
Abstract
The cytochrome b 6 f complex of oxygenic photosynthesis produces substantial levels of reactive oxygen species (ROS). It has been observed that the ROS production rate by b 6 f is 10–20 fold higher than that observed for the analogous respiratory cytochrome bc1 complex. The types of ROS produced (O2•−, 1O2, and, possibly, H2O2) and the site(s) of ROS production within the b 6 f complex have been the subject of some debate. Proposed sources of ROS have included the heme b p , PQ p •− (possible sources for O2•−), the Rieske iron–sulfur cluster (possible source of O2•− and/or 1O2), Chl a (possible source of 1O2), and heme c n (possible source of O2•− and/or H2O2). Our working hypothesis is that amino acid residues proximal to the ROS production sites will be more susceptible to oxidative modification than distant residues. In the current study, we have identified natively oxidized amino acid residues in the subunits of the spinach cytochrome b 6 f complex. The oxidized residues were identified by tandem mass spectrometry using the MassMatrix Program. Our results indicate that numerous residues, principally localized near p-side cofactors and Chl a, were oxidatively modified. We hypothesize that these sites are sources for ROS generation in the spinach cytochrome b 6 f complex.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The cytochrome b 6 f complex acts as a plastoquinol–plastocyanin (cytochrome c553 in cyanobacteria) oxidoreductase and is similar to cytochrome bc1 complexes present in heterotrophic organisms. Moderate resolution crystal structures (≈ 3 Å) are available for b 6 f complexes of both thermophilic cyanobacteria [Mastigocladus (Kurisu et al. 2003) and Nostoc (Baniulis et al. 2009)] and a mesophilic green alga [Chlamydomonas (Stroebel et al. 2003)]. Recently, a 2.5 Å structure of the Nostoc protein has been presented (Hasan and Cramer 2014). This higher-resolution structure allowed the identification of numerous lipids and intra-protein water molecules that were not identifiable in the earlier structures. The b 6 f complex is a symmetric dimer with a molecular mass of 220 kDa containing, within each monomer, eight subunits: Cyt f (PetA), Cyt b6 (PetB), Rieske iron–sulfur protein (PetC), subunit IV (PetD), and four smaller subunits (PetG, PetL, PetM, and PetN). These proteins are associated with seven prosthetic groups: 2 c-type hemes, 2 b-type hemes, 1 Fe2S2 cluster, 1 Chl a, and 1 β-carotene. Additionally, a plastoquinol-binding site is present on the p-side (lumenal side) of the complex and a plastoquinone-binding site is present on the n-side (stromal side) of the complex. In cytochrome bc 1 complexes, linear electron transport occurs via a modified Q-cycle mechanism (Crofts et al. 2003). However, it is unclear if this is the case for the cytochrome b 6 f complex. The presence of the novel heme c n and the observation that the complex can participate in cyclic electron transport, accepting electrons from reduced ferredoxin possibly via ferredoxin-NADP+ oxidoreductase [which appears to be a subunit of the in vivo complex (Zhang et al. 2001)], both argue against a classical modified Q-cycle mechanism. The functions of the Chl a and the β-carotene are unclear and these have been hypothesized to play a structural role in b 6 f assembly or are possibly required for complex stability (Yan et al. 2008).
In thylakoid membranes, reactive oxygen species (ROS) are produced at a number of sites within the linear electron transport chain including PS II, the b 6 f complex and PS I. ROS are formed by the excitation of dioxygen (singlet oxygen, 1O2), the partial reduction of dioxygen (O2•−, O22−, H2O2, and •OH), and the partial oxidation of water (H2O2 and •OH). These are unavoidable byproducts of oxygenic photosynthesis. ROS can oxidatively damage proteins, lipids, and nucleic acids (Das and Roychoudhury 2014) and, consequently, place limits on photosynthetic productivity [estimated to be at least 10% based on PS II photoinhibition, alone (Long et al. 1994)]. It should also be recognized, however, that ROS also serve as signal molecules which can modulate a variety of cellular processes including stress acclimatization, differentiation and development, programmed cell death, and pathogen defense (Mittler 2016).
While ROS generation by PS II has been extensively examined (Kale et al. 2017; Kim and Jung 1992; Pospíšil 2009, 2016), relatively few studies have been performed on the b 6 f complex. A number of cofactors within the complex have, however, been proposed as sites of ROS production. Cytochrome bc 1 -type complexes, in general, produce O2•− (Lanciano et al. 2013), and production of O2•− by the b 6 f complex has been observed by EPR spin-trapping spectroscopy (Sang et al. 2011a). Recently it has been demonstrated that b 6 f complex isolated from both spinach and Mastigocladus produce 20–30× more O2•−, on a per complex basis, than does the yeast cytochrome bc 1 complex (Baniulis et al. 2013). Two potential O2•− production sites were suggested, either the heme b p or PQ p •−. The redox potential of the heme b p appears more negative (Em7 = − 40 to − 90 mV (Furbacher et al. 1989; Rich and Bendall 1980), Em7.5 = − 150 mV [Kramer and Crofts 1994)] than those reported for yeast and mammalian cytochrome bc 1 heme b p [Em7 ≈ − 20 mV, (T’Sai and Palmer 1983)]. This would make dioxygen reduction more feasible in the photosynthetic complex (Sarewicz et al. 2010). It was also hypothesized that dioxygen reduction by PQ p •− might be facilitated by a longer residence time of the semiquinone at the PQ p -binding site (Baniulis et al. 2013). Other investigators have suggested that the Rieske iron–sulfur protein is involved in O2•− production in both cytochrome bc 1 complexes (Genova et al. 2001) and the b 6 f complex (Sang et al. 2011a). In this regard it is interesting that Sang et al. (2011a) reported that while no O2•− was generated from complexes lacking the Rieske cluster, 1O2 was produced. These authors suggested that O2•− was produced by, at least partially, a 1O2-dependent process (Sang et al. 2011a, b).
The possible production of 1O2 by the b 6 f complex is intriguing. As noted above, the complex contains Chl a. The presence of chlorophyll prosthetic groups can be quite hazardous due to the possible production of 1O2 by intersystem crossing. Typically, chlorophylls are found in close proximity to carotenoids that can quench 1O2. The β-carotene in the b 6 f complex, however, is located ≥ 14 Å from the Chl a and too distant to serve as an efficient quencher (Dashdorj et al. 2005; Kim et al. 2005). It has been suggested that quenching of the 1O2 may be facilitated by a putative hydrophobic ROS channel which funnels 1O2 to the carotenoid (Kim et al. 2005). It should also be noted that aromatic residues in the vicinity of the Chl a significantly shorten the fluorescence lifetime (by about 20-fold), which would lower the yield of 3Chl, reducing the probability of 1O2 formation (Dashdorj et al. 2005; Peterman et al. 1998; Yan et al. 2008). Both Chl a and the β-carotene may also function in the assembly of the complex (Cramer et al. 2009). Finally, it has been suggested that iron–sulfur proteins, in general, can serve as blue-light sensitizers for the production of 1O2 (Kim and Jung 1992). These authors have specifically suggested that the Rieske iron–sulfur cluster in the b 6 f complex is a major source of 1O2 in thylakoid membranes (Suh et al. 2000). This hypothesis is controversial, and strong evidence indicating that Chl a is the principal source of 1O2 has been presented (Sang et al. 2010).
It should also be noted that heme c n may directly bind dioxygen. This is suggested by the observation that NO, a dioxygen analogue, binds tightly to the heme (Twigg et al. 2009). The authors presented the possibility that heme c n could function as a plastoquinol oxidase. In this capacity, an aberrant formation of O2•− by a one-electron reduction of dioxygen (or H2O2 by a two-electron reduction) could hypothetically occur. It is also possible that the observed strong binding of NO, itself, is physiologically relevant as NO, often in cooperation with ROS, is involved in a wide variety of signal transduction pathways involving plant response to abiotic stress (Farnese et al. 2016; Mittler 2016).
No in-depth characterization of oxidative modification sites on the b 6 f complex has been performed, and the relative importance and/or contribution of the different proposed ROS production sites (heme b p , PQ p , Fe2S2, Chl a) have not been evaluated. Galetskiy et al. (2011) did report that eleven residues were ROS-modified within the complex but did not provide their locations. Importantly, these authors did not utilize a non-oxidizing denaturing PAGE system (see below) in their study. Consequently, the possibility of protein oxidative modification artifacts due to electrophoretic conditions cannot be excluded.
In our study, we have used high-resolution tandem mass spectrometry to identify the location of oxidized residues within the cytochrome b 6 f complex isolated from field-grown spinach. These “natively” oxidized residues are the product of ROS production in the field environment where the plants may be exposed to a variety of abiotic stressors such as high light intensities, low or high temperatures, or drought (Choudhry et al. 2016; You and Chan 2015). It should be noted that earlier we have used these methods to identify natively oxidized residues in spinach PS II (Frankel et al. 2012, 2013), results which have recently been confirmed and extended for the cyanobacterial photosystem (Weisz et al. 2017).
In the current study, we have mapped the natively oxidized residues identified in field-grown spinach cytochrome b 6 f complex onto the corresponding residues of the Chlamydomonas b 6 f complex structure (Stroebel et al. 2003). Our results indicate that numerous oxidized amino acid residues are located in the vicinity of the p-side cofactors heme b p , the Rieske iron–sulfur protein, and the PQ p -binding site. None were observed in the vicinity of n-side cofactors. Additionally, oxidized residues were located adjacent to the Chl a. Our findings support the hypothesis that the p-side cofactors and Chl a are responsible for most of the ROS produced by the cytochrome b 6 f complex.
Materials and methods
The cytochrome b 6 f complex was isolated from market spinach essentially by the method previously described (Hurt and Hauska 1981). The b 6 f subunits were resolved on a 12.5–20% acrylamide gradient by LiDS-PAGE (Delepelaire and Chua 1979) either using the standard method (see Fig. 1B) or, for mass spectrometry, using a non-oxidizing gel system (Rabilloud et al. 1995). This was required, as standard PAGE is known to introduce numerous protein oxidation artifacts (Sun and Anderson 2004). In this system, after degassing, the gels are polymerized with riboflavin in the presence of diphenyliodonium chloride and toluenesulfinate followed by exposure to UV light. The upper reservoir buffer contained thioglycolate. Previously, we had demonstrated that PS II proteins resolved in this system exhibited much lower levels of artifactual protein oxidation than proteins resolved by standard PAGE (Frankel et al. 2012), confirming the earlier reports of Rabilloud et al. (Rabilloud et al. 1995) and Sun and Anderson (2004), both of which examined other test proteins. For mass spectrometry, electrophoresis was terminated when the stacked proteins first entered the resolving gel. The gel was then stained with Coomassie blue, destained, and the thick protein band containing the stacked b 6 f subunits was excised. This electrophoresis by denaturing LiDS-PAGE provides facile detergent removal during protein band processing prior to proteolysis and potentially yields greater cleavage site accessibility during subsequent protease treatment (specifically with chymotrypsin or pepsin) when compared to “in solution” digestion protocols. The protein bands were then digested using either trypsin, chymotrypsin, or pepsin following standard procedures for “in-gel” proteolysis. Three biological replicates were analyzed for each of the three proteases (chymotrypsin, pepsin, and trypsin), and the union set of these replicates is presented. After protease digestion, the peptides were resolved by HPLC on a C:18-reversed phase column and ionized via electrospray into a Thermo Scientific Orbitrap Fusion Lumos mass spectrometer. The samples were analyzed in a data-dependent mode with one Orbitrap MS1 scan acquired simultaneously with up to ten linear ion trap MS2 scans. Identification and analysis of the peptides containing oxidative modifications were performed using the MassMatrix Program (Xu and Freitas 2009). A library containing the sequences of the eight subunits of the spinach complex plus Ferredoxin-NADP+ oxidoreductase was searched, as was a decoy library which contained these same sequences but in reversed amino acid order. Twelve different types of oxidative modifications were included as possible post-translational modifications. For a putative positive identification of an oxidized residue, the peptide must exhibit a p value of 10−5 or smaller; this value was selected prior to data collection. Peptides meeting this p value threshold were then examined manually, with the quality of the MS2, collision-induced dissociation spectra being confirmed. Additionally, only peptides with charge states of +3 or lower were considered. Finally, the mass error of the precursor ion was required to be ≤ 5.0 ppm and was required to be the product of specific proteolytic cleavage. The identified oxidized amino acid residues were mapped onto the crystal structure of the Chlamydomonas reinhardtii b 6 f complex [PDB: 1Q90, (Stroebel et al. 2003)] using PYMOL (DeLano 2002).

Purification of the Spinach Cytochrome b 6 f Complex. The complex was prepared essentially according to the methods of Hurt and Hauska (Hurt and Hauska 1981). A Sucrose density gradient. Both monomer and dimer bands were observed; the dimer was used for all subsequent studies. B LiDS-PAGE of thylakoids (Thy) and the b 6 f complex dimer. Subunits are labeled to the right, standard proteins to the left. The small subunits, PetG, PetL, PetM, and PetN which have apparent molecular masses in the 2–4 kDa region, are not resolved in this gel system
Results and discussion
Isolation of the spinach b 6 f complex yielded results which were basically indistinguishable from previous reports (Black et al. 1987; Hurt and Hauska 1981; Zhang et al. 2001) for the isolation of the spinach complex (Fig. 1A). Four major polypeptides were identified: PetA, PetB, PetC, and PetD (Fig. 1B). It should be noted that in standard LiDS-PAGE (Fig. 1B), the low molecular mass subunits (2–4 kDa) PetG, PetL, PetM, and PetN are not resolved. A fifth unidentified peptide with an apparent molecular mass of 48 kDa was also observed. This component, which is probably a contaminant, has been sporadically observed in other preparations of the complex (Hauska 2004). Tandem mass spectrometry analysis of the chymotryptic, peptic, and tryptic peptides of the cytochrome b 6 f complex allowed the identification of 55 oxidatively modified residues present on these subunits. The identity of these oxidized residues and the types of modifications observed are presented in Table 1. No oxidative modifications were observed on the small subunits of the complex (PetG, PetL, PetM, and PetN). It should be emphasized that it is highly unlikely that all of the observed modifications would be present on every copy of the complex. Rather, this portfolio of detectable modifications is present within the full population of cytochrome b 6 f complexes present in our biological samples. It should be noted that for this study we isolated the complex from field-grown market spinach. Consequently, the exact growth conditions are unknown. Many studies examining the spinach cytochrome b 6 f complex have utilized comparable biological materials (Baniulis et al. 2013; Baymann et al. 2007; Stofleth 2012; Szymańska et al. 2010).
Figure 2 illustrates the quality of the data used for the identification of oxidized amino acid residues within the cytochrome b 6 f complex. In this figure, the tandem mass spectrometry data collected for the 45E–56L peptic peptide of PetA are illustrated. In Fig. 2A, the data from the unmodified peptide are shown, while in Fig. 2B, data from this peptide bearing oxidized 54M are shown. Both of these were observed in the same biological replicate. The observed mass accuracies for the parent peptic ions were − 0.35 and + 0.34 ppm, respectively. The p value for both of the illustrated peptides was 10−5.5 and are, consequently, among the lowest-quality peptides used in this study (p value range = 10−5.0 – 10−11.1). Even these peptides, however, clearly exhibited nearly complete y- and b-ion series, allowing unequivocal identification of the oxidative mass modification. This result indicates that the use of p values ≤ 10−5 provided very high-quality peptide identifications. Fig. S1 illustrates results for peptides exhibiting the median and lowest p value peptides identified in this study (p values of 10−6.4 and 10−11.1, respectively). One should note that all of the subunits of the cytochrome b 6 f complex are intrinsic membrane proteins. The analysis of such proteins by mass spectrometry is often difficult, with only relatively low sequence coverage being reported in standard “bottom-up” experiments (Kar et al. 2017; Souda et al. 2011; Weisz et al. 2017). In this study, however, we have obtained nearly complete coverage (≥ 90%) for all of the major subunits of the complex using the enzymes trypsin, chymotrypsin, and pepsin for proteolysis. This is illustrated in Fig. 3.

Quality of the mass spectrometry. Shown are the mass spectrometry results for the peptide PetA:45EAVVRIPYDMQ56L in both the unmodified (A) and modified (54M+16) forms (B). A Top, spectrum of the CID dissociation of the unmodified peptide PetA:45EAVVRIPYDMQ56L. Various identified ions are labeled. Bottom, table of all predicted masses for the y- and b-ions generated from this peptide sequence. Ions identified in the CID spectrum (top) are shown in red. The b’++, b’+ y’++, and y’+ ions are generated by the neutral loss of water, while the b*++, b*+ y*++, and y*+ ions are generated from the loss of ammonia. B Top, spectrum of the CID dissociation of the modified PetA:45EAVVRIPYDM (+ 16)Q56L. Various identified ions are labeled. Bottom, table of all predicted masses for the y- and b- ions generated from this peptide sequence. Ions identified in the CID spectrum are shown in red. The ions y3+–y9+ exhibit the + 16 mass modification as does the b10+ ion when compared to the same ions in A. This verifies that 54M contains an oxidative modification. The p values for both the unmodified and modified peptides were 10−5.5

Sequence Alignments of Spinach and Chlamydomonas Cytochrome b 6 f Subunits and Mass Spectrometry Coverage. The subunits of the spinach and Chlamydomonas are very similar, which supports the use of the Chlamydomonas structure for these studies. Alignments were performed with CLUSTAL Omega (Sievers et al. 2011). Similarity scores were calculated using BLAST (Camacho et al. 2009). Combined mass spectrometry coverage of the b 6 f complex subunits, using trypsin, chymotrypsin, and pepsin coupled with Orbitrap analysis, was excellent (≥ 90%). Sequences which were not identified by mass spectrometry are boxed
No crystal structure is currently available for the spinach cytochrome b 6 f complex. However, crystal structures are available for the thermophilic cyanobacteria Mastigocladus laminosus (Kurisu et al. 2003) and Nostoc sp. PCC7120 (Baniulis et al. 2009), as well as the mesophilic eukaryote Chlamydomonas reinhardtii (Stroebel et al. 2003). The sequence similarity between the spinach subunits and the Chlamydomonas subunits is high (Fig. 3), being 82% for PetA, 94% for PetB, 76% for PetC, and 94% for PetD. This high degree of similarity allowed us to rationally map the oxidized amino acids that we observed in the spinach b 6 f complex onto the crystal structure of the Chlamydomonas protein complex. Indeed, 36 of the identified 55 modified residues (68%) were identical in both systems.
Figure 4 presents an overview of the locations of the oxidized residues that we identified within the context of the cytochrome b 6 f complex dimer. The vast majority of the observed oxidized residues were located on the p-side of the complex. This does not appear to be the result of a sampling error since our mass coverage of the n-side residues was 96% (136/142 residues). Surface domains on the PetA and PetC subunits appear to be particularly susceptible to oxidative modification. This is not surprising since the surfaces of these components are exposed to the bulk solvent of the lumen. ROS produced by PS II due to manganese cluster damage (HO• and possibly, H2O2), 1O2 produced at P680*, the production of O2•−, and possibly other ROS species by the b 6 f complex itself, and possibly PS I, may all contribute to the oxidative modification of lumenally exposed domains. Additionally, while there are many ROS detoxification systems localized to the n-side of the thylakoid membrane (Das and Roychoudhury 2014; Tripathy and Oelmuller 2012), only a few lumenal components of putative p-side ROS detoxification systems have been reported (Bermudez et al. 2012; Levesque-Tremblay et al. 2009). Consequently, it is possible that ROS are not detoxified as efficiently in the thylakoid lumen as they are in the chloroplast stroma.

Overview of natively oxidized amino acid residues in the Spinach b 6 f Complex. A Side view of complex from within the plane of the membrane. B Lumenal (p-side) view of the complex. The subunits are shown as follows: PetA (pale green), PetB (pale blue), PetC (pink), PetD (pale yellow), and the small subunits (gray). Oxidatively labeled residues are shown as clusters of spheres colored in darker shades and were mapped onto their corresponding locations on the Chlamydomonas reinhardtii b 6 f structure (Stroebel et al. 2003). Cofactors and TDS are shown in stick representation
In addition to these surface-exposed oxidatively modified residues, a number of oxidized residues were observed which were buried or partially buried within the protein matrix, or present on the surface of the complex but buried within the lipid bilayer of the thylakoid membrane. Our working hypothesis is that amino acid residues that are in the vicinity of ROS production sites would be more prone to oxidative modification than residues that are more distant from these sites. The number of amino acids modified near each of these cofactors in relation to the total number of residues in the vicinity of each cofactor is summarized in Table S1.
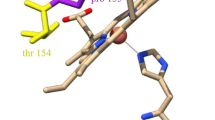
In Fig. 5, we have examined the p-side cofactors, which include heme f, the Rieske iron–sulfur cluster, heme b p , and the lumenal plastoquinol-binding site (PQ p ) which, in this structure, is occupied by the b 6 f inhibitor TDS (tridecyl-stigmatellin). In Fig. 5, a 7.5 Å region surrounding each of the cofactors is shown with oxidized residues represented as spheres and labeled.

Details of the oxidative modifications identified in the vicinity of p-side cofactors. Shown is the protein structure located within 7.5 Å of the p-side cofactors (A) heme f, (B) FeS, and the PQ p -binding pocket, which is occupied by TDS, and (C) heme cytochrome b p . Color coding of the b 6 f subunits is as shown in Fig. 4. Oxidatively modified residues are shown as clusters of spheres in darker shades and are labeled
Figure 5B demonstrates that even though a large number of oxidatively modified residues are located on the cytochrome f subunit, none of these are in close proximity to heme f. This was expected since the production of ROS by this heme was not likely, given its high Em7 [+ 355 mV, (Alric et al. 2005)]. Figure 5B illustrates the location of oxidatively modified residues near the iron–sulfur cluster and the PQ p -binding site. The PetC residues 107C, 108T, 109H, 110L, and 113V were modified, as were 76L and 101M of PetD. The presence of seven oxidatively modified residues in close proximity to these cofactors strongly suggests that either the putative long-lived semiquinone occupying the PQ p -binding site and/or the iron–sulfur cluster is a source of ROS in the complex. The high Em<8 (+ 320 mV) of the iron–sulfur cluster (Cramer et al. 2011; Nitschke et al. 1992) makes it unlikely that this site would be the source of O2•−. Additionally, the ability of the iron–sulfur cluster to act as a photosensitizer for 1O2 production is questionable (Sang et al. 2010). Nevertheless, we cannot rigorously exclude these possibilities at this time. Conversely, the ability of semiquinones to reduce O2 to O2•− is well documented (Mubarakshina and Ivanov 2010). The production of O2•− at the PQ p site may be exacerbated by a hypothesized long residency time of the semiquinone (Baniulis et al. 2013). This long residency time might be due to the presence in the plastoquinol entrance/plastoquinone exit pathway of the phytol tail of the Chl a, which might hinder quinol exchange.
In Fig. 5C, oxidized residues in the vicinity of heme b p are shown. Three residues, 187H and 188T of the PetB and 61M of PetD, were identified as being oxidatively modified. This observation raises the possibility that heme b p may also be a source of ROS, probably O2•−, as was previously hypothesized (Baniulis et al. 2013; Sarewicz et al. 2010; Twigg et al. 2009). Interestingly, 187H is a ligand to the heme iron. It is unclear what, if any, consequences this oxidative modification would have on the redox function of heme b p .
In Fig. 6, the immediate environment surrounding the hemes b n and c n are illustrated. No oxidatively modified residues were observed within 7.5 Å of the b n or c n hemes or the adjacent PQ n -binding pocket. It should be noted that in the Mastigocladus crystal structure (Hasan et al. 2013), the PQ n -binding pocket is occupied by TDS. This observation does not preclude the possibility that heme c n is associated with a putative plastoquinol oxidase activity (Twigg et al. 2009). It does suggest, however, that if an oxidase activity is present that it is efficient and not prone to the production of ROS in sufficient quantities to produce detectable oxidative modifications.

Details of the oxidative modifications identified in the vicinity of n-side cofactors. Shown is the protein structure located within 7.5 Å of the n-side cofactors heme b n and heme c n . The PQ n -binding pocket is indicated by a cyan ellipse. In the Chlamydomonas structure (Stroebel et al. 2003), this site is unoccupied while in the Mastigocladus structure it is occupied with TDS (Hasan et al. 2013)
In Fig. 7, oxidized residues in the vicinity of the Chl a and the β-carotene (Fig. 7A) are shown. Two residues adjacent to the Chl a, 100L and 101M of PetD (i.e., within 7.5 Å), are associated with the Chl a-binding pocket and, in the case of 101M, the PQ p -binding site as well (see above). No oxidized residues were observed near the β-carotene. It had been hypothesized that a hydrophobic channel between the Chl a and the β-carotene exists which could funnel 1O2 from the Chl a to the β-carotene to facilitate quenching (Kim et al. 2005). This hypothetical channel would include residues PetB:36I,95L, 96M, 98I, 99L, and 102F, PetD:133F, and several hydrophobic residues of PetG. None of these residues exhibited oxidative modification. It should be pointed out, however, that we do not have mass spectrometry coverage of PetB:36I and PetD:133F. Consequently, our results, while not precluding the presence of a channel, provide no evidence in support of this hypothesis. Interestingly, two other oxidized PetA residues were observed which are in contact with 100L and 101M; these residues, 96L and 103A (Fig. 7B), are also closely associated with the Chl a-binding pocket although more distant than 7.5 Å from the Chl a (12.1 and 8.8 Å, respectively). These results strongly suggest that ROS, probably 1O2, is produced at the Chl a, as has previously been hypothesized (Sang et al. 2010). It is possible that, since the Chl a-binding pocket is exposed at the surface of the complex but buried in the lipid bilayer, that 1O2 is released directly from the Chl a-binding pocket to the lipid bilayer (Fig. 7B).

Details of the oxidative modifications identified in the vicinity of Chl a and the β-carotene. A Shown is the protein structure located within 7.5 Å of the Chl a and the β-carotene. Color coding of the b 6 f subunits is as shown in Fig. 4. Oxidatively modified residues are shown as clusters of spheres in darker shades and are labeled. Note that no oxidative modifications were observed for the intervening hydrophobic residues located between the Chl a and the β-Carotene. B Surface of the b 6 f complex in the vicinity of the Chl a-binding pocket. Color coding of the b 6 f subunits is as shown in Fig. 4. Oxidatively modified residues are shown as spheres in darker shades and are labeled
Conclusions
In this communication, we have identified numerous oxidized residues in the vicinity of the p-side cofactors heme b p , the Rieske iron–sulfur cluster, the PQ p -binding domain, and adjacent to the Chl a-binding pocket. No oxidized residues were identified in the vicinity of the β-carotene or heme f, or the n-side cofactors heme b n or heme c n . The locations of these modified residues are consistent with our hypothesis that residues in the vicinity of ROS production sites would be prone to oxidative modification. We have not, at this time, determined the type of ROS leading to these observed modifications, the relative importance of the various possible sites in ROS production, or the time course for the appearance of oxidative modifications. These important questions are the subject of future studies.
References
Alric J, Pierre Y, Picot D, Lavergne J, Rappaport F (2005) Spectral and redox characterization of the heme ci of the cytochrome b 6 f complex. Proc Natl Acad Sci (USA) 102:15860–15865
Baniulis D et al (2009) Structure-function, stability, and chemical modification of the cyanobacterial Cytochrome b 6 f Complex from Nostoc sp. PCC 7120. J Biol Chem 284:9861–9869
Baniulis D, Hasan SS, Stofleth JT, Cramer WA (2013) Mechanism of enhanced superoxide production in the cytochrome b 6 f complex of oxygenic. Photosyn Biochem 52:8975–8983
Baymann F, Giusti F, Picot D, Nitschke W (2007) The c i /b H moiety in the b 6 f complex studied by EPR: a pair of strongly interacting hemes. Proc Natl Acad Sci (USA) 104:519–524
Bermudez MA, Galmes J, Moreno I, Mullineaux PM, Gotor C, Romero LC (2012) Photosynthetic adaptation to length of day is dependent on S-sulfocysteine synthase activity in the thylakoid lumen. Plant Physiol 160:274–288
Black MT, Widger WR, Cramer WA (1987) Large-scale purification of active cytochrome b 6 /f complex from spinach chloroplasts. Arch Biochem Biophys 252:655–661
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) Blast+: architecture and applications. BMC Bioinform 10:421–430
Choudhry FK, Rivero RM, Blumwald E, Mittler R (2016) Reactive oxygen species, abiotic stress and stress combination. Plant J 90:856–867
Cramer WA, Savikhin S, Yan J, Yamashita E (2009) The enigmatic chlorophyll a molecule in the cytochrome b 6 f complex. In: Rebeiz C et al (eds) The chloroplast system: Biochemistry and molecular biology, vol 31. Advances in Photosynthesis and Respiration. Springer, Dordrecht, pp 89–92
Cramer WA, Hasan SS, Yamashita E (2011) The Q cycle of cytochrome bc complexes: a structure perspective. Biochim Biophys Acta 1807:788–802
Crofts AR, Shinkarev VP, Kolling DRJ, Hong S (2003) The modified Q-cycle explains the apparent mismatch between the kinetics of reduction of cytochromes c 1 and bH in the bc 1 complex. J Biol Chem 278:36191–36201
Das K, Roychoudhury A (2014) Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front Environ Sci 2:53. https://doi.org/10.3389/fenvs.2014.00053
Dashdorj N, Zhang H, Kim H, Yan J, Cramer WA, Savikhin S (2005) The single chlorophyll a molecule in the cytochrome b 6 f complex: unusual optical properties protect the complex against singlet oxygen. Biophys J 88:4178–4187
DeLano WL (2002) The PyMOL molecular graphics system Software
Delepelaire P, Chua NH (1979) Lithium dodecyl sulfate/polyacrylamide gel electrophoresis of thylakoid membranes at 4 degrees C: characterizations of two additional chlorophyll a-protein complexes. Proc Natl Acad Sci USA 76:111–115
Farnese FS, Menezes-Silva PE, Gusman GS, Oliveira JA (2016) When bad guys become good ones: the key role of reactive oxygen species and nitric oxide in the plant responses to abiotic stress. Front Plant Sci 7:471. https://doi.org/10.3389/fpls.2016.00471
Frankel LK, Sallans L, Limbach PA, Bricker TM (2012) Identification of oxidized amino acid residues in the vicinity of the Mn4CaO5 cluster of Photosystem II: implications for the identification of oxygen channels within. Photosyst Biochem 51:6371–6377. https://doi.org/10.1021/bi300650n
Frankel LK, Sallans L, Limbach PA, Bricker TM (2013) Oxidized amino acid residues in the vicinity of QA and PheoD1 of the Photosystem II reaction center: putative generation sites of reducing-side reactive oxygen species. PLoS ONE 8:e58042
Furbacher PN, Girvin ME, Cramer WA (1989) On the question of interheme electron transfer in the chloroplast cytochrome b 6 in situ. Biochemistry 28:8990–8998
Galetskiy D, Lohscheider JN, Kononikhin AS, Popov IA, Nikolaev EN, Adamska I (2011) Mass spectrometric characterization of photooxidative protein modifications in Arabidopsis thaliana thylakoid membranes. Rapid Commun Mass Spectrom 25:184–190
Genova ML, Ventura B, Giuliano G, Bovina C, Formiggini G, Castelli GP, Lenaz G (2001) The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron–sulfur cluster N2. FEBS Lett 505:364–368
Hasan SS, Cramer WA (2014) Internal lipid architecture of the hetero-oligomeric cytochrome b 6 f complex. Structure 22:1008–1015
Hasan SS, Yamashita E, Baniulis S, Cramer WA (2013) Quinone-dependent proton transfer pathways in the photosynthetic cytochrome b 6 /f complex. Proc Natl Acad Sci (USA) 110:4297–4302
Hauska G (2004) The isolation of a functional cytochrome b 6 f complex: from lucky encounter to rewarding experiences. Photosyn Res 80:277–291
Hurt E, Hauska G (1981) A cytochrome f/b 6 complex of five polypeptides with plastoquinol-plastocyanin-oxidoreductase activity from spinach chloroplasts. Eur J Biochem 117:591–599
Kale R, Hebert AE, Frankel LK, Sallans L, Bricker TM, Pospíšil P (2017) Amino acid oxidation of the D1 and D2 proteins by oxygen radicals during photoinhibition of Photosystem. Proc Natl Acad Sci (USA) 114:2988–2993
Kar UK, Simonian M, Whitelegge JP (2017) Integral membrane proteins: bottom-up, top-down and structural proteomics. Expert Rev Proteom 14:715–723
Kim CS, Jung J (1992) Iron-sulfur centers as endogenous blue light sensitizers in cells: a study with an artificial non-heme iron protein. Photochem Photobiol 56:63–68
Kim H, Dashdorj N, Zhang H, Yan J, Cramer WA, Savikhin S (2005) An anomalous distance dependence of intra-protein chlorophyll-carotenoid triplet energy transfer. Biophys J 89:PL28–PL30
Kramer DM, Crofts AR (1994) Re-examination of the properties and function of the b cytochromes of the thylakoid cytochrome bf complex. Biochim Biophys Acta 1184:193–201
Kurisu G, Zhang H, Smith JL, Cramer WA (2003) Structure of the cytochrome b 6 f complex of oxygenic photosynthesis: tuning the cavity. Science 302:1009–1014
Lanciano P, Khalfaoui-Hassani B, Selamoglu N, Ghelli A, Rugolo M, Daldal F (2013) Molecular mechanisms of superoxide production by complex III: a bacterial versus human mitochondrial comparative case study. Biochim Biophys Acta 1827:1332 – 1339
Levesque-Tremblay G, Havaux M, Ouellet F (2009) The chloroplastic lipocalin AtCHL prevents lipid peroxidation and protects Arabidopsis against oxidative stress. Plant J 60:691–702
Long SP, Humphries S, Falkowski PG (1994) Photoinhibition of photosynthesis in nature. Ann Rev Plant Mol Biol 45:633–662
Mittler R (2016) ROS are good. Trends Plant Sci 7:405–410. https://doi.org/10.1016/j.tplants.2016.08.002
Mubarakshina MM, Ivanov BN (2010) The production and scavenging of reactive oxygen species in the plastoquinone pool of chloroplast thylakoid membranes. Physiol Plant 140:103–110
Nitschke W, Joliot P, Liebl U, Rutherford AW, Hauska G, Muller A, Riedel A (1992) The pH dependence of the redox midpoint potential of the 2Fe2S cluster from cytochrome b 6 f complex (the ‘Rieske centre’). Biochim Biophys Acta 1102:266–268
Peterman EJG et al (1998) Fluorescence and absorption spectroscopy of the weakly fluorescent chlorophyll a in cytochrome b 6 f of Synechocystis PCC6803. Biophys J 75:389–398
Pospíšil P (2009) Production of reactive oxygen species by Photosystem II. Biochim Biophys Acta 1787:1151–1160
Pospíšil P (2016) Production of reactive oxygen species by Photosystem II as a response to light and temperature stress. Front Plant Sci 7:1950. https://doi.org/10.3389/fpls.2016.01950
Rabilloud T, Vincon M, Garin J (1995) Micropreparative one- and two-dimensional electrophoresis. Improvement with new photopolymerization systems. Electrophoresis 16:1414–1422
Rich PR, Bendall DS (1980) The redox potentials for the b-type cytochromes of higher plant chloroplasts. Biochim Biophys Acta 591:153–161
Sang M et al (2010) High-light induced singlet oxygen formation in cytochrome b 6 f complex from Bryopsis corticulans as detected by EPR spectroscopy. Biophys Chem 146:7–12
Sang M et al (2011a) High-light-induced superoxide anion radical formation in cytochrome b 6 f complex from spinach as detected by. EPR spectroscopy Photosynthetica 49:48–54
Sang M et al (2011b) High-light induced superoxide radical formation in cytochrome b 6 f complex from Bryopsis corticulans as detected by EPR spectroscopy. Photochem Photobiol 102:177–181
Sarewicz M, Borek A, Cieluch E, Swierczek M, Osyczka A (2010) Discrimination between two possible reaction sequences that create potential risk of generation of deleterious radicals by cytochrome bc1. Implications for the mechanism of superoxide production. Biochim Biophys Acta 1797:1820–1827
Sievers F et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Sys Biol 7:539. https://doi.org/10.1038/msb.2011.75
Souda P, Ryan CM, Cramer WA, Whitelegge JP (2011) Profiling of integral membrane proteins and their post translational modifications using high-resolution mass spectrometry. Methods 55:330–336
Stofleth JT (2012) Understanding free radicals: Isolating active thylakoid membranes and purifying the cytochrome b 6 f complex for superoxide generation studies. J Pur Undergrad Res 2:64–69
Stroebel D, Choquet Y, Popot JL, Picot D (2003) An atypical haem in the cytochrome b 6 f complex. Nature 426:413–418
Suh H-J, Kim CS, Jung J (2000) Cytochrome b 6 /f complex as an indigenous photodynamic generator of singlet oxygen in thylakoid membranes. Photochem Photobiol 71:103–109
Sun G, Anderson VE (2004) Prevention of artifactual protein oxidation generated during sodium dodecyl sulfate-gel electrophoresis. Electrophoresis 25:959–965
Szymańska R, Dluzewska J, Slesak I, Kruk J (2010) Ferredoxin:NADP+ oxidoreductase bound to cytochrome b 6 f complex is active in plastoquinone reduction: implications for cyclic electron transport. Physiol Plant 141:289–298
T’Sai A, Palmer G (1983) Potentiometric studies on yeast complex III. Biochim Biophys Acta 722:349–363
Tripathy BC, Oelmuller R (2012) Reactive oxygen species generation and signaling in plants. Plant Signal Behav 7:1621–1633
Twigg AI, Baniulis D, Cramer WA, Hendrich MP (2009) EPR detection of an O2 surrogate bound to heme cn of the cytochrome b 6 f complex. J Amer Chem Soc 131:12536–12537
Weisz DA, Gross ML, Pakrasi HB (2017) Reactive oxygen species leave a damage trail that reveals water channels in Photosystem II. Sci Adv 3:eaao3013
Xu H, Freitas MA (2009) MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics 9:1548–1555
Yan J, Dashdorj N, Baniulis D, Yamashita E, Savikhin S, Cramer WA (2008) On the structural role of the aromatic residue environment of the chlorophyll a in the cytochrome b 6 f complex. Biochemistry 47:3654–3661
You J, Chan Z (2015) ROS regulation during abiotic stress in crop plants. Front Plant Sci 6:1092
Zhang H, Whitelegge JP, Cramer WA (2001) Ferredoxin NADP+ oxidoreductase is a subunit of the chloroplast cytochrome b 6 f complex. J Biol Chem 276:38159–38165
Acknowledgements
This work was supported by the United States Department of Energy, Office of Basic Energy Sciences Grant DE-FG02-09ER20310 given to TMB and LKF.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Taylor, R.M., Sallans, L., Frankel, L.K. et al. Natively oxidized amino acid residues in the spinach cytochrome b 6 f complex. Photosynth Res 137, 141–151 (2018). https://doi.org/10.1007/s11120-018-0485-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-018-0485-0