
Abstract
Oxygen evolution by photosystem II (PSII) involves activation by Cl− ion, which is regulated by extrinsic subunits PsbQ and PsbP. In this study, the kinetics of chloride activation of oxygen evolution was studied in preparations of PSII depleted of the PsbQ and PsbP subunits (NaCl-washed and Na2SO4/pH 7.5-treated) over a pH range from 5.3 to 8.0. At low pH, activation by chloride was followed by inhibition at chloride concentrations >100 mM, whereas at high pH activation continued as the chloride concentration increased above 100 mM. Both activation and inhibition were more pronounced at lower pH, indicating that Cl− binding depended on protonation events in each case. The simplest kinetic model that could account for the complete data set included binding of Cl− at two sites, one for activation and one for inhibition, and four protonation steps. The intrinsic (pH-independent) dissociation constant for Cl− activation, K S, was found to be 0.9 ± 0.2 mM for both preparations, and three of the four pK as were determined, with the fourth falling below the pH range studied. The intrinsic inhibition constant, K I, was found to be 64 ± 2 and 103 ± 7 mM for the NaCl-washed and Na2SO4/pH7.5-treated preparations, respectively, and is considered in terms of the conditions likely to be present in the thylakoid lumen. This enzyme kinetics analysis provides a more complete characterization of chloride and pH dependence of O2 evolution activity than has been previously presented.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Photosystem II (PSII) is the membrane-bound protein complex within plants, algae, and cyanobacteria that produces O2 from H2O using the energy from light absorption (Dau and Haumann 2008; McConnell 2008; Najafpour et al. 2012). It is one of the two major photosystems, along with photosystem I, that use a photosynthetic reaction center to harvest light energy from the sun for use by all life forms. In addition, PSII is the primary source of oxygen in the atmosphere, giving it an important dual role in the earth’s life cycle. Photosystem II contains a unique catalytic Mn4Ca center within the oxygen evolving complex (OEC), which includes, in addition to the Mn4Ca cluster, protein residues, cofactors, and channels necessary for O2 production. The OEC extracts four electrons from water in a cycle that involves five major oxidation states of the Mn4Ca cluster, called S-states and designated as S i where i = 0–4. Absorption of a photon at the reaction center accompanies advancement of the OEC from S i to S i+1, until reaching the S4 state, whereupon O2 is formed and the OEC returns to the S0 state. While much progress has been made in understanding the nature of the Mn4Ca cluster and its oxidation state cycle, the mechanism of O2 formation and the regulation of its catalysis are still the subject of much study.
Chloride ion has long been known to be an essential cofactor in the production of oxygen by photosystem II (Coleman 1990; Critchley 1985; Pokhrel et al. 2011; Popelkova and Yocum 2007; Yocum 2008). In higher plant PSII, the extrinsic subunits PsbQ and PsbP, with molecular weights of 17 and 23 kDa, respectively, regulate the access of chloride and calcium ions to the OEC at the lumenal side of PSII. Using dialysis (Lindberg and Andréasson 1996) or short-term high pH treatment (Homann 1985), PSII can be depleted of chloride without disturbance of the protein composition. For these preparations, there always remains significant residual oxygen evolution activity in the absence of Cl− and full activity is restored by the addition of mM concentrations of chloride. On the other hand, removal of the PsbQ and PsbP subunits results in a PSII preparation from which activating chloride can be easily removed by washing in chloride-free buffer (Miyao and Murata 1985; Seidler 1996), such that the residual activity is near zero in the absence of chloride. In addition, these preparations become dependent on calcium for restoration of full activity, indicating that functional Ca2+ is lost from the OEC in the absence of PsbP and PsbQ, although it is tightly retained in their presence. Chloride ions can be replaced by bromide with nearly full support of oxygen evolution activity and by a few other monovalent anions including nitrate and iodide with lower effectiveness (Bryson et al. 2005; Lindberg and Andréasson 1996; Olesen and Andréasson 2003; Ono et al. 1987; Wincencjusz et al. 1999; Yachandra et al. 1986).
Studies of the direct binding of chloride to PSII have been rarely carried out because of the difficulty of measuring chloride in free or bound states. Lindberg and Andréasson used 36Cl− to show that intact PSII (with all extrinsic subunits bound) contained one high-affinity binding site for chloride that exchanged over a period of many hours (Lindberg et al. 1990, 1993). Researchers have otherwise relied on studies of activation of oxygen evolution to evaluate functional chloride binding. Additional studies by Andréasson and coworkers suggested that intact PSII has two inter-convertible Cl− binding sites or two modes of binding to a single site to explain the observation of a high-affinity slow exchange site and a low-affinity rapid exchange site (Lindberg and Andréasson 1996; Olesen and Andréasson 2003). Researchers have often estimated the dissociation constant K d by assuming that it is essentially equal to the Michaelis constant K M for chloride activation, arriving at values in the micromolar to millimolar range. In addition, using flash studies the affinity of PSII for chloride was found to decrease as the S-states advanced (Wincencjusz et al. 1998). Other studies demonstrated a pH dependence of chloride activation, leading to the proposed involvement of amino acid residues such as histidine, glutamate, or aspartate (Homann 1985, 1988a).
X-ray diffraction (XRD) studies of PSII from thermophilic cyanobacteria have shown the presence of two chloride-binding sites (often characterized using bromide in place of chloride) with distances of 6.7 and 7.4 Å from the Mn4Ca cluster in the S1 state (Guskov et al. 2009; Kawakami et al. 2009, 2011; Murray et al. 2008). In the first site (Site 1), located 6.7 Å from the distal manganese Mn4, chloride is coordinated by the side-chain nitrogen of residue D2-Lys317, the backbone nitrogen of D1-Glu333, and two water molecules. In the second site (Site 2), located 7.4 Å from Mn2, chloride is ligated by the backbone nitrogens of CP43-Glu354 and D1-Asn338 and by two water molecules. Because chloride in Site 1 is bound by a charged residue (lysine), whereas in Site 2 none of the ligating residues is charged, Site 1 is likely to bind chloride with higher affinity (Kawakami et al. 2011). Differential binding affinities of the two sites are supported by the observation that one of the XRD studies revealed only Site 1 (Guskov et al. 2009). Each chloride ion appears to be located at the entrance of a hydrophilic channel (Kawakami et al. 2011; Murray et al. 2008), suggesting a role in proton removal. In higher plant PSII, structural identification of a chloride-binding site was carried out using X-ray absorption fine structure (EXAFS) studies, which was facilitated by replacement of chloride with bromide. In this case, a single Cl− was bound per PSII at a distance of 5.0 Å from one Mn of the Mn4Ca cluster (Haumann et al. 2006), somewhat shorter than the distance for Site 1 in cyanobacteria.
We report studies of the chloride effect on oxygen evolution activity in two higher plant PSII preparations lacking the PsbQ and PsbP extrinsic subunits. Use of these preparations allows chloride to be treated as an activating cofactor in terms of enzyme kinetics by reducing residual oxygen evolution activity in the absence of chloride. In one procedure, the two extrinsic subunits were removed by 2 M NaCl treatment (Ono et al. 2001), while in the other they were removed by 50 mM Na2SO4 treatment at mildly elevated pH of 7.5 (Wincencjusz et al. 1997). Over a pH range from 5.3 to 8.0, both PSII preparations showed similar patterns of Cl− activation and at higher concentrations Cl− inhibition. The data are interpreted in terms of a classic enzyme kinetics model of pH dependence of activation involving four protonation events, with inhibition by binding of the activator at a second site. This was found to be the minimal model required to account for each full data set. The analysis presents a more complete picture of chloride dependence than has been previously presented and for the first time provides a characterization of chloride inhibition.
Materials and methods
PSII sample preparation
PSII-enriched thylakoid membrane fragments (referred to as intact PSII) were prepared from fresh market spinach by the method of Berthold, Babcock, and Yocum (Berthold et al. 1981) with minor modifications (Ghanotakis et al. 1984). PSII membranes at a concentration of 3–5 mg of chlorophyll (mgChl) mL−1 were frozen in buffer containing 0.4 M sucrose, 50 mM MES-NaOH, pH 6.0, plus 35 mM NaCl, and stored in liquid N2.
Removal of PsbP and PsbQ subunits from PSII by high NaCl treatment was carried out by incubation in buffer containing 2 M NaCl, followed by a short incubation with 2 mM EDTA to remove Ca2+, essentially as described by Ono et al. (2001). Removal of PsbP and PsbQ subunits from PSII by Na2SO4 treatment was carried out by incubation in buffer containing 50 mM Na2SO4 at pH 7.5, as described by Wincencjusz et al. (1997), except that the incubation was carried out at a concentration of 0.1 mgChl mL−1 for 30 min. The extended treatment time for the latter preparation was used to achieve homogeneity in terms of Ca2+ dependence (see Online Resource). For each preparation, treatment to remove the extrinsic subunits was followed with washing by centrifugation three times in buffer without added Cl−. Preparations were suspended in buffer containing 0.3 M sucrose and 50 mM MES-NaOH, pH 6.0, and stored in liquid N2 until used. Loss of the PsbP and PsbQ subunits was confirmed by SDS–polyacrylamide gel electrophoresis (Kashino et al. 2001) (see Online Resource). The maximum activities of the two preparations were observed to be different, with the NaCl-washed PSII showing lower overall activity. This is probably because the Na2SO4/pH7.5 treatment is generally milder, but may also be related to small differences in conditions of light exposure and/or subsequent washes to remove Cl−.
Oxygen evolution assays
Rates of oxygen evolution activity were measured as initial rates of O2 concentration versus time using a Clark-type O2-sensitive electrode (Yellow Springs Instruments, model 5331) under saturating continuous illumination by visible light (tungsten halogen source). Concentration was calibrated using O2-saturated water, as calculated for the prevailing pressure using Henry’s law. Assays were generally carried out at 25 °C in buffer containing MES or HEPES at the pH indicated, 0.4 M sucrose, with chloride and/or calcium ion added as indicated. PSII was present at a concentration of 20–30 μgChl mL−1, and 1.8 mM phenyl-p-benzoquinone (PPBQ) was added as electron acceptor. The oxygen evolution activity of intact PSII membranes at pH 6.0 with sufficient chloride was 500–600 μmol O2 mgChl−1 h−1.
For the study of the chloride concentration dependence of oxygen evolution activity at different pH values, 25–100 mM MES-NaOH and/or MES-Ca(OH)2 or 25–100 mM HEPES–NaOH and/or HEPES-Ca(OH)2 were used to cover the pH range from 5.3 to 8.0, where Ca(OH)2 was added to provide the desired concentration of Ca2+. The large range of buffer concentrations was necessary for the addition of sufficient calcium ion as Ca(OH)2 when chloride was absent or present in low concentrations. Chloride ions were supplied as NaCl. The background chloride present in the buffers, i.e., that in the absence of added Cl−, was estimated using a Cl− ion-selective electrode (Accumet, half cell, 13-620-519) and found to be 20–40 μM. The Cl−-depleted NaCl-washed PSII or Na2SO4/pH 7.5-treated PSII was added to the assay medium and preincubated for 1–2 min at 25 °C before the measurement.
The pH and Cl− dependence of O2 evolution activity was analyzed in terms of the classic enzyme kinetics model of pH dependence for a substrate or activator (Cl− in this case) involving four protonation events (Cornish-Bowden 1995; Marangoni 2003; Tipton et al. 2009), extending the model to include the binding of a second Cl− along an unproductive or inhibitory path. The chloride activation curves at various pH values were fitted individually, and the resulting parameters were combined for the pH dependence analysis. Details of the development and application of the model are given in “Results” section. Fits to the data were carried out using nonlinear regression analyses with SigmaPlot software (Systat Software, Inc.).
EPR spectroscopy
For electron paramagnetic resonance (EPR) spectroscopy, NaCl-washed PSII as prepared above was washed by centrifugation and resuspended in buffer at the pH and chloride concentration indicated to a final concentration of 11–12 mgChl mL−1. Buffers contained 0.4 M sucrose, 50–78 mM MES (for pH 6.2 and 5.5) or 50 mM HEPES (for pH 7.4). The Ca2+ concentration was adjusted to 20 mM by adding a combination of Ca(OH)2 and CaCl2 for buffers containing 25 mM Cl− or by adding 20 mM CaC12 for buffers containing 200 mM Cl−; the remaining chloride was added as NaCl to the latter. Samples were transferred to 4 mm outer diameter clear fused quartz EPR tubes (Wilmad Glass, Buena, NJ, USA) and dark-adapted on ice for 75 min before freezing in liquid nitrogen for storage.
To check for the presence of the dark-stable multiline signal from the inhibited S2 state, samples were pre-illuminated just before EPR spectroscopy. To do this, samples were thawed quickly in room temperature water for a few seconds and 2 mM PPBQ was added from a 50-mM stock solution with dimethylsulfoxide as solvent. Samples were then illuminated for 5 s at room temperature and dark-adapted on ice for about 25 min before refreezing, after which the spectra of the dark-adapted state were taken. For production of the normal S2 state, the same samples were illuminated at 195 K in a dry ice/ethanol bath for 4.0 min. Finally, to induce the \({\text{S}}_{2} {\text{Y}}_{\text{Z}}^{ \cdot }\) state in inhibited centers, samples were brought to 273 K by transferring first to a dry ice/ethanol bath (195 K), then to an ice water bath (273 K), and illuminated for 30 s. In each case, illumination was carried out with a 300-W halogen lamp using a Pradovit color projector (Leitz), with the light beam directed to a patch of approximately 2 cm2 by passing through 6 cm of 5 mM CuSO4 solution in a cylindrical glass container.
EPR spectroscopy was carried out at X-band (9.65 GHz) using a Bruker Instruments EMX 10/12 EPR spectrometer equipped with an ER4116DM dual mode cavity. Temperature was controlled with an Oxford Instruments ESR 900 liquid He cryostat. Signals were observed at 10.0 K using a microwave power of 20 mW, modulation frequency of 100 kHz, and modulation amplitude of 17 gauss. Peak heights for the S2 state multiline signal were measured using the average of the three peaks downfield from the signal center (at about 2960, 3050, and 3140 gauss).
Measurement of manganese content
Manganese content of PSII was measured as aqueous Mn2+ by EPR spectroscopy, essentially as described by Kuntzleman et al. (2004). Portions of the samples that had been prepared for low-temperature EPR experiments above (those shown in Fig. 3) were adjusted to 2 mgChl mL−1 with the appropriate buffer and illuminated for 10 s in the presence of 2 mM PPBQ to mimic the treatment of EPR samples. Samples were then pelleted in a microfuge and the supernatant buffer removed. The pelleted PSII was resuspended in the buffer corresponding to the control (pH 6.3, 20 mM NaCl) and treated as described for wet-ashing in 0.6 M HCl (Kuntzleman et al. 2004). For EPR measurement of the released Mn2+, samples were placed in capillary tubes and the hexaquo-Mn2+ signal observed at room temperature. Signal heights were compared with those of a set of Mn2+ standards prepared in the same 0.6 M HCl solution. EPR settings included 50 mW microwave power and 10 gauss modulation amplitude. Using untreated, intact PSII, this measurement gave a manganese content of 3.6–4.4 Mn per PSII, assuming 250 chlorophylls per PSII center.
Results
pH dependence of Cl− activation of oxygen evolution activity
In this study, the chloride and pH dependences of oxygen evolution activity were examined in two different preparations of PSII-enriched membrane fragments from which the PsbP and PsbQ extrinsic subunits had been removed. Removal of the two extrinsic subunits facilitates examination of the chloride dependence, because when they are removed activity is near 0% in the absence of chloride. Without their removal, the oxygen evolution activity remains between 30 and 90% of the chloride-containing control depending on the chloride depletion treatment used. One method used in this study to remove the PsbP and PsbQ subunits was the well-known method of incubation with 2 M NaCl in the presence of EDTA (Miyao and Murata 1984; Ono et al. 2001), which is effective in removing the essential Ca2+ ion as well as the two extrinsic subunits; this preparation is referred to here as NaCl-washed PSII. The second method used to remove the PsbP and PsbQ subunits was short-term incubation with 50 mM Na2SO4 at elevated pH of 7.5 (Homann 1988b; Wincencjusz et al. 1997); this preparation is referred to here as Na2SO4/pH 7.5-treated PSII. This preparation was also found to be Ca2+-dependent under our treatment conditions (see Online Resource).
The residual oxygen evolution activity in the absence of added Cl−, measured in the presence of saturating concentrations of Ca2+, was somewhat different in the two types of PSII preparations. In Na2SO4/pH 7.5-treated PSII, the residual activity was highest at low pH values, showing about 11% of the maximum restored activity, but was unobservable at pH >7. This is comparable with observations reported by Homann (1988a), although in our case the residual oxygen evolution activity was much lower. In NaCl-washed PSII, the residual activity was essentially unobservable (<5% maximum activity) at all pH values.
Chloride dependence of oxygen evolution activity was studied in the presence of Ca2+ for both the NaCl-washed and Na2SO4/pH 7.5-treated PSII preparations. Preliminary titrations to determine saturating amounts of Ca2+ at different pH values were carried out to avoid complexity in the Cl− dependence study. The titrations revealed that the highest requirement for Ca2+ was at low pH, with apparent K CaM ~ 1.5 mM at pH 6. The requirement for Ca2+ decreased with increasing pH, becoming about 50 μM at pH 7.9 (data not shown). The concentrations of Ca2+ used for the chloride dependence studies were chosen based on these results.
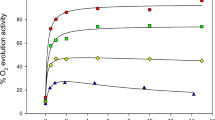
Using NaCl-washed and Na2SO4/pH 7.5-treated PSII preparations, which both lacked the PsbP and PsbQ subunits, the activation of oxygen evolution by chloride was studied at pH values between 5.3 and 8.0 (Figs. 1, S3). Using a broad range of chloride concentrations, similar trends were observed for the two preparations, although the NaCl-washed PSII showed lower overall activity. Activation by Cl− was observed at all pH values studied, and as the pH increased, the chloride concentration required to reach maximal activity also increased. In addition, at low pH values inhibition of oxygen evolution activity was observed as the chloride concentration increased, but this effect disappeared at pH values above 7. To test whether inhibition could be attributed to competition between Na+ and Ca2+, titrations were carried out in the presence of different concentrations of Ca2+ using the Na2SO4/pH 7.5-treated PSII preparation. The Cl− dependence of the kinetics of oxygen evolution activity was found to be the same in the presence of different Ca2+ concentrations in the range of 2–20 mM.

Chloride dependence of the rate of oxygen evolution by NaCl-washed PSII at various pH values. a Chloride concentration up to 100 mM; b extended chloride concentration range up to 500 mM; pH values are 5.36 (filled circle), 5.50 (filled triangle), 6.19 (filled square), 6.43 (filled diamond), 6.71 (circle), 7.39 (triangle), 7.65 (square), 7.92 (diamond), and 8.01 (inverted open triangle). Lines represent fits to individual curves using the substrate inhibition model of Scheme 1 given by Eq. 1. O2 evolution assays were carried out in the presence of 1.8 or 10 mM Ca2+ in MES-Ca(OH)2 or HEPES-Ca(OH)2 buffer systems
Analysis of the pH dependence of kinetic parameters
As an overview, the analysis of the pH and chloride dependence effects on O2 evolution activity was carried out in two stages. First, the chloride dependence curves at various pHs were analyzed according to a substrate inhibition model that did not take into account pH dependence. This simple analysis was based on two Cl− binding sites, one activating and the other inhibitory, and gave apparent values of kinetic constants for maximum velocity \((V_{\hbox{max} }^{\prime } ),\) chloride activation \((K_{\text{S}}^{\prime } ),\) and chloride inhibition \((K_{\text{I}}^{\prime } ).\) Next, the results of these individual fits were analyzed to determine the pH dependence of the apparent kinetic constants involved. For this, a more complete model was developed based on a classic pH dependence model of enzyme kinetics involving four protonation events, modified to include binding of the second chloride. The possibility of more than one way to build in the pH dependence of the second chloride was considered. Using the simplest complete model, the analysis resulted in final intrinsic or pH-independent kinetic constants for maximum velocity (V max), activation (K S), inhibition (K I), and the various protonation events they depend on (\(K_{{{\text{P}}_{1} }} ,K_{{{\text{P}}_{2} }} ,K_{{{\text{PCl}}_{1} }}\), and \(K_{{{\text{PCl}}_{2} }}\)).
In general, the substrate inhibition model is based on the binding of two substrate molecules, one that promotes activity and another that is inhibitory (Cornish-Bowden 1995; Schulz 1994). The drop in activity at higher substrate concentrations is only observed if the inhibitory substrate molecule binds second; thus, the model resembles uncompetitive inhibition kinetics in the sense that the inhibitor binds to the enzyme–substrate complex. This simple kinetics model for the case of chloride activation/inhibition of PSII is given in Scheme 1, where P represents PSII and Cl represents chloride. The rate equation, based on equilibrium binding of Cl− in each step except for product formation, is given by:
where \(K_{\text{S}}^{\prime }\) is the apparent dissociation constant for activating chloride, \(K_{\text{I}}^{\prime }\) is the apparent inhibition constant or dissociation constant for inhibitory chloride, and \(V_{\hbox{max} }^{\prime }\) is the apparent maximum velocity at that pH. The constant \(K_{\text{S}}^{\prime }\), the dissociation constant of PCl1 into P and Cl−, was introduced into the equation through an assumption of pre-equilibrium before product formation. However, it is often taken as the Michaelis constant K M, which also incorporates the product formation rate constant k P, based on the steady-state assumption for the two steps from Cl− binding to product formation. In the analysis of the data, the two constants are indistinguishable.

Kinetics model for substrate inhibition of oxygen evolution by chloride, in which the activator chloride is treated as substrate. \(K_{\text{S}}^{\prime }\) and \(K_{\text{I}}^{\prime }\) are pH-dependent chloride activation and inhibition dissociation constants, respectively
From the fits to the individual Cl− dependence curves, the values for \(V_{\hbox{max} }^{\prime }\), \(K_{\text{S}}^{\prime }\), and \(K_{\text{I}}^{\prime }\) were determined for each pH. While inhibition by high chloride concentrations was evident at low pH values, it became insignificant at higher pH values, as shown by very high values of \(K_{\text{I}}^{\prime }\) with high errors. For fitting purposes, the inhibition term ([Cl]2/\(K_{\text{I}}^{\prime }\)) in the denominator was dropped when the error in \(K_{\text{I}}^{\prime }\) was on the order of magnitude of \(K_{\text{I}}^{\prime }\) itself, since this reflected an inability to determine a value for \(K_{\text{I}}^{\prime }\). In this way, a series of \(K_{\text{S}}^{\prime }\) values between pH 5.3 and pH 8.0 and a series of \(K_{\text{I}}^{\prime }\) values between pH 5.3 and pH 6.7 were obtained. These fits are shown as the solid lines in Figs. 1 and S3.
Once pH-dependent kinetic constants were determine for the individual curves, the pH dependence of the constants was analyzed. The model used was based on the classic model of the pH dependence of enzymatic activity (Cornish-Bowden 1995; Marangoni 2003; Tipton et al. 2009), which involves substrate binding, two protonation events for the enzyme with no substrate bound, and two protonation events for the enzyme with substrate bound. While many protonation events are no doubt involved in the formation of product, those included in the model are considered to define the observable pH dependence behavior. As applied to Cl− activation (Scheme 2), the path toward products requires deprotonation of the acidic proton (H2) and protonation by the basic proton (H1). Activator Cl− binds to the singly protonated form of enzyme, PH1, to form the active species PH1Cl1. (Binding of activator or substrate to the other protonated states of the enzyme is of course conceivable, but usually omitted in the simple pH dependence model.) The two steps that involve protonation of P to form PH1 followed by binding of Cl− to form PH1Cl1 correspond to the model of Homann (1985, 1988a), in which protonation precedes chloride activation. Here, however, further protonation events are included for a more complete description. Note also that although dissociation constants \(K_{{{\text{P}}_{1} }}\) and \(K_{{{\text{PCl}}_{ 1} }}\) appear to refer to the same proton (H2), these in general would represent completely independent protonation events. The same is true for dissociation constants \(K_{{{\text{P}}_{ 2} }}\) and \(K_{{{\text{PCl}}_{ 2} }}\).

Model of pH dependence of activation by chloride based on the classic pH dependence model, including binding of a second inhibitory chloride. The binding of the inhibitory chloride to PH1Cl1H2 is the simplest possibility. K S and K I are the pH-independent chloride activation and inhibition constants, respectively. \(K_{{{\text{P}}_{1} }}\) and \(K_{{{\text{P}}_{2} }}\) are the acidic and basic protonation constants, respectively, before binding of activator chloride and \(K_{{{\text{PCl}}_{1} }}\) and \(K_{{{\text{PCl}}_{2} }}\) are acidic and basic protonation constants, respectively, after binding of activator chloride. Protonation constants are designated according to common convention for the classic pH dependence model (Cornish-Bowden 1995; Marangoni 2003)
To include the inhibitory effect of Cl− corresponding to substrate inhibition, the pH dependence model was extended to include the binding of a second Cl−. There are two requirements for the incorporation of the second Cl− into the model, based on the experimental observations. First, as noted above, the Cl− associated with inhibition must bind after the activating Cl− for the data to show a decrease in activity as Cl− concentration increases. Next, the second Cl− must bind after a protonation event to account for the observation of greater inhibition at lower pH. The simplest model that incorporates these requirements is shown in Scheme 2, in which the second Cl− binds to the species PH1Cl1H2 of the classic pH dependence model. Other possible models can also account for the characteristics of inhibition, and may be more correct, but in any other model additional protonation events must be introduced. One such model in which an additional proton (H3) precedes binding of the second Cl− is considered in Scheme S1 of the Online Resource.
The rate equation describing the model presented in Scheme 2 is shown in Eq. 2. The equation is the same as that derived for the classic pH dependence model, but includes an additional term in the denominator that accounts for the substrate inhibition behavior.
where [H] and [Cl] are proton and chloride concentrations, respectively, and equilibrium constants correspond to those defined in Scheme 2. V max is defined formally as k P[P]tot; however, note that in our analysis, the rates are actually given per chlorophyll concentration, so in practice do not depend on enzyme (P) concentration. Comparison with Eq. 1 reveals expressions for the pH dependence of \(V_{\hbox{max} }^{\prime }\), \(K_{\text{S}}^{\prime }\), and \(K_{\text{I}}^{\prime }\) in terms of the pH-independent constants given in the model. Expressions involving \(V_{\hbox{max} }^{\prime }\) and \(K_{\text{S}}^{\prime }\) are found to be the same as those found for the classic pH dependence analysis and are given by Eqs. 3a–3c. In addition, Scheme 2 leads to an expression for the pH dependence of the inhibition constant \(K_{\text{I}}^{\prime }\), given by Eq. 3d.
Application of the pH dependence model with inhibition
To determine the pH dependence of chloride activation of O2 evolution, the pH dependence of values of \(V_{\hbox{max} }^{\prime }\) and \(V_{\hbox{max} }^{\prime }\)/\(K_{\text{S}}^{\prime }\) found from fits of the individual chloride dependence curves was analyzed according to Eqs. 3a and 3b. The pH dependence of \(K_{\text{S}}^{\prime }\) involves all four protonation constants, so Eq. 3c was not used for data fitting purposes.
For the NaCl-washed PSII samples, the analysis of \(V_{\hbox{max} }^{\prime }\) versus pH (Fig. 2a) led to values for \({\text{p}}K_{{{\text{PCl}}_{1} }}\) and \({\text{p}}K_{{{\text{PCl}}_{2} }}\) of 5.1 and 8.3, respectively, as well as a pH-independent value for V max (280 μmol mgChl−1 h−1). Observation of the plot of \(V_{\hbox{max} }^{\prime }\)/\(K_{\text{S}}^{\prime }\) versus pH (Fig. 2b) showed that the data did not define the acidic protonation constant \(K_{{{\text{P}}_{1} }}\), i.e., \({\text{p}}K_{{{\text{P}}_{1} }}\) must be <5. For this reason, the analysis employed a modified version of Eq. 3b, since the ratio [H]/\(K_{{{\text{P}}_{1} }}\) must be negligible:

Analysis of the pH dependence of apparent kinetic constants from chloride dependence curves for NaCl-washed PSII: a \(V_{\hbox{max} }^{\prime }\) versus pH, b \(V_{\hbox{max} }^{\prime }\)/\(K_{\text{S}}^{\prime }\) versus pH; and c \(K_{\text{I}}^{\prime }\) versus pH. Apparent kinetic constants were taken from the substrate inhibition fits to individual plots of chloride dependence of O2 evolution, shown in Fig. 1. Lines represent fits to Eq. 3a (a), Eq. 3b′ (b), and Eq. 3d (c)
Results of the fit to Eq. 3b′ gave a value of \({\text{p}}K_{{{\text{P}}_{2} }}\) of 6.2 and a value for the ratio V max/K S of 331 μmol mgChl−1 h−1 mM−1. Using the value previously found for V max, K S was found to be 0.85 mM.
To determine the value of K I, the pH dependence data of \(K_{\text{I}}^{\prime }\) were fitted to Eq. 3d using the values previously determined for \(K_{{{\text{PCl}}_{1} }}\) and \(K_{{{\text{PCl}}_{2} }}\) (Fig. 2c). The value for the pH-independent inhibition constant K I was thereby found to be 64 mM.
A similar analysis was carried out to characterize the pH dependence of the values of \(V_{\hbox{max} }^{\prime }\), \(K_{\text{S}}^{\prime }\), and \(K_{\text{I}}^{\prime }\) derived from the Cl− dependence curves of Na2SO4/pH 7.5-treated PSII (Online Resource, Figure S4). The values obtained were similar to those found for NaCl-washed PSII, although K I was found to be significantly higher. The kinetic parameters found from all pH dependence analyses of both PSII samples are shown together in Table 1.
Recovery of activity after buffer treatments
The possibility was considered that the inhibitory effect of high chloride concentration at low pH was due to irreversible damage to PSII, such as release of manganese or loss of other subunits or cofactors. To test this possibility, samples of NaCl-washed PSII were prepared in various buffers at selected pH and NaCl concentrations corresponding to representative points on the chloride dependence curves seen in Fig. 2. These included pHs of 5.5, 6.3, and 7.4 and Cl− concentrations of 25 and 200 mM. Within minutes after preparation of the sample in the test buffer, each sample was assayed in the same buffer it was prepared in to confirm that the activity approximately matched that observed in the chloride dependence assay (Table 2). At pH 6.3, a slight drop in activity of 5–10% was observed in the presence of 200 mM NaCl compared to 25 mM NaCl, as has been observed in previous Cl− dependence studies. At pH 5.5, an overall drop in activity was observed, with an additional decrease of about 25% when the concentration of NaCl was increased from 25 to 200 mM NaCl. At pH 7.4, the reverse was seen, with the higher activity observed in 200 mM NaCl, where it was over 85% of the control activity. These observations correlate well with the trends observed in Fig. 1.
After measuring the activity in the test buffer, each sample was allowed to incubate in the test buffer on ice for 90–100 min. It was then assayed in the control buffer at pH 6.3 + 25 mM NaCl to assess the loss of activity due to the buffer treatment (Table 2). The control sample showed essentially no loss in activity, since the 2% decrease in activity was within the error of the measurement. Similarly, the samples prepared in buffer at pH 6.3 + 200 mM NaCl and in buffer at pH 7.4 showed no significant decrease in activity. The samples prepared in buffer at pH 5.5 showed over 90% of the control activity. Thus, although a decrease in activity of 7–9% resulted from incubation in the test buffer, each sample regained at least 80% of the activity it had lost when assayed in the test buffer. In addition, incubation in the higher concentration of 200 mM NaCl, where inhibition by Cl− is more pronounced, did not lead to a significantly greater loss in activity than incubation in 25 mM NaCl. Thus, the inhibitory effect of high Cl− concentrations at the lower pH cannot be attributed to irreversible damage caused by the conditions.
EPR spectroscopy
NaCl-washed PSII samples that correspond to key points of the chloride and pH dependence study of activities were examined by EPR spectroscopy. Treatment at pH 5.5 with 25 mM NaCl corresponded to the maximum activity at this pH, while pH 5.5 with 200 mM NaCl corresponded to a region of inhibition at this pH. Treatment at pH 7.4 with 200 mM NaCl corresponded to the region of maximum activity at this pH, at which little or no inhibitory effect was observed. These were compared to the control treatment at pH 6.2 with 25 mM NaCl, which represents standard conditions. All samples also contained 20 mM Ca2+.
The NaCl-washed PSII samples were first checked for the appearance of the dark-stable multiline signal in the S1 state, after pre-illumination and dark adaptation. Evidence of this signal, which would be indicative of the loss of Ca2+ from the manganese cluster, was not observed (not shown).
Examination of the S2 state multiline signal produced by illumination at 195 K in NaCl-washed PSII samples (Fig. 3a) indicated that at pH 5.5/25 mM NaCl, the signal was nearly as strong as in the control sample at pH 6.2/25 mM NaCl, showing about 90% of the peak height of the control. At pH 5.5/200 mM NaCl and pH 7.4/200 mM NaCl, on the other hand, the multiline signal from NaCl-washed PSII was reduced, with both samples showing about 60% of the control peak height. The relative signal heights did not correlate well with the relative levels of activity. Based on the constants found from the data fits, the activities are expected to be 74% at pH 5.5/25 mM NaCl, 45% at pH 5.5/200 mM NaCl, and 97% at pH 7.4/200 mM NaCl. Samples prepared independently in similar buffers, shown in Table 2, showed this trend in activities, although the experimental activities were 5–10% lower relative to the control. Rather than following the trend in activity levels, the multiline signal heights seemed to depend on the amount of NaCl, with the greater signal height observed at the lower NaCl concentration. It was also noted that the g = 4.1 signal appeared to be more pronounced in the two samples containing 200 mM NaCl, although this was not as clear because of baseline inconsistencies.

EPR spectroscopy of NaCl-washed PSII showing: a the S2-state signals produced by illumination at 195 K; b same region after illumination at 0 °C to test for possible formation of a signal from the \({\text{S}}_{2} {\text{Y}}_{\text{Z}}^{ \cdot }\) state. Samples were prepared as described in “Materials and methods” section in buffer at pH 5.5 with 200 mM NaCl (trace a), pH 5.5 with 25 mM NaCl (trace b), pH 6.2 with 25 mM NaCl (trace c), or pH 7.4 with 200 mM NaCl (trace d). Samples contained 2 mM PPBQ. EPR conditions included 20 mW power, 17 gauss modulation amplitude, and temperature of 10 K
The same NaCl-washed PSII samples were subsequently illuminated at 0 °C to test for the production of a broad radical (Fig. 3b), which could suggest the presence of the \({\text{S}}_{2} {\text{Y}}_{\text{Z}}^{ \cdot }\) radical due to interference with electron transfer to Tyr Z. Only a small amount of radical of this type was seen for the two samples at pH 5.5.
After illumination at 0 °C, a noticeable negative Mn2+ signal was observed for the sample at pH 7.4/200 mM NaCl. This indicates that a Mn2+ signal present in the dark-adapted state decreased after illumination, i.e., that unbound Mn2+ decreased due to illumination. For each sample, a Mn2+ signal was observed in the dark-adapted spectrum (after pre-illumination); although the pH 7.4/200 mM NaCl sample showed the most Mn2+ in the dark, the others showed between 60 and 95% as much. (The presence of a Mn2+ EPR signal is not unusual, particularly for NaCl-washed PSII that has undergone subsequent treatment.) Comparison of the Mn2+ signal in the pH 7.4/200 mM NaCl sample before illumination and after illumination at 0 °C indicated that the negative Mn2+ signal after illumination had about 75% the height of the positive signal in the dark. This suggests that the majority of the unbound Mn2+ present in the dark was reincorporated during turnovers for this sample.
Because of the observation of Mn2+ in the EPR spectra of the dark-adapted samples, the loss of manganese from the Mn4Ca cluster during treatment was assessed by measuring the total Mn. Aliquots of the samples prepared for EPR spectroscopy above (after storage in liquid nitrogen) were briefly illuminated at room temperature in the presence of the electron acceptor PPBQ to mimic EPR sample conditions. Samples were then prepared and wet-ashed as described in “Materials and Methods” section to released Mn as aqueous Mn2+. Measurement of aqueous Mn2+ by EPR spectroscopy revealed that all four samples contained 2.6–2.8 (±0.2) Mn per PSII center. Although this amount is noticeably lower than the 4.0 Mn per PSII that would be found if all PSII centers contained undamaged Mn4Ca clusters, no sample showed a significantly greater loss of Mn that could be attributed to different buffer conditions. Rather the loss of manganese is probably related to the general loss of activity that resulted from the removal of PsbP and PsbQ by NaCl washing and subsequent treatments.
Discussion
Overview of the kinetics analysis
In this study, the oxygen evolution activity by higher plant PSII was investigated over a range of pHs between 5 and 8 and extended chloride concentrations. Because of the difficulty in depleting intact PSII of chloride when the PsbP (23 kDa) and PsbQ (17 kDa) subunits are bound, these subunits were removed to facilitate the chloride dependence measurements. Two different methods, washing with 2 M NaCl in the presence of EDTA and treatment with 50 mM Na2SO4 at pH 7.5, were used to remove PsbP and PsbQ along with activating Ca2+ ion. The use of both preparations helped to ensure that the results were general to PSII lacking these subunits.
The enzyme kinetics model used here to analyze the chloride- and pH-dependent oxygen evolution rates was developed according to several requirements to explain the observations. Two types of binding sites for chloride were needed, one promoting activity and one associated with an unproductive or inhibitory path. To lead to the decrease in activity at higher concentrations, the inhibitory chloride must bind after the activating chloride as for substrate inhibition (Cornish-Bowden 1995; Schulz 1994). (Binding of inhibitory substrate before the functional substrate would result in a decrease in the effective enzyme concentration and lower overall activity, but activity would not decrease with increasing concentration.) Because both activation and inhibition were seen to be more pronounced as pH decreased, the model required a protonation step prior to each chloride-binding step. We found in early analyses of the pH dependence of activation that additional protonation events were needed to account for the pH dependence of \(V_{\hbox{max} }^{\prime }\). This requirement was satisfied by the classic pH dependence model of enzyme kinetics (Cornish-Bowden 1995; Marangoni 2003; Tipton et al. 2009), which involves four protonation steps built upon the simple Michaelis–Menten enzyme kinetics scheme. To account for the pH dependence of the inhibitory chloride, the simplest possibility would be for the second Cl− to bind after protonation of the activated species PH1Cl1 by H2, a proton that is already a part of the classic four-proton pH dependence model. Thus, Scheme 2 is the simplest possible model that includes both the classic four-proton pH dependence of activation and the pH-dependent substrate inhibition. Any other model would include additional binding steps for Cl− and/or H+ and may be more accurate, but little would be gained from application of a more complex model, since the data sets allow the determination of a limited number of constants. An example is seen in Scheme S1 of the Online Resource, which includes an additional protonation step.
To analyze the chloride and pH dependence data, each chloride dependence curve was first fitted to the simple substrate inhibition model (Eq. 1) to obtain apparent \(V_{\hbox{max} }^{\prime }\), \(K_{\text{S}}^{\prime }\), and \(K_{\text{I}}^{\prime }\) values. Then, the apparent parameters were fitted to the pH-dependent expressions for \(V_{\hbox{max} }^{\prime }\), \(V_{\hbox{max} }^{\prime }\)/\(K_{\text{S}}^{\prime }\), and \(K_{\text{I}}^{\prime }\) as determined from the rate equation for Scheme 2 to obtain the four protonation constants, \(K_{{{\text{P}}_{1} }} ,K_{{{\text{P}}_{2} }} ,K_{{{\text{PCl}}_{1} }} ,K_{{{\text{PCl}}_{2} }} ,\) as well as inherent or pH-independent values of V max, K S, and K I.
Activation constant, K S
The pH-independent constant for Cl− activation, K S, was found to be about 0.9 mM for both NaCl-washed and Na2SO4/pH 7.5-treated PSII. In the optimal experimental pH range of 6.2–6.5, both calculated and observed values of apparent \(K_{\text{S}}^{\prime }\) were 1–4 mM for the two PSII preparations. This is consistent with other studies in our laboratory, which typically find apparent \(K_{\text{S}}^{\prime }\) or \(K_{\text{M}}^{\prime }\) values of 1–3 mM for NaCl-washed PSII at pH 6.3, with variation that appears to depend on the preparation. Other values reported in the literature for the chloride dissociation constant K S or the Michaelis constant K M for chloride activation are generally in the mM range for PSII lacking PsbP and PsbQ, with lower values often reported for intact PSII. In an early study that helped to confirm the requirement of PSII for chloride, the apparent \(K_{\text{M}}^{\prime }\) was found to be 0.9 mM in Cl−-depleted thylakoid membranes (Kelley and Izawa 1978). In his study of the pH dependence of chloride activation, Homann estimated the pH-independent KM to be 70 μM (Homann 1985) using intact PSII depleted of chloride by brief treatment at pH 9.6–9.7; in a later study, the affinity for chloride was found to be about tenfold lower in PSII after removal of the PsbP and PsbQ subunits (Homann 1988a), consistent with findings by other researchers. In another study, when intact PSII was depleted of Cl− by extensive dialysis, two dissociation constants for Cl− of 20 μM and 0.5 mM were found, which was interpreted in terms of a single site with interconverting binding affinities (Lindberg and Andréasson 1996). Popelkova and coworkers determined the \(K_{\text{M}}^{\prime }\) for Cl− to be 0.9 mM in NaCl-washed PSII at pH 6 (Popelkova et al. 2008). In a study of PSII in which the PsbP and PsbQ subunits had been removed by Na2SO4 treatment at pH 7.5, \(K_{\text{S}}^{\prime }\) for chloride was estimated to be about 6.5 mM from the half-maximal level of O2 evolution activity (Wincencjusz et al. 1998); this study also examined the S-state dependence of Cl− binding affinity, finding that it decreased as the S-states advanced.
The current structural information about the Cl− binding sites is mainly based on XRD studies of PSII from thermophilic cyanobacteria in the S1 state. The observation that Cl− is not required in the S1 state but rather has its effects in the S2 state and higher suggests that the currently known Cl− sites from XRD may not provide a full description of the functional Cl− sites, particularly since the structures may be compromised by damage to the crystals from high beam intensities. However, given the structural information that is currently available, the site of chloride activation is most likely to be the same as or close to Cl− Site 1 identified in XRD studies of thermophilic cyanobacteria PSII. At this site, Cl− is ligated by D2-Lys317 (side), D1-Glu333 (main), and probably D1-Asn181 (side) (Guskov et al. 2009; Kawakami et al. 2011; Murray et al. 2008). Because this site includes ligation by a charged amine group, it is expected to have the higher affinity for Cl− ion compared with Site 2, in which Cl− is ligated by neutral amide groups from CP43-Glu354 (main), D1-Asn338 (main), and possibly D1-Phe339 (main) (Kawakami et al. 2011; Murray et al. 2008; Rivalta et al. 2011). Modeling studies have shown that Site 2 has an affinity that is quite a bit lower than that of Site 1, with a difference in ΔG of binding between the two sites estimated to be 10 kcal mol−1 (Rivalta et al. 2011). The presence of a single high-affinity site in higher plant PSII was indicated in earlier studies using 36Cl−, in which about 0.6 Cl− per PSII bound with high affinity to intact spinach PSII (Lindberg et al. 1990, 1993), and is further supported by studies in which a single bound Cl− (or Br−) was revealed by XRD (Guskov et al. 2009) and EXAFS (Haumann et al. 2006). While the kinetic data presented here are unable to distinguish how many activating Cl− sites are present, it is fairly certain that there is one with high affinity.
The second known Cl−-binding site, Site 2, identified near the Mn4Ca cluster in XRD studies would be expected to influence the chloride dependence of O2 evolution, but its influence is evidently not resolved. It is more likely to be an activating than an inhibitory chloride site because it appears to have a structural role similar to that of Site 1, since each chloride is coordinated by the backbone nitrogen of a glutamate residue (D1-Glu333 in Site 1 and CP43-Glu354 in Site 2) that concurrently coordinates two Mn through the carboxylate side chain. The lower affinity of Site 2 for Cl− may make it difficult to resolve by kinetics or binding studies. The possibility that a high-affinity site is mainly responsible for chloride activation with secondary modulation by a low-affinity site has been suggested in previous studies (Boussac et al. 2012; van Vliet and Rutherford 1996).
pH dependence constants for Cl− binding
The pH dependence of the activation constant \(K_{\text{S}}^{\prime }\) is influenced by all four protonation constants in the kinetics model of Scheme 2, as seen in Eq. 3c. Two of these, \(K_{{{\text{P}}_{1} }}\) and \(K_{{{\text{P}}_{2} }} ,\) represent protonation and deprotonation of the species before Cl− binding, PH1. The value of \({\text{p}}K_{{{\text{P}}_{2} }}\) was found to be 6.2–6.3, while the value of \({\text{p}}K_{{{\text{P}}_{1} }}\) was found to be out of the pH range employed or below 5. In the studies of Homann (1985, 1988a), the first to employ a pH dependence model of Cl− activation, only a single protonation event was assumed to be involved in Cl− binding. This protonation step preceded Cl− binding and corresponds to the step given by \(K_{{{\text{P}}_{2} }}\) in Scheme 2, but the difference in models used in the two studies (one vs. four protonation events) means that the two values of K a would not be equivalent. In Homann’s first study, which used PSII depleted of Cl− by treatment at pH ~10, the protonation step was found to have a pK a of 6 (Homann 1985). However, this value was reconsidered in the later study because of concern over the possible loss of the extrinsic PsbP and PsbQ subunits, and it was concluded that the pK a was probably below 5 in both intact and NaCl-washed PSII (Homann 1988a).
Another approach to determining the pH dependence associated with chloride binding was taken by Lindberg et al. (1993), who used the isotope 36Cl− to observe a single high-affinity Cl− binding site in intact PSII. The maximum level of binding was observed at pH as low as 5.5, indicating that the acidic pK a was below this pH. This suggests a similarity with the chloride activation site observed here, for which \({\text{p}}K_{{{\text{P}}_{1} }}\) was <5, although the conditions for the studies differ notably because of the presence of the PsbP and PsbQ subunits in the Lindberg study and the observation of high-affinity chloride binding (i.e., with K d ~ 20 μM). Our observation of a value of 6.2–6.3 for \({\text{p}}K_{{{\text{P}}_{2} }}\) suggests that a decrease in 36Cl− binding would be expected at and above this pH, but such was not observed in the 36Cl− binding study. Instead, a decrease in binding with an apparent pK a around 7.5 was observed, but it is possible that this decrease in binding affinity was due to partial loss of the PsbP and PsbQ extrinsic subunits at the higher pH, as discussed by the authors.
Since \({\text{p}}K_{{{\text{P}}_{1} }}\) is <5, it is consistent with a glutamate or aspartate residue that must be deprotonated for chloride to bind functionally. A negatively charged carboxylate would not be involved directly in Cl− binding, but may be involved in binding through indirect means. An example may be D1-Glu333, which coordinates two manganese of the cluster (Mn3 and Mn4) through its carboxylate side chain and Cl− at Site 1 through its backbone amide nitrogen cluster (Guskov et al. 2009; Kawakami et al. 2009, 2011; Murray et al. 2008). If this residue were responsible for \(K_{{{\text{P}}_{1} }}\), it would suggest that it must be deprotonated and coordinated to manganese in order to associate with Cl−.
The fourth protonation constant, with \({\text{p}}K_{{{\text{P}}_{2} }}\) of 6.2–6.3, represents a residue that must be protonated for Cl− binding or function, and its value suggests a histidine residue, which is expected to have a pK a around 5.5–7. Since there is no directly ligating histidine residue for Cl− at Site 1, it would also have its effect indirectly. One possibility may be the residue D1-His332, which ligates manganese (Mn1) and is adjacent to the Cl−-ligating residue D1-Glu333. Another may be D1-His 337, which is hydrogen-bonded to a bridging oxygen of the Mn4Ca cluster. It should be noted that although the model shows that protonation of the residue precedes Cl− binding, it does not require that the residue go through a protonation–deprotonation cycle during catalysis. The simplest interpretation is that the residue is protonated throughout the catalytic cycle with a probability depending on the pH or local effective pH. On the other hand, if the Cl− relevant residue is involved in a proton transfer network, it probably does go through a protonation–deprotonation cycle that affects the Cl− affinity or function during the catalytic cycle.
Although an obvious candidate for a participating protonation constant would be D2-Lys317, which directly ligates Cl− at Site 1, it is unlikely to be the residue associated with pKP2 (or with \({\text{p}}K_{{{\text{PCl}}_{2} }}\)). Lysine residues are expected to have pK as close to 10–10.5, and although a pK a may shift in a hydrophobic environment, such a large shift would not be favored by the solvent-exposed environment near Site 1. D2-Lys317 is probably protonated throughout and well above the pH range studied and in that case would not appear as any of the proton dissociation constants determined here.
pH dependence constants after Cl− binding
The two pK as that define the plots of \(V_{\hbox{max} }^{\prime }\) versus pH (Figs. 2a, S4A), \(K_{{{\text{PCl}}_{1} }}\) and \(K_{{{\text{PCl}}_{2} }}\), correspond to the protonation and deprotonation of PH1Cl1, which is the Cl−-activated enzyme that precedes product formation. The plot of \(V_{\hbox{max} }^{\prime }\) versus pH would correspond to a pH dependence curve determined experimentally using PSII saturated with activating Cl− and in the absence of inhibitory Cl−. The pK as were found to be 4.9–5.1 for \({\text{p}}K_{{{\text{PCl}}_{1} }}\) and 7.9–8.3 for \({\text{p}}K_{{{\text{PCl}}_{2} }}\), respectively. These define a relatively broad pH dependence curve compared with experimental curves of activity versus pH found in the literature, perhaps because of the use of \(V_{\hbox{max} }^{\prime }\) values, but the difference in PSII preparations may be a factor also. For example, in one study using intact PSII from spinach, the apparent pK as for the pH dependence curve were determined to be 5.5 and 7.6 (Haddy et al. 1999). Other examples of pH dependence from a variety of PSII preparations (Commet et al. 2012; Homann 1988a; Kuntzleman and Haddy 2009; Schiller and Dau 2000; Vass and Styring 1991) show curves that are less broad than the \(V_{\hbox{max} }^{\prime }\) versus pH plots presented here, for the most part, although a fairly low value of around 5 for the acidic pK a is observed in a couple cases. The pK a values for protonation associated with the various S-state transitions were determined using EPR spectroscopy by Bernát et al. (2002). pK as of 4.0 and 9.4 were found for the S2-to-S3 transition, 4.5 and 8.0 for the S3-to-S0 transition, and 4.7 for the S0-to-S1 transition; the S1-to-S2 transition was found to be pH-independent, as was the basic pK a for the S0-to-S1 transition. The highest of the acidic pK as (4.7) and the lowest of the basic pK as (8.0) would be expected to define the values for complete catalytic turnovers; although the acidic value reported here for \({\text{p}}K_{{{\text{PCl}}_{1} }}\) is somewhat higher, the basic value reported here for \({\text{p}}K_{{{\text{PCl}}_{2} }}\) matches quite well.
With a value of 4.9–5.1, \({\text{p}}K_{{{\text{PCl}}_{1} }}\) may represent a carboxylate group from glutamate or aspartate. For example, D1-Asp61 has been proposed as a residue involved in the proton transfer network, which Cl− at Site 1 also participates in (Debus 2014; Dilbeck et al. 2012). The value of 7.9–8.3 for \({\text{p}}K_{{{\text{PCl}}_{2} }}\) does not correspond well with the expected pK as for histidine or lysine (5.5–7 and 10–10.5, respectively); however, pK as can shift by up to several pH units within a hydrophobic protein environment.
A study by Commet et al. (2012) explored the effect of the PsbO subunit on the pH dependence and Cl− activation of oxygen evolution, interpreting the results in terms of a role for the PsbO subunit in protecting the OEC from inhibition by OH−. For the various preparations and PsbO mutants tested, the decrease in activity at higher pH was relieved by high Cl− concentrations up to 100 mM. This is similar to the observation made here that higher Cl− concentrations are required for optimal activity as pH increases (i.e., the apparent \(K_{\text{S}}^{\prime }\) for Cl− increases with pH). Commet and coworkers interpreted their results to mean that the elevated Cl− concentration effectively competes with inhibitory OH−; in our study, the result was interpreted to mean that the higher Cl− concentration overcomes the decrease in affinity for Cl− due to decreased protonation of the associated residue. These two interpretations are probably not distinguishable by kinetics studies alone.
Inhibition constant, K I
While the Cl− activation constant is in the low mM range, the inhibitory effect of Cl− was found to take place at much higher concentrations. Using the model of Scheme 2, the intrinsic or pH-independent Cl− inhibition constant, K I, was found to be 64 mM for NaCl-washed PSII and 103 mM for Na2SO4/pH 7.5-treated PSII. This indicates that the affinity in NaCl-washed PSII was somewhat higher than in Na2SO4/pH 7.5-treated PSII. In both cases, in the optimal pH range of 6–6.5, the apparent \(K_{\text{I}}^{\prime }\) was 1000s of mM, placing it in a range that is not of significance under normal assay conditions. However, the inhibitory effect may be of significance under intrathylakoid conditions, as will be discussed below.
Although the Cl− inhibition observed in this study is characterized by a single inhibition constant, this does not imply that the inhibitory effect is actually due to binding of Cl− to a single specific site. As noted already, the model used to account for the kinetics observations is a minimal one since use of a more complex model cannot reveal additional constants. Thus, the constant for inhibition may represent the effects of multiple sites with high dissociation constants.
A third Cl− binding site observed by XRD (Kawakami et al. 2009, 2011) may suggest a possible explanation of the inhibitory Cl− binding observed here. This binding site was observed in the 1.9 Å resolution structure to be about 25 Å away from the Mn4Ca cluster within a channel containing a hydrogen-bond network leading to the lumenal surface. With six water molecules as direct ligands, this Cl− has relatively low affinity and suggests a possible mode of inhibition. In the absence of PsbP and PsbQ, Cl− apparently has unhindered access to its functional site, presumably through channels that are also shared with water and protons. Exchange of Cl− is probably ongoing under these conditions, but even in the presence of the extrinsic subunits, Cl− may dissociate and rebind within a local region. The exchange of Cl− may involve relatively transient or low-affinity binding at a series of sites along the access channel. Inhibition may occur if occupation of these sites becomes too high, either through protonation that leads to higher affinity or through an excessively high concentration of Cl−. This may then impede the movement of Cl− and/or protons, resulting in an inhibitory effect.
A second possibility is suggested by the characteristics of the anions I− and NO2 −, which show substrate inhibition of O2 evolution with activation followed by inhibition from a second binding site. Iodide behaves as a substrate inhibitor, with K I of 1–4 mM in NaCl-washed PSII (Bryson et al. 2005) and nitrite has been observed to show similar behavior (our unpublished observations). Analysis of nitrite as an uncompetitive inhibitor with respect to chloride activation (which corresponds to the inhibitor site for a substrate inhibitor) revealed KI values of about 1 and 8 mM (Pokhrel and Brudvig 2013). The inhibitory effects of I− and NO2 − are believed to involve reduction in the higher S-states (Wincencjusz et al. 1999). Also, evidence has been found for binding of I− at Y D and Y Z , probably through reaction with the radical tyrosyl species, under conditions of illumination (Ikeuchi and Inoue 1987; Ikeuchi et al. 1988; Takahashi et al. 1986). These results imply that inhibition by I− and NO2 − takes place from sites near the Mn4Ca cluster. For these anions, in contrast to chloride, the inhibitory effect becomes evident at relatively low concentrations. On this basis, it would seem unlikely that the inhibitory sites observed for I− and NO2 − can also be identified with the site of inhibition by Cl−. However, the inhibition constants for iodide and nitrite may be influenced by their reactivity with the higher S-states and removal through oxidation, whereas chloride would not experience oxidation under these circumstances. This suggests the possibility that Cl− may bind at a non-activating water accessible site near the Mn4Ca cluster.
Tests for unrecoverable inhibition
Over the range of pHs and Cl− concentrations used, there was minimal irreversible damage to the PSII. This was determined by longtime exposure of PSII to selected pH and chloride concentrations, with the result that most activity was recovered upon return to the control buffer (Table 2). Only 5–10% activity was lost due to treatment of PSII at pH 5.5 for a time that far exceeded the exposure time used in the assays for the pH- and Cl−-dependent curves. In addition, the use of the higher concentration of 200 mM NaCl would have been expected to result in much greater unrecoverable activity if inhibition were associated with irreversible loss of activity, but this was not observed. In other studies, we have carried out similar tests of PSII samples treated at pH 5.5, also without observation of a significant loss of activity [e.g., Haddy and Ore (2010)]. Thus, the inhibitory effect of high Cl− concentration at lower pH cannot be explained by unrecoverable loss of activity.
The EPR experiments did not reveal a correlation between the S2 state multiline signal and the activity level under various buffer conditions. Rather, the signal height seemed to be influenced mainly by the chloride concentration, with a decrease to about 60% of the control signal in the presence of 200 mM NaCl at both pH 5.5 and pH 7.4. The absence of an effect of pH on the multiline signal would be consistent with the results of Bernát et al. (2002), who examined the pH dependence of the S-state transitions by EPR spectroscopy. However, the meaning of the decrease in signal height due to the presence of 200 mM NaCl is not clear. It may mean that the transition from the S1 to the S2 state is the point in the cycle at which the OEC is sensitive to the higher NaCl concentration, at least for the sample at pH 5.5.
In the EPR spectrum of the PSII sample at pH 7.4 in 200 mM NaCl, the negative Mn2+ signal observed after illumination at 0 °C (Fig. 3b) is interesting, since it indicates reincorporation of manganese during advancement of the S-states. The difference EPR spectrum compared a mixture of S-states present after illumination with the dark-adapted state, most likely the S1 state. The presence of a Mn2+ signal was evidently not related to greater irreversible loss of manganese from this sample compared to any other, based on the manganese content and the relatively high level of activity (90% of the control). Thus, the observation seems to indicate that Mn dissociates more easily (and is reduced) in the dark-adapted state, but is still available for oxidation and reincorporation into an active catalytic center under continuous illumination. The fact that chloride in excess of 100 mM promotes activity at the elevated pH further suggests that Cl− stabilizes the binding of manganese at the Mn4Ca cluster, at least in the higher S-states.
Conditions within the thylakoid
While the conditions under which inhibition is observed may seem extreme (low pH, high chloride concentration), they may not be far from the physiological conditions within the thylakoid lumen. The pH within the thylakoid is known to be on the acidic side due to the proton gradient needed for ATP synthesis. Under conditions of illumination, the pH difference is commonly thought to be 3.0–3.5 pH units, with the pH of the lumen as low as 5. However, it has been argued that the lumen pH under normal conditions is more likely to be relatively moderate (5.8–6.5), with a gradient of 2.0 pH units (Kramer et al. 1999). This places the minimum pH at about 5.8, although it may be less under stress conditions. The concentration of chloride within the thylakoid lumen is not well known. However, the Cl− concentration within the chloroplast (mainly stroma) was found to be in the range of 30–120 mM for higher plants with many of the measurements for spinach near 100 mM (Demmig and Gimmler 1983; Harvey et al. 1981; Robinson and Downton 1984; Robinson et al. 1983). Under various conditions, the Cl− concentration in spinach chloroplasts remained relatively unchanged, suggesting that the concentration is well regulated (Robinson and Downton 1984).
While the concentration of chloride in the lumen is not known, it is thought that rapid flux of Cl− and other ions across the thylakoid membrane is necessary for adjustment of the membrane proton motive force (between ΔpH and Δψ) at least transiently upon illumination, by electrically balancing excess positive charge in the lumen. Recently characterized chloride transporters, CLCe (Lv et al. 2009; Marmagne et al. 2007) and VCCN1 (Herdean et al. 2015), are specific to the thylakoid membrane and are thought to serve in this capacity (Pfeil et al. 2014; Spetea and Schoefs 2010). Thus, the lumen Cl− concentration is probably modulated in a way that depends on the photosynthetic requirements. However, since Cl− is not thought to contribute much to the Δψ, which itself is relatively small, it is likely that the internal and external concentrations of Cl− are similar under constant illumination. Therefore, high concentrations of chloride in the range of 100 mM are quite plausible.
Given these considerations, it is reasonable to suggest that normal intrathylakoid conditions under continuous illumination would include a pH of about 5.8 and a chloride concentration of about 100 mM. In this case, a small amount of chloride inhibition of oxygen evolution would be observed; however, if the pH were to drop to 5.5, the inhibitory effect would become much more significant. Catalytic activity of the OEC under these conditions would not cause much irreversible damage, but the higher light intensity leading to this situation may through photoinhibitory effects. This suggests that the inhibition by high Cl− concentration may serve in a protective way when the pH drops too much, by slowing down oxygen evolution activity and the associated electron transfer that can cause photoinhibition.
References
Bernát G, Morvaridi F, Feyziyev Y, Styring S (2002) pH dependence of the four individual transitions in the catalytic S-cycle during photosynthetic oxygen evolution. Biochemistry 41:5830–5843
Berthold DA, Babcock GT, Yocum CF (1981) A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. FEBS Lett 134:231–234
Boussac A, Ishida N, Sugiura M, Rappaport F (2012) Probing the role of chloride in photosystem II from Thermosynechococcus elongatus by exchanging chloride for iodide. Biochim Biophys Acta 1817:802–810
Bryson DI, Doctor N, Johnson R, Baranov S, Haddy A (2005) Characteristics of iodide activation and inhibition of oxygen evolution by photosystem II. Biochemistry 44:7354–7360
Coleman WJ (1990) Chloride binding proteins: mechanistic implications for the oxygen-evolving complex of photosystem II. Photosynth Res 23:1–27
Commet A, Boswell N, Yocum CF, Popelka H (2012) pH optimum of the photosystem II H2O oxidation reaction: effects of PsbO, the manganese-stabilizing protein, Cl− retention, and deprotonation of a component required for O2 evolution activity. Biochemistry 51:3808–3818
Cornish-Bowden A (1995) Fundamentals of enzyme kinetics. Portland Press, London
Critchley C (1985) The role of chloride in photosystem II. Biochim Biophys Acta 811:33–46
Dau H, Haumann M (2008) The manganese complex of photosystem II in its reaction cycle—basic framework and possible realization at the atomic level. Coord Chem Rev 252:273–295
Debus RJ (2014) Evidence from FTIR difference spectroscopy that D1-Asp61 influences the water reactions of the oxygen-evolving Mn4CaO5 cluster of photosystem II. Biochemistry 53:2941–2955
Demmig B, Gimmler H (1983) Properties of the isolated intact chloroplast at cytoplasmic K+ concentrations. Plant Physiol 73:169–174
Dilbeck PL, Hwang HJ, Zaharieva I, Gerencser L, Dau H, Burnap RL (2012) The D1-D61 N mutation in Synechocystis sp. PCC 6803 allows the observation of pH-sensitive intermediates in the formation and release of O2 from photosystem II. Biochemistry 51:1079–1091
Ghanotakis DF, Babcock GT, Yocum CF (1984) Structural and catalytic properties of the oxygen-evolving complex: correlation of polypeptide and manganese release with the behavior of Z+. Biochim Biophys Acta 765:388–398
Guskov A, Kern J, Gabdulkhakov A, Broser M, Zouni A, Saenger W (2009) Cyanobacterial photosystem II at 2.9-Å resolution and the role of quinones, lipids, channels and chloride. Nat Struct Mol Biol 16:334–342
Haddy A, Ore BM (2010) An alternative method for calcium depletion of the oxygen evolving complex of photosystem II as revealed by the dark-stable multiline EPR signal. Biochemistry 49:3805–3814
Haddy A, Hatchell JA, Kimel RA, Thomas R (1999) Azide as a competitor of chloride in oxygen evolution by photosystem II. Biochemistry 38:6104–6110
Harvey DMR, Hall JL, Flowers TJ, Kent B (1981) Quantitative ion localization within Suaeda maritima leaf mesophyll cells. Planta 151:555–560
Haumann M et al (2006) Bromide does not bind to the Mn4Ca complex in its S1 state in Cl−-depleted and Br−-reconstituted oxygen-evolving photosystem II: evidence from X-ray absorption spectroscopy at the Br K-edge. Biochemistry 45:13101–13107
Herdean A et al (2015) A voltage-dependent chloride channel fine-tunes photosynthesis in plants. Nat Commun 7:11654. doi:10.1038/ncomms11654
Homann PH (1985) The association of functional anions with the oxygen-evolving center of chloroplasts. Biochim Biophys Acta 809:311–319
Homann PH (1988a) The chloride and calcium requirement of photosynthetic water oxidation: effects of pH. Biochim Biophys Acta 934:1–13
Homann PH (1988b) Structural effects of Cl− and other anions on the water oxidizing complex of chloroplast photosystem II. Plant Physiol 88:194–199
Ikeuchi M, Inoue Y (1987) Specific 125I labeling of D1 (herbicide-binding protein). FEBS Lett 210:71–76
Ikeuchi M, Koike H, Inoue Y (1988) Iodination of D1 (herbicide-binding protein) is coupled with photooxidation of 125I− associated with Cl−-binding site in Photosystem-II water-oxidation system. Biochim Biophys Acta 932:160–169
Kashino Y, Koike H, Satoh K (2001) An improved sodium dodecyl sulfate-polyacrylamide gel electrophoresis system for the analysis of membrane protein complexes. Electrophoresis 22:1004–1007
Kawakami K, Umena Y, Kamiya N, Shen J-R (2009) Location of chloride and its possible functions in oxygen-evolving photosystem II revealed by X-ray crystallography. Proc Natl Acad Sci USA 106:8567–8572
Kawakami K, Umena Y, Kamiya N, Shen J-R (2011) Structure of the catalytic, inorganic core of oxygen-evolving photosystem II at 1.9 Å resolution. J Photochem Photobiol, B 104:9–18
Kelley P, Izawa S (1978) The role of chloride ion in photosystem II: I. Effects of chloride ion on photosystem II electron transport and on hydroxylamine inhibition. Biochim Biophys Acta 502:198–210
Kramer DM, Sacksteder CA, Cruz JA (1999) How acidic is the lumen? Photosynth Res 60:151–163
Kuntzleman TS, Haddy A (2009) Fluoride inhibition of photosystem II and the effect of removal of the PsbQ subunit. Photosynth Res 102:7–19
Kuntzleman T, McCarrick R, Penner-Hahn JE, Yocum CF (2004) Probing reactive sites within the photosystem II manganese cluster: evidence for separate populations of manganese that differ in redox potential. Phys Chem Chem Phys 6:4897–4904
Lindberg K, Andréasson L-E (1996) A one-site, two-state model for the binding of anions in photosystem II. Biochemistry 35:14259–14267
Lindberg K, Wydrzynski T, Vänngård T, Andréasson L-E (1990) Slow release of chloride from 36Cl-labeled photosystem II membranes. FEBS Lett 264:153–155
Lindberg K, Vänngård T, Andréasson L-E (1993) Studies of the slowly exchanging chloride in photosystem II of higher plants. Photosynth Res 38:401–408
Lv Q, Tang R, Liu H, Gao X, Li Y, Zheng H, Zhang H (2009) Cloning and molecular analysis of the Arabidopsis thaliana chloride channel gene family. Plant Sci 176:650–661
Marangoni AG (2003) Enzyme kinetics: a modern approach. Wiley, Hoboken
Marmagne A et al (2007) Two members of the Arabidopsis CLC (chloride channel) family, AtCLCe and AtCLCf, are associated with thylakoid and Golgi members, respectively. J Exp Bot 58:3385–3393
McConnell IL (2008) Substrate water binding and oxidation in photosystem II. Photosynth Res 98:261–276
Miyao M, Murata N (1984) Calcium ions can be substituted for the 24-kDa polypeptide in photosynthetic oxygen evolution. FEBS Lett 168:118–120
Miyao M, Murata N (1985) The Cl− effect on photosynthetic oxygen evolution: interaction of Cl− with 18-kDa, 24-kDa and 33-kDa proteins. FEBS Lett 180:303–308
Murray JW et al (2008) X-ray crystallography identifies two chloride binding sites in the oxygen evolving centre of photosystem II. Energy Environ Sci 1:161–166
Najafpour MM et al (2012) Biological water-oxidizing complex: a nano-sized manganese-calcium oxide in a protein environment. Photosynth Res 114:1–13
Olesen K, Andréasson L-E (2003) The function of the chloride ion in photosynthetic oxygen evolution. Biochemistry 42:2025–2035
Ono T-A, Nakayama H, Gleiter H, Inoue Y, Kawamori A (1987) Modification of the properties of S2 state in photosynthetic O2-evolving center by replacement of chloride with other anions. Arch Biochem Biophys 256:618–624
Ono T-A, Rompel A, Mino H, Chiba N (2001) Ca2+ function in photosynthetic oxygen evolution studied by alkali metal cation substitution. Biophys J 81:1831–1840
Pfeil BE, Schoefs B, Spetea C (2014) Function and evolution of channels and transporters in photosynthetic membranes. Cell Mol Life Sci 71:979–998
Pokhrel R, Brudvig GW (2013) Investigation of the inhibitory effect of nitrite on photosystem II. Biochemistry 52:3781–3789
Pokhrel R, McConnell IL, Brudvig GW (2011) Chloride regulation of enzyme turnover: application to the role of chloride in photosystem II. Biochemistry 50:2725–2734
Popelkova H, Yocum CF (2007) Current status of the role of Cl− ion in the oxygen-evolving complex. Photosynth Res 93:111–121
Popelkova H, Commet A, Kuntzleman T, Yocum CF (2008) Inorganic cofactor stabilization and retention: the unique functions of the two PsbO subunits of eukaryotic photosystem II. Biochemistry 47:12593–12600
Rivalta I et al (2011) Structural-functional role of chloride in photosystem II. Biochemistry 50:6312–6315
Robinson SP, Downton WJS (1984) Potassium, sodium, and chloride content of isolated intact chloroplasts in relation to ionic compartmentation in leaves. Arch Biochem Biophys 228:197–206
Robinson SP, Downton WJS, Millhouse JA (1983) Photosynthesis and ion content of leaves and isolated chloroplasts of salt-stressed spinach. Plant Physiol 73:238–242
Schiller H, Dau H (2000) Preparation protocols for high-activity photosystem II membrane particles of green algae and higher plants, pH dependence of oxygen evolution and comparison of the S2-state multiline signal by X-band EPR spectroscopy. J Photochem Photobiol B 55:138–144
Schulz AR (1994) Enzyme kinetics: from diastase to multi-enzyme systems. Cambridge University Press, Cambridge
Seidler A (1996) The extrinsic polypeptides of photosystem II. Biochim Biophys Acta 1277:35–60
Spetea C, Schoefs B (2010) Solute transporter in plant thylakoid membranes. Commun Integr Biol 3:122–129
Takahashi Y, M-a Takahashi, Satoh K (1986) Identification of the site of iodide photooxidation in the photosystem II reaction center complex. FEBS Lett 208:347–351
Tipton KF, McDonald AG, Dixon HBF (2009) Effects of pH on enzymes. In: Purich DL (ed) Contemporary enzyme kinetics and mechanisms, 3rd edn. Elsevier, Amsterdam
van Vliet P, Rutherford AW (1996) Properties of the chloride-depleted oxygen-evolving complex of photosystem II studied by electron paramagnetic resonance. Biochemistry 35:1829–1839
Vass I, Styring S (1991) pH-dependent charge equilibria between tryosine-D and the S states in photosystem II. Estimation of relative midpoint redox potentials. Biochemistry 30:830–839
Wincencjusz H, van Gorkom HJ, Yocum CF (1997) The photosynthetic oxygen evolving complex requires chloride for its redox state S2-to-S3 and S3-to-S0 transitions but not for S0-to-S1 or S1-to-S2 transitions. Biochemistry 36:3663–3670
Wincencjusz H, Yocum CF, van Gorkom HJ (1998) S-state dependence of chloride binding affinities and exchange dynamics in the intact and polypeptide-depleted O2 evolving complex of photosystem II. Biochemistry 37:8595–8604
Wincencjusz H, Yocum CF, van Gorkom HJ (1999) Activating anions that replace Cl− in the O2-evolving complex of photosystem II slow the kinetics of the terminal step in water oxidation and destabilize the S2 and S3 states. Biochemistry 38:3719–3725
Yachandra VK, Guiles RD, Sauer K, Klein MP (1986) The state of manganese in the photosynthetic apparatus. 5. The chloride effect in photosynthetic oxygen evolution. Biochim Biophys Acta 850:333–342
Yocum CF (2008) The calcium and chloride requirements of the O2 evolving complex. Coord Chem Rev 252:296–305
Acknowledgements
This work was supported by grants from the National Science Foundation (MCB-0950285, MCB-0111356) and the Camille and Henry Dreyfus Foundation (TH-00-12). We thank Dr. A. M. Tyryshkin for insightful discussion.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Baranov, S., Haddy, A. An enzyme kinetics study of the pH dependence of chloride activation of oxygen evolution in photosystem II. Photosynth Res 131, 317–332 (2017). https://doi.org/10.1007/s11120-016-0325-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-016-0325-z