physiol 127(3):740–748
Melis A, Zhang L, Forestier M, Ghirardi ML, Seibert M (2000) Sustained photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the green alga Chlamydomonas reinhardtii. Plant Physiol 122(1):127–136
Meuser JE, D’Adamo S, Jinkerson RE, Mus F, Yang W, Ghirardi ML, Seibert M, Grossman AR, Posewitz MC (2012) Genetic disruption of both Chlamydomonas reinhardtii [FeFe]-hydrogenases: insight into the role of HYDA2 in H(2) production. Biochem Biophys Res Commun 417(2):704–709. https://doi.org/10.1016/j.bbrc.2011.12.002
Mosebach L, Heilmann C, Mutoh R, Gabelein P, Steinbeck J, Happe T, Ikegami T, Hanke G, Kurisu G, Hippler M (2017) Association of ferredoxin:NADP + oxidoreductase with the photosynthetic apparatus modulates electron transfer in Chlamydomonas reinhardtii. Photosynth Res. https://doi.org/10.1007/s11120-017-0408-5
Mulder DW, Ortillo DO, Gardenghi DJ, Naumov AV, Ruebush SS, Szilagyi RK, Huynh B, Broderick JB, Peters JW (2009) Activation of HydA(DeltaEFG) requires a preformed [4Fe-4S] cluster. Biochemistry 48(26):6240–6248. https://doi.org/10.1021/bi9000563
Mulder DW, Boyd ES, Sarma R, Lange RK, Endrizzi JA, Broderick JB, Peters JW (2010) Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydA(DeltaEFG). Nature 465(7295):248–251. https://doi.org/10.1038/nature08993
Mus F, Cournac L, Cardettini V, Caruana A, Peltier G (2005) Inhibitor studies on non-photochemical plastoquinone reduction and H(2) photoproduction in Chlamydomonas reinhardtii. Biochim Biophys Acta 1708(3):322–332. https://doi.org/10.1016/j.bbabio.2005.05.003
Mus F, Dubini A, Seibert M, Posewitz MC, Grossman AR (2007) Anaerobic acclimation in Chlamydomonas reinhardtii: anoxic gene expression, hydrogenase induction, and metabolic pathways. J Biol Chem 282(35):25475–25486. https://doi.org/10.1074/jbc.M701415200
Nawrocki WJ, Santabarbara S, Mosebach L, Wollman FA, Rappaport F (2016) State transitions redistribute rather than dissipate energy between the two photosystems in Chlamydomonas. Nat Plants 2:16031. https://doi.org/10.1038/nplants.2016.31
Onda Y, Matsumura T, Kimata-Ariga Y, Sakakibara H, Sugiyama T, Hase T (2000) Differential interaction of maize root ferredoxin:NADP(+) oxidoreductase with photosynthetic and non-photosynthetic ferredoxin isoproteins. Plant Physiol 123(3):1037–1046
Ong SE, Mann M (2005) Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol 1(5):252–262. https://doi.org/10.1038/nchembio736
Peden EA, Boehm M, Mulder DW, Davis R, Old WM, King PW, Ghirardi ML, Dubini A (2013) Identification of global ferredoxin interaction networks in Chlamydomonas reinhardtii. J Biol Chem 288(49):35192–35209. https://doi.org/10.1074/jbc.M113.483727
Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC (1998) X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science 282(5395):1853–1858
Porra RJ, Thompson WA, Kriedemann PE (1989) Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim Biophys Acta 975(3):384–394. https://doi.org/10.1016/S0005-2728(89)80347-0
Quinn JM, Barraco P, Eriksson M, Merchant S (2000) Coordinate copper- and oxygen-responsive Cyc6 and Cpx1 expression in Chlamydomonas is mediated by the same element. J Biol Chem 275(9):6080–6089
Quinn JM, Eriksson M, Moseley JL, Merchant S (2002) Oxygen deficiency responsive gene expression in Chlamydomonas reinhardtii through a copper-sensing signal transduction pathway. Plant Physiol 128(2):463–471. https://doi.org/10.1104/pp.010694
Roessler PG, Lien S (1984a) Activation and de novo synthesis of hydrogenase in chlamydomonas. Plant Physiol 76(4):1086–1089
Roessler PG, Lien S (1984b) Purification of hydrogenase from Chlamydomonas reinhardtii. Plant Physiol 75(3):705–709
Rupprecht J, Hankamer B, Mussgnug JH, Ananyev G, Dismukes C, Kruse O (2006) Perspectives and advances of biological H2 production in microorganisms. Appl Microbiol Biotechnol 72(3):442–449. https://doi.org/10.1007/s00253-006-0528-x
Sawyer A, Winkler M (2017) Evolution of Chlamydomonas reinhardtii ferredoxins and their interactions with [FeFe]-hydrogenases. Photosynth Res. https://doi.org/10.1007/s11120-017-0409-4
Sawyer A, Bai Y, Lu Y, Hemschemeier A, Happe T (2017) Compartmentalisation of [FeFe]-hydrogenase maturation in Chlamydomonas reinhardtii. Plant J 90(6):1134–1143. https://doi.org/10.1111/tpj.13535
Schottler MA, Kirchhoff H, Weis E (2004) The role of plastocyanin in the adjustment of the photosynthetic electron transport to the carbon metabolism in tobacco. Plant Physiol 136(4):4265–4274. https://doi.org/10.1104/pp.104.052324
Steinbeck J, Nikolova D, Weingarten R, Johnson X, Richaud P, Peltier G, Hermann M, Magneschi L, Hippler M (2015) Deletion of proton gradient regulation 5 (PGR5) and PGR5-Like 1 (PGRL1) proteins promote sustainable light-driven hydrogen production in Chlamydomonas reinhardtii due to increased PSII activity under sulfur deprivation. Front Plant Sci 6:892. https://doi.org/10.3389/fpls.2015.00892
Stripp ST, Goldet G, Brandmayr C, Sanganas O, Vincent KA, Haumann M, Armstrong FA, Happe T (2009) How oxygen attacks [FeFe] hydrogenases from photosynthetic organisms. Proc Natl Acad Sci USA 106(41):17331–17336. https://doi.org/10.1073/pnas.0905343106
Swanson KD, Ratzloff MW, Mulder DW, Artz JH, Ghose S, Hoffman A, White S, Zadvornyy OA, Broderick JB, Bothner B, King PW, Peters JW (2015) [FeFe]-hydrogenase oxygen inactivation is initiated at the H cluster 2Fe subcluster. J Am Chem Soc 137(5):1809–1816. https://doi.org/10.1021/ja510169s
Terashima M, Specht M, Naumann B, Hippler M (2010) Characterizing the anaerobic response of Chlamydomonas reinhardtii by quantitative proteomics. Mol Cell Proteomics 9(7):1514–1532. https://doi.org/10.1074/mcp.M900421-MCP200
Terauchi AM, Lu SF, Zaffagnini M, Tappa S, Hirasawa M, Tripathy JN, Knaff DB, Farmer PJ, Lemaire SD, Hase T, Merchant SS (2009) Pattern of expression and substrate specificity of chloroplast ferredoxins from Chlamydomonas reinhardtii. J Biol Chem 284(38):25867–25878. https://doi.org/10.1074/jbc.M109.023622
Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44(22):11033. https://doi.org/10.1093/nar/gkw880
Volgusheva A, Styring S, Mamedov F (2013) Increased photosystem II stability promotes H2 production in sulfur-deprived Chlamydomonas reinhardtii. Proc Natl Acad Sci USA 110(18):7223–7228. https://doi.org/10.1073/pnas.1220645110
Winkler M, Kuhlgert S, Hippler M, Happe T (2009) Characterization of the key step for light-driven hydrogen evolution in green algae. J Biol Chem 284(52):36620–36627. https://doi.org/10.1074/jbc.M109.053496
Yacoby I, Pochekailov S, Toporik H, Ghirardi ML, King PW, Zhang S (2011) Photosynthetic electron partitioning between [FeFe]-hydrogenase and ferredoxin:NADP+-oxidoreductase (FNR) enzymes in vitro. Proc Natl Acad Sci USA 108(23):9396–9401. https://doi.org/10.1073/pnas.1103659108
Yacoby I, Tegler LT, Pochekailov S, Zhang S, King PW, Riggs PD (2012) Optimized expression and purification for high-activity preparations of Algal [FeFe]-Hydrogenase. PLoS ONE 7(4):e35886
Yang W, Wittkopp TM, Li X, Warakanont J, Dubini A, Catalanotti C, Kim RG, Nowack EC, Mackinder LC, Aksoy M, Page MD, D’Adamo S, Saroussi S, Heinnickel M, Johnson X, Richaud P, Alric J, Boehm M, Jonikas MC, Benning C, Merchant SS, Posewitz MC, Grossman AR (2015) Critical role of Chlamydomonas reinhardtii ferredoxin-5 in maintaining membrane structure and dark metabolism. Proc Natl Acad Sci USA 112(48):14978–14983. https://doi.org/10.1073/pnas.1515240112
Zalutskaya Z, Minaeva E, Filina V, Ostroukhova M, Ermilova E (2017) Regulation of sulfur deprivation-induced expression of the ferredoxin-encoding FDX5 gene Chlamydomonas reinhardtii in aerobic conditions. Plant Physiol Biochem 123:18–23. https://doi.org/10.1016/j.plaphy.2017.11.024
Acknowledgements
We are grateful to Prof. Dr. Thomas Happe from the University of Bochum for providing the plasmid pJJ18. M.H. acknowledges support from the German Science Foundation (DFG, HI 739/13-1).
Funding
This study was funded by the German Science Foundation (DFG, HI 739/13-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Nikolova, D., Heilmann, C., Hawat, S. et al. Absolute quantification of selected photosynthetic electron transfer proteins in Chlamydomonas reinhardtii in the presence and absence of oxygen. Photosynth Res 137, 281–293 (2018). https://doi.org/10.1007/s11120-018-0502-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-018-0502-3