
Abstract
Non-photochemical quenching (NPQ) is a mechanism responsible for high light tolerance in photosynthetic organisms. In cyanobacteria, NPQ is realized by the interplay between light-harvesting complexes, phycobilisomes (PBs), a light sensor and effector of NPQ, the photoactive orange carotenoid protein (OCP), and the fluorescence recovery protein (FRP). Here, we introduced a biophysical model, which takes into account the whole spectrum of interactions between PBs, OCP, and FRP and describes the experimental PBs fluorescence kinetics, unraveling interaction rate constants between the components involved and their relative concentrations in the cell. We took benefit from the possibility to reconstruct the photoprotection mechanism and its parts in vitro, where most of the parameters could be varied, to develop the model and then applied it to describe the NPQ kinetics in the Synechocystis sp. PCC 6803 mutant lacking photosystems. Our analyses revealed that while an excess of the OCP over PBs is required to obtain substantial PBs fluorescence quenching in vitro, in vivo the OCP/PBs ratio is less than unity, due to higher local concentration of PBs, which was estimated as ~10−5 M, compared to in vitro experiments. The analysis of PBs fluorescence recovery on the basis of the generalized model of enzymatic catalysis resulted in determination of the FRP concentration in vivo close to 10% of the OCP concentration. Finally, the possible role of the FRP oligomeric state alteration in the kinetics of PBs fluorescence was shown. This paper provides the most comprehensive model of the OCP-induced PBs fluorescence quenching to date and the results are important for better understanding of the regulatory molecular mechanisms underlying NPQ in cyanobacteria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Oxygenic photosynthetic organisms encounter a significant problem when they tame the energy of light and convert it into more usable forms such as the energy of chemical bonds. This problem deals with potentially hazardous excessive light conditions, in which the photosynthetic apparatus needs to be safely protected by dissipation of the excessive light energy. In cyanobacteria, the light energy is absorbed by water-soluble light-harvesting complexes, phycobilisomes (PBs), and normally reaches the reaction centers of the photosystem II on the initial steps of photosynthesis. Under supra-optimal light conditions, due to the strictly controlled process of non-photochemical quenching (NPQ), it becomes possible to reduce the amount of energy being delivered to the reaction centers (Kirilovsky 2015). This is thought to substantially prevent formation of damaging oxidation products, reactive oxygen species (Bartosz 1997; Sedoud et al. 2014), but requires fine-tuned controlling of the components of NPQ. In vitro experiments (Gwizdala et al. 2011), aimed at recapitulation of processes known to occur in vivo, revealed that regulated photoprotective mechanism in cyanobacteria is realized by the set of three basic components: (1) light-harvesting antennae (PBs), (2) a sensor of elevated light levels and an effector which interacts with PBs and quenches their excessive fluorescence (the orange carotenoid protein, OCP), (3) the fluorescence recovery protein, FRP, which is required for abrogation of the OCP action and decouples the quenched OCP/PBs system. Although different aspects of structural and functional organization of PBs, OCP, and FRP, have been successfully studied (Kerfeld et al. 2003; Sutter et al. 2013; Liu et al. 2013; Leverenz et al. 2015; Gupta et al. 2015; Sluchanko et al. 2017), the complete picture of NPQ regulation is far from being fully understood, and some details of how this ternary OCP–PBs–FRP system operates require further intensive research.
It was proposed that as the major NPQ mediator the OCP exhibits a modular structure, with the two domains, N- and C-terminal (NTD and CTD), that hold a carotenoid molecule in the interdomain cavity (Kerfeld et al. 2003). The NTD and CTD, connected by a flexible linker, are believed to have a distinct, albeit still debatable, specialization: the NTD is suggested to be responsible for PBs binding, while the CTD is responsible for the interaction with FRP (Leverenz et al. 2015). Despite the structures of individual components are relatively well studied (Kirilovsky and Kerfeld 2016), the field desperately requires structural knowledge about OCP/FRP and OCP/PBs complexes and currently relies on often indirect biochemical and biophysical information.
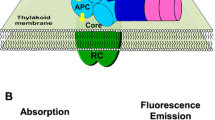
Figure 1 summarizes our current understanding of the OCP, PBs, and FRP involvement in the PBs fluorescence quenching process from the kinetics point of view. During the first stage (see Fig. 1), upon absorption of a blue–green light quantum by the OCP’s carotenoid chromophore, inactive orange OCP form (OCPO) is converted to an active red state (OCPR) (Wilson et al. 2008). During the second stage, the OCPR forms a stable complex with the PB, leading to an efficient quenching of the excess excitation energy (Tian et al. 2011, 2012; Maksimov et al. 2014) and ultimately reducing the amount of excitation delivered to the reaction centers of photosystem II and protecting from formation of hazardous reactive oxygen species. Alternatively, OCPR can spontaneously reconvert back to its inactive OCPO, or form a complex with the FRP (stage 7 in Fig. 1), which accelerates the conversion of the OCPR to OCPO (stage 6 in Fig. 1) significantly (Boulay et al. 2010; Sluchanko et al. 2017). Finally, under dark/low light conditions, the OCP dissociates from PBs with a rate k d. The FRP may also bind to the OCP–PB complex (stage 3 in Fig. 1), thus accelerating the OCP detachment from PBs (stage 5 in Fig. 1) and restoring an efficient energy flow from PBs to photosystem II reaction centers.

Current understanding of the OCP-induced PBs fluorescence quenching process in cyanobacteria mediated by the interplay of the OCP, FRP, and PBs. The following stages are indicated: (1) the OCP photoactivation and spontaneous reconversion, (2) the OCPR binding to PB, (3) binding of the FRP dimer to the OCPR–PB complex, (4) monomerization of the FRP and stabilization of the OCP–PB–FRP complex, (5) the FRP-assisted detachment of the OCP from PB, (6) the FRP-assisted conversion of the OCP to its stable orange state, (7) binding of the FRP dimer to the OCPR, (8) monomerization of the FRP and stabilization of the FRP–OCP complex. See text for details
An important factor in the context of the PBs fluorescence kinetics is the oligomeric state of the involved components, the OCP and FRP in particular. For example, it was shown that the FRP forms stable dimeric structures (Sutter et al. 2013; Sluchanko et al. 2017) but forms complexes with the OCP at a 1:1 stoichiometry, implying dissociation of the FRP dimers upon interaction with the OCP (Sluchanko et al. 2017). Most likely, the interaction between the OCP and FRP involves (i) initial binding of the FRP dimer to the OCPR with a consequent detachment of the FRP monomer (stage 7 in Fig. 1), which may have a different binding affinity to the OCPR, and (ii) further stabilization of the FRP/OCP complex (stage 8 in Fig. 1). The same stages of the FRP monomerization could be assumed for its interaction with the OCPR–PB complexes (stages 3 and 4 in Fig. 1). There are also observations in favor of the OCP self-association, especially at higher protein concentrations. It was suggested that the OCP may also exist in the dimeric form (Zhang et al. 2013), and in this case the OCP monomerization caused by its photoactivation must be taken into account when describing kinetics of NPQ. However, it seems likely that at low (micromolar) concentrations the OCPO and OCPR are both monomeric (Maksimov et al. 2016; Sluchanko et al. 2017).
As two steps are required for PBs fluorescence quenching (Fig. 1), under exposure to strong light, two stages can be detected in PBs fluorescence quenching upon exposure to strong light—light-dependent and light-independent (or “dark”) (Gorbunov et al. 2011). The light-dependent phase is detected on the onset of illumination and reflects binding of the activated OCPR to PBs. Its rate is determined by the slower of the two processes: the OCPO to OCPR conversion (high light intensities) or binding of the OCPR to PB (low light intensities). The “dark” phase of fluorescence quenching is observed after termination of strong illumination and reflects the binding of a portion of the activated OCPR to PBs, which leads to further quenching of PBs fluorescence. The typical rate of the dark phase PBs fluorescence quenching (k dark) is ~0.1 s−1 (Gorbunov et al. 2011; Kuzminov et al. 2012). The fluorescence recovery kinetics has been studied in less detail; however, it was shown that this process is strongly non-linear with its rate depending on the initial quenching level (Maksimov et al. 2015b). However, a detailed biophysical model of processes, underlying PBs fluorescence kinetics during NPQ, is lacking.
This study is aimed at comparative kinetic analysis of the PBs fluorescence quenching processes in cyanobacteria in vivo and in vitro, as well as at determination of relative concentrations of the components involved in the regulated photoprotective mechanism in vivo. Here we demonstrate that the ratios of the OCP, FRP, and PBS in vivo, as well as the origin of processes responsible for quenching, are significantly different than that in vitro, suggesting the predominant role of the intracellular microenvironment on the rates of protein diffusion and interaction.
Materials and methods
In vivo experiments
The wild-type cells of the cyanobacterium Synechocystis sp. PCC 6803 were grown in modified BG-11 medium at 30 °C constant white light illumination of 40 µmol photons m−2 s−1. A ΔPSI/ΔPSII (ΔpsaAB/ΔpsbDIC/ΔpsbDII) mutant of Synechocystis sp. PCC 6803, lacking photosystems I and II (Ermakova-Gerdes et al. 2015), was cultivated in BG-11 medium, containing glucose (15 mM), spectinomycin (25 µg/ml), erythromycin (20 µg/ml), and chloramphenicol (20 µg/ml) at light exposure of 5 µmol photons m−2 s−1 as described in (Kuzminov et al. 2014; Maksimov et al. 2015b).
The samples were excited with 630 nm light, and fluorescence emission was measured in a 90° scheme using a Maya2000 Pro spectrometer (Ocean Optics), allowing fluorescence kinetic measurements with 0.1 s resolution. Non-photochemical quenching was induced simultaneously with a 3000 µmol photons m−2 s−1 455 nm LED.
Fluorescence recovery kinetics measured after prolonged illumination, which exceeded the times required to reach the maximal quenching (>200 s), all had similar shape. This indicates the absence of irreversible photodegradation in the system during the experiments.
In vitro experiments
The OCP, FRP, and PBs preparations for the in vitro experiments were obtained as described previously (Sluchanko et al. 2017). N-terminally 6xHis-tagged wild-type orange carotenoid protein (OCP) from Synechocystis sp. PCC 6803 was expressed in E. coli NEB DH5α Turbo cells carrying the whole machinery for carotenoid synthesis and OCP/FRP were purified by a combination of immobilized metal-affinity and size-exclusion chromatography (SEC) as described in (Maksimov et al. 2016). Protein concentrations were determined on a Lambda-25 spectrophotometer (Perkin-Elmer, USA) using molar extinction coefficients ε λ = 280 nm = 15,220 M−1 cm−1 for FRP and ε λ = 495 nm = 63,000 M−1 cm−1 for the OCP. PBs purification and concentration measurement was conducted according to the procedures described in (Stadnichuk et al. 2013).
Experiments on PB fluorescence quenching were conducted in a 0.75 M phosphate buffer at pH 7.4, the temperature was fixed to 25 °C. A M455L3 (Thorlabs, USA) 900 mW light-emitting diode with maximum emission at 455 nm was used for blue–green illumination of the samples (i.e., OCP photoswitching and PBs quenching induction). The steady-state fluorescence measurements were performed using FluoroMax-4 spectrofluorimeter (Horiba Jobin Yvon, Japan-France).
Results and discussion
OCP photoconversion and interaction with FRP in vitro
Although it includes three basic components, the model of OCP-induced PBs quenching in cyanobacteria (Fig. 1) considers several transient complexes: OCPR, OCPR–FRP, OCPR–PB, and FRP–OCPR–PB, whose populations are governed by more than 10 parameters. In vitro experiments allow us to simplify this model by dividing it into several blocks of processes, which can be studied independently: (i) OCP photoconversion, (ii) OCP–FRP interactions, (iii) OCP–PB interaction, and (iv) FRP–OCP–PB interaction. The number of parameters in each sub-model is greatly reduced, thus permitting us to improve the precision of retrieving characteristics of each process.
First, we examined the in vitro kinetics of light-induced alterations of the OCP within the two-component system of OCP and FRP. Under illumination with strong blue–green light, the OCP exhibits a transition to its active state and the rate of this process is proportional to light intensity F and photoconversion cross-section σ:
After the offset of illumination, reconversion to the OCPO state occurs with the rate k r:
The combination of these two processes leads to the following equation describing the reversible photoconversion kinetics of the OCP (Maksimov et al. 2015a):
where R is the concentration of the OCPR normalized to [OCP]T. Hereinafter, [OCP]T, [FRP]T, and [PBs]T are the total concentrations of the OCP, FRP, and PBs, respectively.
Using this equation, kinetics of light-induced OCP transition was analyzed at different ionic strength, namely, 0.15 and 0.75 M phosphate buffer. The latter buffer was used further in the in vitro experiments to maintain PBs stability. In accordance with (Gwizdala et al. 2011), we observed that an increased ionic strength had a significant influence on both conversion level and rates, namely, only 10% of the OCP could be photoactivated in 0.75 M phosphate buffer (Fig. S1). The OCPR relaxation rate in the 0.75 M phosphate buffer was k r = 0.04 s−1. The photoconversion cross section was similar for both ionic strengths and equal to σ = (2.0 ± 0.2) × 10−19 cm2, that is in accordance with literature data (Maksimov et al. 2015a).
In the presence of the FRP, the following processes must be taken into account to quantify the kinetics of OCP conversion: FRP binding to OCPR, which occurs with a rate constant k FRP,
and the subsequent decay of the OCPR–FRP complex (i.e., the accelerated FRP-assisted conversion of OCPR to OCPO) with a back conversion rate k C:
This model is represented as a scheme in Fig. 2a and leads to the following system of equations describing the kinetics of light-induced OCP transitions in the presence of the FRP:

a The scheme of OCP–FRP interaction, b the OCP conversion kinetics measured at different FRP concentrations. The conversion level in the absence of FRP was taken as 1. Black lines correspond to the approximation with Eq. (6). [OCP]T = 4 × 10−6 M
where R and K are the concentrations of the OCPR and OCPR–FRP complexes normalized to [OСP]T, β = [FRP]T/[OCP]T, and \(k_{{\text{FRP}}}^*={k_{{\text{FRP}}}}{\left[ {{\text{OCP}}} \right]_{\text{T}}}\).
Figure 2b shows the OCP photoconversion kinetics at different β values, i.e.. at different [FRP]T. By fitting the measured kinetics to the model (6), we have obtained the following parameters: \(k_{{\text{FRP}}}^*\) = 0.1 s−1, k C = 0.15 s−1. These results clearly suggest that the presence of the FRP accelerates the OCPR conversion to OCPO in darkness by a factor of ~4, which is in agreement with the literature data (Boulay et al. 2010; Gwizdala et al. 2011, 2013; Sluchanko et al. 2017). Interestingly, (Sluchanko et al. 2017) estimated the value of the dissociation constant for the OCPR–FRP complex (K d ≈ 2 μM) based on data obtained for the purple OCP-W288A mutant, which was shown to be an analogue of the red form of OCP. Given that by definition K d is the inverse ratio of binding (k FRP) and OCPR–FRP back conversion (k C) rate constants, the obtained values result in a 0.15 s−1/(0.1 s−1/4 × 10−6 M) = 6 μM value of K d, which is in a qualitative agreement with the data of (Sluchanko et al. 2017). Therefore, modeling of the kinetic curves allowed us to obtain independent estimations of equilibrium constants.
Kinetics of PB fluorescence quenching in vitro
As shown in (Gwizdala et al. 2011), reconstitution of the OCP-mediated photoprotective mechanism in cyanobacteria is possible in vitro using three components: OCP, PBs, and FRP. However, modeling of the initial phase of photoprotection, i.e., PBs fluorescence quenching, can also be done on the two-component system of OCP and PBs.
For the two-component OCP–PBs system, the light-induced PBs quenching kinetics can be fully described by the model that takes into account the following processes: OCP photoconversion and relaxation, described by Eqs. (1) and (2), respectively; binding of the OCPR to PBs and formation of the OCPR–PB complexes in the 1:1 stoichiometry (Wilson et al. 2006) with a rate constant k PB:
and decay of the OCPR—PB complexes to the OCPO and free PB with the rate constant k d:
Under the assumption of complete PBs fluorescence quenching upon their interaction with the OCPR, the measured fluorescence signal is proportional to the number of free phycobilisomes, i.e., ([PBs]T − [OCPR–PBs]). The processes are presented in Fig. 3a and can be described by the system of two differential equations:

a The scheme of PBs fluorescence quenching by the OCP. b Light-induced PBs fluorescence quenching obtained at different [OCP]T/[PBs]T ratio at total PBs concentration [PBs]T = 5 × 10−11 M and their fits with model (1). The curves were normalized to maximum
Here, R and C stand for the concentrations of OCPR and OCPR–PBs complexes normalized to [PBs]T. Importantly, Eqs. (9) contain the term α = [OCP]T/[PBs]T, which can be used for estimation of the relative OCP concentration from a set of fluorescence quenching curves. Also, the rate constant \(k_{{\text{PB}}}^*={k_{{\text{PB}}}}{\left[ {{\text{PBs}}} \right]_{\text{T}}}\). We note that the term that describes interaction between OCPR and PB in the model (9), is different from that described in (Gorbunov et al. 2011) as it takes into account the depletion of free PBs with time caused by their occupation with the OCPR. Description of the dark phase of PB fluorescence quenching can be readily achieved with Eqs. (9) by setting F to zero.
Figure 3b shows the kinetics of PBs fluorescence quenching under exposure to strong light (3000 μM s−1 cm−2 455 nm) obtained in the samples with different [OCP]T/[PBs]T ratios. On the offset of the illumination, a “dark” phase of PBs fluorescence quenching is observed (Fig. S2), which is due to interaction of the OCP molecules, activated during the light phase, with PBs (Gorbunov et al. 2011). Fluorescence quenching was measured for varying [OCP]T/[PBs]T for three fixed [PBs]T values until fluorescence signal reached a steady-state, that corresponded to a stationary concentration of OCPR–PBs complexes. As shown in Fig. 3b, an efficient PBs fluorescence quenching was observed only at a significant excess of the OCP over PBs, [OCP]T > [PBs]T, that is in agreement with the results of (Gwizdala et al. 2011; Sluchanko et al. 2017).
The maximum possible number of OCPR–PBs complexes is [PBs]T if the OCPR decoupling is neglected. Under such conditions, zero fluorescence would be observed for 100% quenching efficiency, however, only a 50–60% quenching occurred in our experiments. This relatively small amplitude of fluorescence quenching could be due to the presence of a fraction of free phycocyanine rods, which are a part of PBs, in the solution, which cannot be quenched by the OCP (Gwizdala et al. 2011; Tian et al. 2012). This suggestion is supported by the fact that the spectral maximum of PBs fluorescence emission without the OCP was at 659 nm—in comparison to the previously published steady-state fluorescence emission spectra (Gwizdala et al. 2011), the observed spectrum was blue-shifted by more than 10 nm. It suggests that the PBs used in the in vitro experiments could be partly dissociated, and a mixture of PBs species was investigated. Moreover, incomplete quenching could be caused by less than 100% excitation energy transfer from PBs to the His-tagged OCP due to the geometry of OCPR–PBs complex. To take this fact into account while modeling the quenching kinetics, we used the following expression for fluorescence intensity estimation:
where ϕ is the fluorescence quantum yield of the OCPR–PBs complexes. To assess the value of ϕ, the dependence of stationary fluorescence levels on [PBs]T/[OCP]T was obtained for each series of measurements and then extrapolated to zero, where fluorescence corresponded to an infinite OCP concentration, i.e., to the impact of the non-fully quenched complexes.
We performed the global approximation of PBs fluorescence quenching kinetics, including both light and dark phases, recorded at 10 varying values of α = [OCP]T/[PBs]T with the biophysical model (Eqs. 9, 10) for three different [PBs]T concentrations. As the OCPR reconversion rate k r = 0.04 s−1 and the photoconversion cross-section σ were determined in independent experiments (Fig. 2b), the fitting procedure resulted in determination of two remaining parameters, \(k_{{\text{PB}}}^*\) and k d (Table 1).
Our results (Table 1) revealed the following trends. First, the rate \(k_{{\text{PB}}}^*\), which determines the OCP interaction with free PB, is proportional to [PB]T. This dependence is indicative of a predominant role of diffusion in this process. Second, the k d values are virtually independent on [PBs]T. This rate of k d (~5 × 10−5 s−1) is extremely slow and is in agreement with the previous observations of slow PBs fluorescence recovery (~10−3 s−1) in the absence of the FRP (Boulay et al. 2010; Gwizdala et al. 2011). Finally, approximately the same maximum quenching levels (~55%) were observed in different series of experiments, possibly indicating heterogeneity in PBs preparation.
We also note that the apparent rate of the dark phase of fluorescence quenching (k dark), obtained by monoexponential approximation, was independent of [PBs]T (Fig. S2). As this rate is determined by two processes, namely, mutual diffusion of the OCP and PBs and OCP binding to PBs, one could expect the concentration dependence of k dark on [PBs]T similar to that for \(k_{{\text{PB}}}^*\). However, it can be shown that the rate of dark phase is governed by the \(k_{{\text{PB}}}^*/{k_{\text{r}}}\) ratio, and as \(k_{{\text{PB}}}^*<<{k_{\text{r}}}\) in case of in vitro experiments, quenching kinetics is governed by the OCPR relaxation, and diffusion processes are not pronounced. It was reported previously (Gorbunov et al. 2011; Kuzminov et al. 2012) that k dark for Synechocystis sp. PCC 6803 is ~0.1 s−1, that is similar to our data obtained in vitro (Table 1); however, as will be shown below, the rate of the dark phase in vivo is dependent on the quenching level and is not limited by k r.
In the in vitro experiments with the three-component system (PBs, OCP, and FRP), the FRP addition resulted in a dramatic reduction of PBs fluorescence quenching efficiency (Fig. S3A). However, addition of FRP to the OCP–PBs system significantly complicates the model (9), as FRP is involved into four processes: binding to the OCPR [Eq. (4)], reconversion of the OCPR to OCPO [Eq. (5)], binding to the OCPR–PBs complex with the rate constant k FRP–PB
and a subsequent FRP-accelerated decay of the OCPR–PB complex with a rate constant k d–PBs:
As shown above, the rate of the OCPR binding to PB is k PB ≈ 2 × 105 M−1 s−1 (Table 1), while the rate of the OCPR binding to FRP is an order of magnitude lower, k FRP ≈ 2 × 104 M−1 s−1. Considering that k FRP–PB and k FRP are of the same order of magnitude, the role of FRP in PBs fluorescence quenching in vivo can be described as follows. During the in vitro experiments, [OCP]T >> [PBs]T, hence, FRP binding to the OCPR is highly efficient, i.e., the corresponding rate exceeds the rate of the OCP binding to PBs, and an addition of low (<<[OCP]T) concentrations of the FRP significantly reduces the quenching level (Fig. S3B). Assuming that in vivo [OCP]T ~ [PBs]T, and taking into account that k FRP << k FRP–PB, it can be expected that the FRP binding to the OCP can be neglected when processing the quenching curves in vivo.
Kinetics of PB fluorescence quenching in vivo
PB fluorescence quenching kinetics obtained for intact cells of the Synechocystis sp. PCC 6803 ΔPSI/ΔPSII mutant at different illumination intensities are presented in Fig. 4.

Similarly to the in vitro experiments, the in vivo PBs fluorescence quenching kinetics measured in the ΔPSI/ΔPSII mutant of Synechocystis sp. PCC 6803 were fitted to Eqs. (9, 10), but the fitting procedure itself was essentially different. While for the in vitro experiments the [OCP]T/[PBs]T ratio (α) was fixed, it was unknown for cells, and the reconversion rate for OCPR could be determined in independent measurements, while it had to be varied in vivo. As the in vitro experiments were performed at specific conditions required to maintain the PBs structure (in a 0.75 M buffer, pH 7.4), properties of microenvironment could be significantly different for a cell, thus setting limitations on a direct extrapolation of the parameters obtained in vitro for the in vivo case. Hence, we performed global fitting of in vivo PBs quenching curves (Fig. 4) which exhibit different dependencies on the fitting parameters. Totally, ~30 quenching curves were simultaneously fitted for each experiment. Table 2 shows parameters used in the fitting procedure and summarize the retrieved values.
Our results revealed the following striking differences between PBs fluorescence quenching processes in vitro and in vivo. First, the comparison of OCPR–PBs interaction rate constants obtained in vitro and in vivo allows estimation of the effective [PBs]T in cell as ~10−5 M, that is four orders higher than the maximum [PBs]T value used in the in vitro experiments, and local intracellular concentrations of PBs in cell could be even higher. While it was demonstrated above (Table 1) that the OCPR–PBs interaction in vitro is governed by free diffusion, as its rate is proportional to [PBs]T, OCPR–PBs interaction in the cell can be influenced by other processes such as slower diffusion or increased viscosity, thus making [PBs]T estimation from kinetic data speculative. However, the number of PBs per cell can be estimated as ~1000 from the cryo electron microscopy data (Wilson et al. 2006), that for a volume of 1 μm3 gives a ~10−5 M concentration. As a result, a similar quenching level is reached in both cases, but in vitro an excess of the OCP over PBs is required due to low local concentrations, and the quenching efficiency for a single OCPR–PBs complex is relatively low. In contrast, in cells almost complete quenching in OCPR–PBs complexes takes place, and high local concentrations make the overall quenching efficient even at [OCP]T/[PBs]T < 1. Finally, almost 10 times higher value of k d was obtained from fluorescence quenching curves in vivo, that is explained by the FRP-assisted acceleration of PBs fluorescence recovery.
When processing the PBs fluorescence quenching phase, we made an assumption about the absence of interactions with the FRP, which resulted in a satisfactory approximation of the data. During the PBs recovery phase, such an assumption is invalid, and FRP-related processes must be taken into account, thus increasing the number of parameters and making the modeling more complex. However, one of the main goals of this work was to verify whether assessment of PBs, OCP, and FRP concentrations is possible on the basis of mathematical description of PBs fluorescence kinetics in cyanobacteria cells; hence, it was necessary to suggest a simplified model of fluorescence recovery, as the FRP, in opposite to the OCP, cannot be quantified solely from the quenching kinetics.
Kinetics PBs fluorescence recovery in vivo
Figure 5a demonstrates the time courses of PBs fluorescence recovery in darkness observed in vivo in Synechocystis sp. PCC 6803 ΔPSI/ΔPSII mutant cells on the offset of illumination. These curves were obtained for different durations pre-exposure to strong light and thus different initial levels of PB fluorescence quenching prior to recovery.

a The time course of fluorescence recovery in intact cells of the ΔPSI/ΔPSII mutant at different initial levels of fluorescence quenching. b The dependence of fluorescence recovery rate on the OCPR–PBs complex concentration (PB fluorescence quenching level) obtained at different temperatures. c, d The dependence of the initial enzyme cycling velocity ν 0 and enzyme cycling velocity reduction η on the initial level of PBs fluorescence quenching
The dependence of the recovery rate on the initial quenching level was strongly non-linear (Fig. 5). The higher was the initial amplitude of fluorescence quenching, the slower was the subsequent fluorescence recovery. This non-linearity cannot be explained by a commonly used first-order kinetics approach, which assumes that the recovery rate is independent on the initial concentration of quenchers. Moreover, our analysis of the time course of the rate of fluorescence recovery d[PBs]/dt (Fig. 5b) revealed that this dependence is non-monotonous, suggesting the presence of several processes that influence the fluorescence recovery rate.
To quantify the non-linear pattern of the PBs fluorescence recovery, we applied an approach that was previously used for description of the kinetics of catalytic reactions. The process of recovery can be presented as a sequence of the following two reactions: (i) OCPR–PB complex (substrate, S) slowly decays to OCPO and free PBs (product, P), (ii) this conversion is accelerated by the FRP (F) by forming a ternary OCPR–PB–FRP complex (SF). Collectively, these processes can be represented by the standard scheme of enzymatic catalysis:
In order to describe the system behavior and elucidate the origin of fluorescence recovery non-linearity, we further applied the generalized approach for enzyme kinetics investigation (Cao and Enrique 2013) that allows one to fit the time course of product formation with the following equation:
where ν 0 represents the initial product formation rate, whereas η indicates non-linearity of the curve caused by the reduction in the enzyme cycling velocity.
Figure 5с, d shows the dependence of the retrieved ν 0 and η values obtained by fitting PBs fluorescence recovery kinetics to Eq. (14) on the initial amplitude of quenching, which is proportional to concentration of the substrate (i.e., OCPR—PB complexes).
The observed saturation of the initial velocity (Fig. 5с) corresponds to the “enzyme” (FRP) saturation by the substrate, which is consistent with the suggested excess of OCP molecules over the FRP in vivo. Above the initial quenching levels of 30%, the maximum rate of recovery is constant and equal to 14 s−1. This is due to the fact that all FRPs are attached to OCPR–PB complexes, thus providing the maximum possible rate of recovery. As the initial quenching level and thus the initial substrate concentration decrease, the maximum recovery rate goes down. For instance, at the initial quenching of 7%, ν 0 was equal to 7 s−1, i.e., ~50% of the maximum recovery rate. Under such conditions, the substrate concentration becomes comparable to the FRP concentration, and [FRP]T can be estimated as the concentration of OCP–PBs complexes at the corresponding quenching level. As the maximum quenching level corresponds to the [OCP]T/[PBs]T ratio, and 7% quenching corresponds to ~10% of the maximum quenching, [FRP]T can be estimated as ~0.1·[OCP]T.
As the initial rate of fluorescence recovery reaches a plateau at moderate quenching amplitudes, the rate of this process is determined by the turnover rate of the enzyme. The values of the enzyme cycling velocity reduction exceed the reciprocal observation time τ: η > τ −1 (τ −1 is shown with a horizontal dashed line in Fig. 5b), hence the fluorescence recovery process is strongly non-linear. When η decreases with substrate concentration, the non-linearity mainly comes from the substrate depletion (Cao and Enrique 2013). However, fluorescence recovery is non-linear even in the substrate concentration region corresponding to ν 0 saturation. This fact indicates that substrate depletion is not the only source of non-linearity in the system.
To further investigate the FRP functioning in vivo, we examined the dependence of fluorescence recovery rate on the OCPR–PBs concentration (Fig. 5b). The measured curves exhibit sigmoidal shape and two deflection points, that, according to the enzyme catalysis literature, corresponds to allosteric functioning of an enzyme. This pattern means that an enzyme is capable of changing its properties, such as its oligomeric state, during the time course of product formation. Another feature of the allosteric influence is that it becomes less pronounced at elevated temperatures (Teipel and Koshland 1969), that is also shown in Fig. 5b.
The FRP normally exists in the dimeric form, while upon interaction with the OCPR it forms monomers (Sluchanko et al. 2017). We propose the following working model to explain the non-linear kinetics of PBs fluorescence recovery. Dimers and monomers of the FRP have different affinity to the OCPR–PBs complex. In this case, the FRP, mainly existing in dimeric form, forms complexes with the OCPR–PBs with lower affinity, and after that all dimeric FRP splits into bound and free monomeric fractions. We also note that the sigmoidal shape of PBs fluorescence recovery curve, which is observed at high quenching levels (see e.g., the curve corresponding to 56% quenching in Fig. 5a), can be explained as a consequence of dimer–monomer transition of the FRP, which accompanies its interaction with the OCP–PBs complexes. Following this, free FRP is mainly in the monomeric form, and the FRP binding to the OCP–PBs complexes occurs with the higher rates, which correspond to the FRP monomers. Finally, when PBs fluorescence approaches its complete recovery, dimers of the FRP start to reappear, thus slowing down the reaction rate again. This hypothesis explains the behavior of PBs fluorescence recovery (Fig. 5b) and strong non-linearity (Fig. 5c); however, further research is necessary to prove this model.
Conclusions
In this work, we presented a biophysical model, which allows for the description of experimental PBs fluorescence kinetics following cyanobacteria illumination with a strong blue–green light. This process is governed by the interplay between three components, PBs, OCP, and FRP, as well as their transient complexes. Using the possibility to reconstruct the OCP-mediated photoprotective mechanism and its parts in vitro, we performed an independent testing of the suggested model on the OCP–FRP and OCP–PBs systems. It was shown that that the dissociation constant for the OCPR–FRP complex estimated from the modeling data as the ratio of OCP and FRP interaction rate constants was equal to 6 μM, that is in a good agreement with the experimental data (Sluchanko et al. 2017). The rate constant of OCPR binding to PBs was shown to be proportional to [PBs]T, as expected for the bimolecular diffusion-limited interaction. Application of the biophysical model to the analysis of PBs fluorescence quenching kinetics in vivo provided for the estimations of relative OCP concentration ([OCP]T/[PBs]T ≈ 0.6) and the local PBs concentration ([PBs]T ~ 10−5 M) in cells, as well as the efficiency of a single PB fluorescence quenching by the OCPR (98%). Next, description of PBs fluorescence recovery kinetics on the basis of the generalized model of enzymatic catalysis (Cao and Enrique 2013) provided for the estimation of the FRP concentration in cells as [FRP]T/[OCP]T ≈ 0.1. Our proposed approach allows one to determine relative OCP, FRP, and PBs concentrations in vivo, and can be used to quantify the NPQ characteristics in different strains of cyanobacteria, as well as in cyanobacteria grown/living in different environmental conditions. Indeed, the time course of non-photochemical quenching is determined by [PBS]T, [FRP]T, or [OCP]T concentrations. In vivo, the OCP to FRP ratio is regulated by the slr1963 (OCP) and slr1964 (FRP) gene expression levels (Boulay et al. 2010), which may vary depending on the environmental conditions. Hence, the approach, suggested in this work, could be used for the examination of fine tuning of the molecular machinery responsible for photoprotection in cyanobacteria.
References
Bartosz G (1997) Oxidative stress in plants. Acta Physiol Plant 19(1): 47–64
Boulay C, Wilson A, D’Haene S, Kirilovsky D (2010) Identification of a protein required for recovery of full antenna capacity in OCP-related photoprotective mechanism in cyanobacteria. Proc Natl Acad Sci USA 107(25):11620–11625
Cao W, Enrique M (2013) Quantitative full time course analysis of nonlinear enzyme cycling kinetics. Sci Rep 3:2658
Ermakova-Gerdes S, Shestakov S, Vermaas WFJ (2015) Development of a photosystem I-less strain of Synechocystis sp. PCC 6803 for analysis of mutations in the photosystem II proteins D2 and CP43. In: Mathis P (ed) Photosynthesis: from biology to biosphere, vol 1. Kluwer, Dordrecht, pp 483–486
Gorbunov MY, Kuzminov FI, Fadeev VV, Kim, JD, Falkowski PG (2011) A kinetic model of non-photochemical quenching in cyanobacteria. BBA Bioenerg 1807(12):1591–1599
Gupta S, Guttman M, Leverenz RL, Zhumadilova K, Pawlowski EG, Petzold CJ et al Kerfeld CA (2015) Local and global structural drivers for the photoactivation of the orange carotenoid protein. Proc Natl Acad Sci USA 112(41):E5567–E5574
Gwizdala M, Wilson A, Kirilovsky D (2011) In vitro reconstitution of the cyanobacterial photoprotective mechanism mediated by the orange carotenoid protein in Synechocystis PCC 6803. Plant Cell 23(7):2631–2643
Gwizdala M, Wilson A, Omairi-Nasser A, Kirilovsky D (2013) Characterization of the Synechocystis PCC 6803 fluorescence recovery protein involved in photoprotection. BBA Bioenerg 1827(3):348–354
Kerfeld CA, Sawaya MR, Brahmandam V, Cascio D, Ho KK, Trevithick-Sutton CC, Krogmann DW, Yeates TO (2003) The crystal structure of a cyanobacterial water-soluble carotenoid binding protein. Structure 11(1):55–65
Kirilovsky D (2015) Modulating energy arriving at photochemical reaction centers: orange carotenoid protein-related photoprotection and state transitions. Photosynth Res 126(1):3–17
Kirilovsky D, Kerfeld CA (2016) Cyanobacterial photoprotection by the orange carotenoid protein. Nat Plants 2:16180
Kuzminov FI, Karapetyan NV, Rakhimberdieva MG, Elanskay IV, Gorbunov MY, Fadeev VV (2012) Investigation of OCP-triggered dissipation of excitation energy in PSI/PSII-less Synechocystis sp. PCC 6803 mutant using non-linear laser fluorimetry. BBA Bioenerg 1817(7):1012–1021
Kuzminov FI, Bolychevtseva YV, Elanskaya IV, Karapetyan NV (2014) Effect of APCD and APCF subunits depletion on phycobilisome fluorescence of the cyanobacterium Synechocystis PCC 6803. J Photochem Photobiol B 133:153–160
Leverenz RL, Sutter M, Wilson A, Gupta S, Thurotte A, de Carbon CB et al. (2015) A 12 Å carotenoid translocation in a photoswitch associated with cyanobacterial photoprotection. Science 348(6242):1463–1466
Liu H, Zhang H, Niedzwiedzki DM, Prado M, He G, Gross ML, Blankenship RE (2013) Phycobilisomes supply excitations to both photosystems in a megacomplex in cyanobacteria. Science 342(6162): 1104–1107
Maksimov EG, Schmitt FJ, Shirshin EA, Svirin MD, Elanskaya IV, Friedrich T et al. (2014) The time course of non-photochemical quenching in phycobilisomes of Synechocystis sp. PCC6803 as revealed by picosecond time-resolved fluorimetry. BBA Bioenerg 1837(9):1540–1547
Maksimov EG, Shirshin EA, Sluchanko NN, Zlenko DV, Parshina EY, Tsoraev GV et al. (2015a) The signaling state of orange carotenoid protein. Biophys J 109(3):595–607
Maksimov EG, Klementiev KE, Shirshin EA, Tsoraev GV, Elanskaya IV, Paschenko VZ (2015b) Features of temporal behavior of fluorescence recovery in Synechocystis sp. PCC6803. Photosynth Res 125(1–2):167–178
Maksimov EG, Moldenhauer M, Shirshin EA, Parshina EA, Sluchanko NN, Klementiev KE, Tsoraev GV, Tavraz NN, Willoweit M, Schmitt FJ, Breitenbach J, Sandmann G, Paschenko VZ, Friedrich T, Rubin AB (2016) A comparative study of three signaling forms of the orange carotenoid protein. Photosynth Res 130(1):389–401
Sedoud A, López-Igual R, Rehman A, Wilson A, Perreau F, Boulay C et al. (2014) The cyanobacterial photoactive orange carotenoid protein is an excellent singlet oxygen quencher. Plant Cell 26(4):1781–1791
Sluchanko NN, Klementiev KE, Shirshin EA, Tsoraev GV, Friedrich T, Maksimov EG (2017) The purple Trp288Ala mutant of Synechocystis OCP persistently quenches phycobilisome fluorescence and tightly interacts with FRP. BBA Bioenerg 1858(1):1–11
Stadnichuk IN, Yanyushin MF, Bernat G, Zlenko DV, Krasilnikov PM, Lukashev EP, Maksimov EG, Paschenko VZ (2013) Fluorescence quenching of the phycobilisome terminal emitter LCM from the cyanobacterium Synechocystis sp. PCC 6803 detected in vivo and in vitro. J Photochem Photobiol B 125:137–145
Sutter M, Wilson A, Leverenz RL, Lopez-Igual R, Thurotte A, Salmeen AE et al. (2013) Crystal structure of the FRP and identification of the active site for modulation of OCP-mediated photoprotection in cyanobacteria. Proc Natl Acad Sci USA 110(24):10022–10027
Teipel J, Koshland DE (1969) Significance of intermediary plateau regions in enzyme saturation curves. Biochemistry 8(11):4656–4663
Tian L, van Stokkum IH, Koehorst RB, Jongerius A, Kirilovsky D, van Amerongen H (2011) Site, rate, and mechanism of photoprotective quenching in cyanobacteria. J Am Chem Soc 133(45):18304–18311
Tian L, Gwizdala M, van Stokkum IH, Koehorst RB, Kirilovsky D, van Amerongen H (2012) Picosecond kinetics of light harvesting and photoprotective quenching in wild-type and mutant phycobilisomes isolated from the cyanobacterium Synechocystis PCC 6803. Biophys J 102(7):1692–1700
Wilson A et al (2006) A soluble carotenoid protein involved in phycobilisome-related energy dissipation in cyanobacteria. Plant Cell 18(4):992–1007
Wilson A, Punginelli C, Gall A, Bonetti C, Alexandre M, Routaboul JM et al. (2008) A photoactive carotenoid protein acting as light intensity sensor. Proc Natl Acad Sci USA 105(33):12075–12080
Zhang H, Liu H, Niedzwiedzki DM, Prado M, Jiang J, Gross ML, Blankenship RE (2013) Molecular mechanism of photoactivation and structural location of the cyanobacterial orange carotenoid protein. Biochemistry 53(1):13–19
Acknowledgements
The work was supported by the Russian Foundation for Basic Research (Grant No. 16-05-01110), the German Ministry for Education and Research (to T.F.; WTZ-RUS grant 01DJ15007), the Russian Science Foundation (grant №14-17-00451), and the German Research Foundation—Cluster of Excellence “Unifying Concepts in Catalysis” (to T.F.). E.G.M. thanks the Russian Foundation for Basic Research (project No. 15-04-01930A), the Russian Ministry of Education and Science (project MK-5949.2015.4), the Dynasty Foundation Fellowship, RFBR, and Moscow City Government according to the research project No. 15-34-70007 «mol_а_mos» for partial support of this work. N.N.S. was supported by a scholarship from the President of Russian Federation (SP-367.2016.4). M.Y.G. was supported by NASA Ocean Biology and Biogeochemistry Program (Grant NNX16AT54G). We thank Kevin Wyman for comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shirshin, E.A., Nikonova, E.E., Kuzminov, F.I. et al. Biophysical modeling of in vitro and in vivo processes underlying regulated photoprotective mechanism in cyanobacteria. Photosynth Res 133, 261–271 (2017). https://doi.org/10.1007/s11120-017-0377-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-017-0377-8