
Abstract
Global transcriptome analyses were used to assess the interactive effects of short-term stress versus long-term acclimation to high light (HL), low temperature (LT) and excitation pressure in Arabidopsis. Microarray analyses indicated that exposure to stress resulted in two times as many modulated transcripts in both, high-light-treated and low-temperature-treated plants, compared to plants that were fully acclimated to either one of these conditions. We showed that 10.9 % of all transcripts were regulated in the same way by both stress conditions, and hence, were categorized as excitation pressure regulated, rather than regulated by either high-light or low-temperature stress per se. This group of chloroplast redox-sensitive genes included various photosynthetic genes as well as genes known to be associated with cold acclimation (cbf3, cor15A, cor15B) and gibberellic acid (GA) metabolism and signalling (ga2ox1, gai). Chemical inhibition of the photosynthetic electron transport by either DCMU or DBMIB indicated that although the plastoquinone pool contributes significantly to redox regulation of the transcriptome (8.6 %), it appears that PSI represents the major source of redox signals (89 %), whereas PSII appears to contribute only 3.1 %. A comparison of the gene expression profiles between stress and acclimated plants indicated that 10 % of the genes induced by a short, 1-h stress were also associated with long-term acclimation to high excitation pressure. This included the APETALA2/ETHYLENE-RESPONSIVE-BINDING PROTEIN family, the MYB domain- and MYB-related transcription factor family as well as the GRAS transcription factor family important in GA signalling confirming that acclimation to stress is a time-nested phenomenon. We suggest that acclimation to photosynthetic redox imbalance extends beyond the chloroplast and the leaf cell to systemic ROS signalling. This is discussed in terms of the control of plant phenotype through regulation of the nuclear encoded cbf regulon and GA metabolism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
To ensure survival and maximum fitness, it is necessary for plants to exhibit the capacity to sense and subsequently respond to changes in their abiotic environment such as irradiance, temperature, water and nutrient availability. Any sudden, short-term change in the environment typically results in plant stress which reflects a disruption in cellular homeostasis. In contrast, plant acclimation is the re-establishment of a new homeostatic state which usually necessitates growth and development in response to an initial stress condition and may lead to an altered phenotype (Hopkins and Hüner 2009). Thus, it has been suggested that plant acclimation represents a temporal continuum between an initial, stress event and a new, final steady-state condition established through the complex integration of myriad biochemical and molecular networks (Stitt and Hurry 2002). The new homeostatic state represents the acclimated state. Consequently, Falkowski and Chen (2003) suggest that acclimation to an environmental stress such as high light (HL) is a time-nested phenomenon. We suggest that this notion can be extended to the acclimation response to any stress including LT.
HL stress occurs when plants are exposed to an irradiance that exceeds the capacity of photosynthesis to utilize the absorbed light energy for the reduction of major electron acceptors such as either CO2 or NO3 − (Melis 1991; Demmig-Adams and Adams 1992; Osmond 1994; Anderson et al. 1995). Such a condition leads to the accumulation of closed PSII reaction centres due to an imbalance between the rates of PSII photochemistry and the rates at which PSII-generated electrons are ultimately consumed by metabolic sinks such as CO2 and NO3 − assimilation. Excitation pressure is a quantitative measure of the relative proportion of closed (P680+ Pheo Q −A ) versus open PSII reaction centres (P680 Pheo QA) which can be estimated either as 1 − qP (Dietz et al. 1985; Schreiber et al. 1994; Hüner et al. 1998; Rosso et al. 2009) or 1 − qL (Kramer et al. 2004). Both parameters are considered to estimate the relative redox state of QA, the first stable, immobile, quinone electron acceptor bound to PSII reaction centres. Thus, an increase in irradiance induces an increase in PSII closure estimated as excitation pressure (1 − qP or 1 − qL) (Rosso et al. 2009; Dahal et al. 2012). From light response curves for excitation pressure, one can estimate the quantum yield for PSII closure, the inverse of which is the quantum requirement for PSII closure (Rosso et al. 2009; Dahal et al. 2012).
Prolonged exposure to excess irradiance can lead to photodamage of PSII reaction centres when the light-dependent rate of D1 damage exceeds the rate of PSII repair (Krause 1988; Baker 1991; Demmig-Adams and Adams 1992; Osmond 1994; Aro et al. 1993; Long et al. 1994; Horton et al. 1996; Melis 1999; Murata et al. 2012) and the rate at which excess light can be dissipated safely as heat through nonphotochemical quenching processes (NPQ) (Demmig-Adams and Adams 1992; Horton et al. 1996; Niyogi 1999; Jahns and Holzwarth 2012; Verhoeven 2014; Derks et al. 2015). Although PSII is considered to be the major site of photodamage, PSI is also susceptible to excess irradiance (Terashima et al. 1998; Ivanov et al. 1998, 2015). Furthermore, exposure to LT stress exacerbates the effects of HL stress (Krause 1988; Baker 1991; Hüner et al. 1993). HL and LT stress enhance the susceptibility for PSII closure which is reflected in a decrease in the quantum requirement for PSII closure (Rosso et al. 2009).
In contrast to HL stress, photoacclimation is the process by which terrestrial plants, algae and cyanobacteria modulate the structure and function of the of the photosynthetic apparatus in response to short and long-term changes in irradiance (Anderson 1986; Falkowski and Laroche 1991; Melis 1991; Fujita et al. 1994; Anderson et al. 1995; Falkowski and Chen 2003; Horton et al. 2008; Li et al. 2009; Dietz 2015). Redox signals are generated in mature chloroplasts due to the over-reduction of the photosynthetic electron transport chain (PETC) and transduced to the nucleus through retrograde regulation to affect nuclear photosynthetic gene expression in response to the absorption of excess light energy (Chory 1997; Stoher and Dietz 2008; Woodson and Chory 2008; Chi et al. 2013; Dietz 2015). This alters the structure, composition and function of the photosynthetic apparatus to ensure energy balance and the maintenance of cellular homeostasis in a fluctuating light environment (Sukenik et al. 1987; Falkowski and Laroche 1991; Escoubas et al. 1995; Anderson et al. 1995; Pogson et al. 2008; Foyer et al. 2012; Dietz 2015). The establishment of such a state of energy balance in response to excess absorbed light energy is called photostasis which minimizes the potential of the chloroplast to generate potentially damaging reactive oxygen species (ROS) (Melis 1998; Hüner et al. 2003). It appears that the redox state of PQ pool (Allen 1993; Escoubas et al. 1995; Maxwell et al. 1995; Durnford and Falkowski 1997; Pfannschmidt 2003; Foyer and Noctor 2009; Fernandez and Strand 2008; Petrillo et al. 2014) as well as and the reducing-side of PSI (Dietz 2003, 2008; Stoher and Dietz 2008; Dietz and Pfannschmidt 2011; Queval and Foyer 2012) are crucial components in the generation of redox signals as a consequence of chloroplast energy imbalance which regulate retrograde signal transduction.
Acclimation to temperature change mimics photoacclimation (Maxwell et al. 1994; Machalek et al. 1996; Hüner et al. 1998, 2003, 2012, 2013). For example, plants and algae exposed to LT but moderate irradiance exhibit an equivalent closure of PSII reaction centres measured as steady-state, excitation pressure as plants exposed to HL but warm temperatures (Hüner et al. 1998). This phenomenon occurs because LT increases excitation pressure due to a reduction in the rates of energy consumption by the metabolic sinks with minimal effects on PSII photochemistry (Hüner et al. 2003; Ensminger et al. 2006; Wilson et al. 2006; Hüner and Grodzinski 2011). To maintain photostasis, winter cultivars of rye and wheat as well as Brassica napus and Arabidopsis minimize changes in pigmentation and polypeptide composition of the PETC but stimulate photosynthetic capacity measured as light saturated rates of CO2 assimilation. This appears to be due to a combination of increased photosynthetic apparatus per unit leaf area coupled with the upregulation of photosynthetic carbon metabolism and enhanced source-sink export in response to acclimation to high excitation pressure (HEP) induced either by HL or by LT (Gray et al. 1996, 1997; Savitch et al. 1997, 2002; Leonardos et al. 2003; Dahal et al. 2012, 2014). Furthermore, the up-regulation of sink capacity is translated into increased biomass accumulation and stimulation of seed yield in winter wheat (Dahal et al. 2012, 2014). Thus, in contrast to either HL or LT stress, photoacclimation and LT acclimation decrease susceptibility to PSII closure by enhancing the capacity for energy consumption through the upregulation of carbon metabolism which results in an increase in the quantum requirement to close PSII reaction centres which minimizes a dependence on NPQ (Rosso et al. 2009; Dahal et al. 2012, 2014). Consequently, assessment of the quantum requirement to close 50 % of PSII reaction centres represents a very sensitive, quantitative measure which can be used to differentiate plant stress from plant acclimation in Arabidopsis thaliana (Rosso et al. 2009), winter cereals and Brassica napus (Dahal et al. 2012, 2014).
In addition to changes in photosynthetic performance, biomass accumulation and freezing tolerance, cold acclimation of overwintering annual plant species such as winter rye, winter wheat, Brassica napus as well as Arabidopsis thaliana induces a dwarf growth habit which is governed by a family of transcription factors called C-repeat-binding factors (CBFs) (Jaglo-Ottosen et al. 1998; Gilmour et al. 2000; Penfield 2008; Thomashow 2010; Medina et al. 2011). Historically, the dwarf phenotype associated with cold acclimation has been presumed to be governed by LT only (Levitt 1980; Steponkus 1984). However, growth of winter wheat and winter rye at moderate temperatures but HL (20 °C/800 μmol photons m−2 s−1) exhibited a similar dwarf phenotype as those plants grown under typical cold acclimation conditions of LT but moderate irradiance (5 °C/250 μmol photons m−2 s−1) (Gray et al. 1997; Hüner et al. 1998). However, when rye plants were cold acclimated by growth at 5 °C but at a low irradiance, an elongated phenotype similar to that of control plants grown at 20 °C/250 μmol photons m−2 s−1 was observed (Gray et al. 1996, 1997; Hüner et al. 1998). These results were explained by the fact that the growth conditions that generated the elongated phenotype were characterized by a comparable low excitation pressure (LEP), whereas the growth conditions that generated the dwarf phenotype were characterized by a comparable HEP (Gray et al. 1997; Hüner et al. 1998). Furthermore, excitation pressure has also been shown to govern the extent of variegation of several variegated mutants of Arabidopsis including immutans, spotty, var1 and var2 (Rosso et al. 2009).
To account for the enhanced photosynthetic performance coupled with the regulation of the dwarf phenotype of cold-acclimated overwintering plants by excitation pressure, we recently proposed a model whereby redox input signals from leaf chloroplasts manifested as modulation of the redox state of the PQ pool (Kurepin et al. 2013; Hüner et al. 2014), are transduced to the nucleus via retrograde regulation that stimulate the expression of CBFs, a member of the APETALA2/ETHYLENE-RESPONSIVE-BINDING PROTEIN (AP2/EREBP) transcription factor family in Arabidopsis (Licausi et al. 2013). The model proposes that redox regulation of CBF expression not only governs photosynthetic performance, cytosolic carbon metabolism, respiration and biomass production (Dahal et al. 2012, 2014) but also governs the observed phenotypic plasticity associated with cold acclimation (Kurepin et al. 2013). The latter suggestion is consistent with the literature (Peng et al. 1997, 1999; Peng and Harberd 1997; Hussain and Peng 2003) whereby CBFs, activate GA2ox genes which decreases the levels of growth-active gibberellic acids (GAs) and maintains levels of DELLA proteins such that growth and stem elongation are repressed to generate a dwarf phenotype (Feng et al. 2008; Kurepin et al. 2013). To test this proposed model that attempts to account for the apparent system-wide, phenotypic response to chloroplast redox imbalance, we performed global transcriptome expression analyses of Arabidopsis thaliana exposed to either a short-term stress induced by a sudden shift to either HL or LT and compared the stress responses to plants acclimated to either HL or LT through long-term growth and development. As a control, we also compared the effects of abiotic environmental modulation of excitation pressure with the effects of chemical modulation of excitation pressure under constant environmental conditions. This allowed us to address the following questions regarding the role of chloroplast redox sensing/signalling in the response of Arabidopsis to stress versus acclimation to excitation pressure induced either by HL or LT. First, what are the major redox sensors/regulators within the PETC involved plant stress responses to excitation pressure? Second, are the genes modulated by short-term HEP stress different from those associated with long-term growth and development at HEP to establish the fully acclimated state, that is, photostasis? Last, does excitation pressure regulate CBF expression and its regulon as well as GA metabolism to account for the dwarf phenotype (Gray et al. 1997; Hüner et al. 1998)?
Materials and methods
Growth conditions
Stress experiment
Arabidopsis thaliana (Columbia) seeds were surface sterilized with 20 % (v/v) bleach and 0.05 % (v/v) Tween 20, sown on moistened and autoclaved soil and imbibed at 4 °C in the dark for 3 days. On the third day the seeds were shifted to a growth cabinet (GCW15, Environmental Growth Chambers, Chagrin Falls, Ohio) where they were exposed to a growth temperature of 25 °C and a light intensity of 50 µmol photons m−2 s−1 (25/50) with a 8-h/16-h (light/dark) photoperiod at 50 % humidity until mid log-phase of vegetative growth. Plants were watered every second day with half strength Hoagland’s solution.
All subsequent treatments were performed 4 h into the photoperiod and lasted for 1 h. Control plants (25 °C/50 µmol photons m−2 s−1; 25/50) were shifted to either 25 °C with 750 µmol photons m2 s−1 (HL, 25/750) or 5 °C with 50 µmol photons m−2 s−1 (LT, 5/50), respectively. During the inhibitor treatments, detached leaves were vacuum infiltrated with either H2O for control, 10 µM 3 DCMU or 6 µM DBMIB and incubated for 1 h. After each treatment, the plant material was immediately frozen in liquid nitrogen and stored at −80 °C until further processing. Control grown plants (25/50) were incubated with the respective inhibitor for 1 h after infiltration at either 25 °C/50 µmol photons m−2 s−1 or in darkness at 25 °C to correct for any pleiotropic effects of the inhibitors.
Acclimation experiment
Arabidopsis thaliana (Columbia) seeds were treated with 20 % (v/v) bleach and 0.05 % (v/v) Tween 20 for surface sterilization, sown on moistened and autoclaved soil and imbibed at 4 °C in the dark for 3 days. On the third day the seeds were shifted to a growth cabinet (GCW15, Environmental Growth Chambers, Chagrin Falls, Ohio) where they were exposed to their respective growth temperature of either 25 or 12 °C and light intensity of either 50 or 450 µmol photons m−2 s−1 (25/50; 25/450; 12/50 and 12/450, respectively) with a 8-h/16-h (light/dark) photoperiod at 50 % humidity until mid-log phase of vegetative growth. This ensured that plants were at a comparable developmental age despite the differing growth conditions. Plants were watered every second day with half strength Hoagland’s solution. Plants were harvested during mid-photoperiod, rapidly frozen in liquid nitrogen and stored at −80 °C until further processing.
Growth rates
Growth of Arabidopsis were estimated nondestructively as described in detail previously by measuring total rosette leaf area as a function of time (Rosso et al. 2009). Rosette leaf area was measured using a dissecting microscope (LeicaWild M3B) at ×4, ×10, and ×40 magnification attached to a CCD camera. Digital photos were taken, and rosette leaf area was analysed using imaging analysis software (Northern Eclipse Image Analysis Software 7.0; Empix Imaging). Rosette leaf area was measured by tracing and measuring the area of each leaf per plant. The image analysis software was calibrated with an object of known size for each magnification, and the number of pixels was divided by the appropriate conversion factor. Exponential growth rates of Arabidopsis rosette leaf expansion were calculated by linear regression analysis on log-transformed data of leaf area (mm2) versus time. One-way analysis of variance (ANOVA) was performed to determine statistical significance between genotypes (P ≤ 0.05) followed by a Bonferroni test to test for differences between group means at a 95 % confidence interval (Microcal Origin Lab 7.5; Origin Lab).
Modulated chlorophyll a fluorescence measurements
Plants were dark adapted for 20 min in order to perform steady-state fluorescence measurements using a Heinz Walz Imaging PAM (Effeltrich, Germany). The dark-adapted leaves of acclimated (25/50; 25/450; 12/50 and 12/450) and stressed plants (control, HL and LT) and detached leaves (H2O, DBMIB and DCMU infiltrated) were pulsed with an 800 ms pulse of saturating blue light (λ = 470 nm; 6000 µmol photons m−2 s−1), supplied by the Imaging PAM photodiode (IMAG-L; Heinz Walz) as a light source. The fluorescence parameters were calculated according to Schreiber et al. (1994). The temperature and measuring light intensity of all plants matched their respective growth conditions, except the HL- and LT-treated plants, which were measured at the light and temperature conditions applied by their respective stress treatment (HL: 25 °C/750 µmol photons m2 s−1; LT: or 5 °C with 50 µmol photons m−2 s−1). For the acclimated plants, light response curves for excitation pressure were generated at their respective growth temperature (either 12 or 25 °C). Excitation pressure, measured as the relative reduction state of QA of PSII reaction centres, was estimated as either, 1 − qP (Dietz et al. 1985; Hüner et al. 1998, 2003) or 1 − qL (Kramer et al. 2004; Baker 2008) and used as an estimate for the relative reduction state of the PETC. Statistical differences were assessed using a one-way ANOVA (P = 0.05) coupled with a Bonferroni test to determine significant differences between group means (Microcal Origin Lab 7.5; Origin Lab).
P700 measurements
The redox state of P700 measured as ΔA 820–860 was monitored under ambient O2 and CO2 conditions on detached leaves in vivo using a dual wavelength emitter–detector ED-P700DW unit and PAM-102 units (Heinz Walz, Germany), as previously described by Ivanov et al. (2006). Far-red light was provided by a FL-101 light source (λ max = 715 nm, 10 W m−2, Schott filter RG 715) and used to fully oxidize P700 to P700+. Subsequent single turnover (ST, 14 µs) and multiple turnover (MT, 50 ms) saturating light flashes were applied with XMT-103 and XST-103 power control units, respectively, in order to reduce P700+ to P700. The redox state of P700/P700+ was assessed as the change of absorbance at 820 nm and the signals were recorded using an oscilloscope card (PC-SCOPE T6420, Intelligente Messtechnik GmbH, Backnang, Germany) installed in an IBM-PC. The peak area under the oxidation curve following the ST and MT flashes and the steady-state P700+ signal was used to assess the inhibition of intersystem PETC by DBMIB and DCMU.
RNA extraction
Leaf material of three individual plants from each growth condition was pooled into one sample, ground to a fine powder using liquid nitrogen and RNA was extracted using the RNeasy Plant Minikit (Qiagen). Residual DNA was digested on-column utilizing the matching RNase-free DNase kit (Qiagen). Three biological replications were performed, meaning for each experimental condition we used three different samples, each containing the leaves of 3 plants.
RNA quality assessment, probe preparation and GeneChip hybridization
The quality of the extracted RNA was examined using the Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Palo Alto, CA) and the RNA 6000 Nano kit (Caliper Life Sciences, Mountain View, CA). Biotinylated complementary RNA (cRNA) was generated from 500 ng of total RNA following the Affymetrix GeneChip 3′ IVT Express Kit Manual (Affymetrix, Santa Clara, CA). A total of 10 μg of labelled cRNA was hybridized to the Affymetrix Arabidopsis ATH1 Genome Arrays for 16 h at 45 °C as described in the Affymetrix GeneChip 3′ IVT Express Kit Manual (Affymetrix, Santa Clara, CA). The first step of GeneChip staining was performed by using Streptavidin–Phycoerythrin, the second step by an antibody solution and then finally another Streptavidin–Phycoerythrin solution, with all liquid handling being performed by a GeneChip Fluidics Station 450. GeneChips were then scanned by means of the Affymetrix GeneChip Scanner 3000 7G (Affymetrix, Santa Clara, CA) using Command Console v1.1.
Probe level (.CEL file) data were generated using the Affymetrix Command Console v1.1. Probes were summarized to the gene level data in Partek Genomics Suite v6.5 (Partek, St. Louis, MO) using the RMA algorithm (Irizarry et al. 2003). Partek was used to determine gene level ANOVA P values, fold changes and false discovery rate (FDR).
Quantitative real-time RT-PCR
First strand cDNA was generated using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, CA). Real-time PCR was then performed using TaqMan Gene Expression Assays (Applied Biosystems, CA) for each of the displayed genes (Table S1) 15 ng of cDNA and the 7900HT Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using the standard run conditions recommended by the manufacturer (50 °C: 2 min; 95 °C: 10 min; ×40 (95 °C: 15 s; 60 °C: 1 min). The total reaction volume was 20 µl and the relative cDNA levels were calculated using the relative standard curve method according to the manufacturer’s recommendations. The expression of each gene was normalized to the expression of act2.
Functional analysis of differentially regulated genes
The MapMan program was used in order to classify and display genes into metabolic pathway groups and to quantify the genes active in cellular responses (Thimm et al. 2004). The Affymetrix probeset ID’s were matched utilizing the appropriate gene annotation files for the ATH1 gene chip and either displayed in the context of the major metabolic pathways, or cellular responses.
Results
Short-term stress
Assessment of the relative reduction state of the PETC
For the stress experiment, we compared the effects of a short-term (1 h) shift of 45–50 day old control plants grown at 25 °C/50 μmol photons m−2 s−1 (25/50) (Fig. 1a; diamonds) to either HL (750 μmol photons m−2 s−1) at 25 °C or LT (5 °C) at 50 μmol photons m−2 s−1. Since maximum, exponential growth rates of Arabidopsis to irradiance was saturated at 450 μmol m−2 s−1 at 25 °C (Fig. 1a; Table 1), a shift to an irradiance of 750 μmol m−2 s−1 at 25 °C was considered sufficient to induce photosynthetic redox stress. To verify this, we performed chlorophyll a fluorescence induction measurements to assess the extent of excitation pressure induced by a short-term shift to either HL or LT stress. The results illustrated in Fig. 2 indicate that growth of plants at 25/50 (control) induced a relatively LEP (1 − qP = 0.15), whereas exposure of control plants to either HL (750 μmol photons m−2 s−1) at 25 °C or LT (5 °C) at 50 μmol photons m−2 s−1 for 1 h induced a comparable fourfold increase in excitation pressure (HL: 1 − qP = 0.57; LT: 1 − qP = 0.59) relative to controls (Fig. 2a). Thus, a 1-h shift of control plants exposed to the HL stress at 25 °C generated a comparable excitation pressure to a 1-h shift of control plants to LT stress (5 °C) at 50 μmol photons m−2 s−1.

Arabidopsis growth curves. The growth of Arabidopsis (Col-0) plants grown at different light (50, 150, and 450 µmol photons m−2 s−1) and temperature (a 25 °C and b 12 °C) regimes were quantified as total rosette leaf area throughout the plants’ vegetative development utilizing digital images. These data show representative results from two independent experiments, and each data point represents the average from 5 to 15 individual plants ± SE

a Quantification of excitation pressure in Arabidopsis leaves after various stress treatments. Excitation Pressure was measured as 1 − qP in all treatments. Control plants and plants infiltrated with DCMU, DBMIB and water were measured at 25 °C and 50 µmol photons m−2 s−1, while plants exposed to HL and LT were measured at 25 °C and 750 µmol photons m−2 s−1 and 5 °C and 50 µmol photons m−2 s−1, respectively. Data represent the mean ± SE calculated from three to six different plants per treatment. Letters represent statistically significant differences between means at the 95 % confidence interval. b Intersystem electron transport in response to inhibitor treatment. P700 traces measured as ΔA 820 in detached leaves that were vacuum infiltrated with either DBMIB and DCMU or water and incubated for 1 h at 25 °C and at 50 µmol photons m−2 s−1. After a steady-state level of P700+ was achieved by illumination with far-red light (FR), a saturating white light single turnover flash (ST) and a multiple turnover (MT) flash pulse were applied. Each trace shows a representative result chosen from three individual measurements from three distinct plants
P700 measurements were performed to assess the effects of the DCMU and DBMIB on the intersystem PETC (Fig. 2b). After exposure of a control leaf infiltrated with H2O to far red light (FR, Fig. 2b), the ΔA 820–860 signal increased rapidly due to the photo-oxidation of P700 to P700+. Application of a single-turnover (ST) and a multiple turnover (MT) saturating light flash resulted in a sudden but transient decrease in the ΔA 820–860 signal indicating the transient reduction of P700+ to P700 due to electrons generated by PSII. Although infiltration with H2O did not impair intersystem electron transport (Fig. 2a, H2O), leaves infiltrated with either 10 μM DCMU or 6 μM DBMIB inhibited the transient reduction of P700+ to P700 by the ST and MT saturating flashes (Fig. 2b). However, as expected, the absolute ΔA 820–860 signal in the presence of DCMU was approximately 25 % lower than that observed in the presence of DBMIB due to the contribution of stromal-reducing equivalents to the reduction of P700+ in the presence of DCMU but not in the presence of DBMIB (Asada et al. 1993; Ivanov et al. 2006). Thus, these concentrations of DCMU and DBMIB were sufficient to completely block intersystem photosynthetic electron transport. This is consistent with the Chl a fluorescence data which indicated that excitation pressure was at the theoretical maximum (1 − qP = 1.0) in the presence of either 10 μM DCMU or 6 μM DBMIB (Fig. 1a) indicating that all PSII reaction centres were completely closed at these concentrations of inhibitors. The absolute values of excitation pressure varied minimally when the relative reduction state of QA was measured as 1 − qP (Dietz et al. 1985; Schreiber et al. 1994; Hüner et al. 1998) or as 1 − qL (Kramer et al. 2004; Baker 2008) (see Supplemental Fig. S1).
Differential gene expression
Our experimental design entailed the assessment of 15 different conditions each in triplicate. First, a principal component analysis plot of the Robust Multi-array Average (RMA) was used to assess the consistency and reproducibility of the primary microarray data generated by the 45 Affymatrix ATH1 genome arrays (Supplemental Fig. S2). As expected for the 15 different conditions used to generate the primary microarray data, each group of triplicate microarrays clustered together indicating minimal discrepancy within each of the 15 conditions. Second, to assess an appropriate cut-off level for global gene expression analyses, we examined differentially regulated genes at different fold-change cut-off levels (1.5x, 2.0x, 2.5x and 3.0x) on a functional level using MapMan (Thimm et al. 2004). As expected, the higher fold cut-off reduced the number of genes differentially expressed under HL and LT stress as well as the number of genes associated with the intersection of both HL and LT stressed samples for both up- and down-regulated genes (Supplemental Fig. S3A, B). While choosing various fold-change values as potential cut-off levels did affect the total number of genes that were differentially expressed, the ratios between up- and down-regulated genes and the intersections between the compared treatments remained the same, indicating that the choice of a cut-off level of 1.5 fold did not skew the results in any particular direction (Supplemental Figs. S3A–C, S4A–F). Based on these results (Fig. 3a–c, Supplemental Fig. S4A–F), a cut-off value of 1.5x with a statistical significance of P = 0.01 was chosen for all subsequent gene expression analyses.

Number of genes regulated by HL and LT. Venn diagrams displaying the total number of genes (a) up- or (b) down-regulated in total after shift to high light (HL) and low temperature (LT) in comparison with control plants (25/50). c Heatmap of 2489 stress-regulated transcripts. Blue colour represents down-regulated genes and red colour represents up-regulated genes. Genes were considered changed at a fold-change value ≥1.5x and P ≤ 0.01
Quantitative real-time RT-PCR was performed in order to verify the microarray expression analysis for the transcripts for (spa1, dreb1a, nda1 and tub8) for microarrays used for short-term stress (Supplemental Fig. S9) as well as for long-term acclimation (Supplemental Fig. S10; Table S1). All four transcripts showed the same trend in both the Affymetrix ATH-1 microarray and the qPCR experiment for the majority of experimental treatments.
Changes in global transcript abundance in response to HL and LT stress
The shift to either HL or LT resulted in the radical reprogramming of gene expression including both up-regulated and down-regulated genes, compared to the control (Fig. 3a–c). Out of the 22,800 probes monitored, ca. 19.9 % were differentially expressed as a result of the HL treatment, or, more precisely, 2073 transcripts were up-regulated (Fig. 3a) and 2456 were down-regulated (Fig. 3b). This is congruent with previous experiments performed by Ruckle et al. (2012) which found that 20 % of the Arabidopsis transcriptome is regulated by light. However, under the LT stress treatment, approximately 29.2 % of the genes were differentially expressed that reflected 2493 up-regulated (Fig. 3a) and 4174 down-regulated transcripts (Fig. 3b). Thus, HL and LT stress in Arabidopsis resulted in a greater proportion of down- regulated than up-regulated transcripts. Furthermore, LT caused a 20 % greater up-regulation (Fig. 3a, c) and a 70 % greater down-regulation than the HL stress treatment (Fig. 3b, c).
The following mRNAs that are typically down-regulated by HL were captured by our HL treatment: lhcb2.1: −1.6x; lhcb2.3: −14.6x; lhcb3: −2.0x; lhcb5: −1.8x; lhcb4.2: −7.9x; lhcb6: −3.1x; lhca1: −1.6x; lhca2: −1.5x; lhca4: −2.6x; lhca6: −2.6x; elip1 9.6x; elip2: 7.1x; petE1: −4.3x. In addition, several transcripts that are known to be cold-induced, such as the cold-regulated (cor) genes and the cbf transcription factors were all up-regulated in our LT shift experiment (cor15a: 2.1x; cor15b: 2.5x; cor47: 6.2x; cbf1: 8.4x; cbf2: 9.8x; cbf3/dreb1a: 10.4x; cbf4: 2.3x). The expected changes in expression of these internal control genes were used to validate our experimental design for the imposition of a short-term stress.
Changes in global transcript abundance in response to the reduction state of the PETC
Approximately 10.9 % of all measured transcripts showed the same altered gene expression in both, the HL and LT treatment of which 817 were up-regulated (Fig. 3c) and 1672 down-regulated (Fig. 3c). Since both HL and LT stressed plants were exposed to comparable excitation pressure (1 − qP) (Fig. 2), we concluded that the overlap between HL and LT gene populations reflected those genes whose expression was regulated by excitation pressure rather than either HL or LT per se. For example, several of the transcripts that were annotated to be cold induced such as cbf3, cor15A and cor15b were also found up-regulated by the HL stress and transcripts annotated to be HL regulated responded in the same way to the LT stress treatment (i.e. lhcb4.1 (down); lhcb6 (down); lhca6 (down); elip2 (up); petE1 (down). In addition, other genes affected by excitation pressure were associated with phytochrome sensing/signalling and included the two phytochromes phyA (down) and phyE (up), as well as the phytochrome associated transcripts pap2 (down), pks2 (down), pif4 (down) and bas1 (down). Furthermore, excitation pressure also appeared to regulate the expression of transcripts associated with GA metabolism. ga2ox1 was up-regulated, whereas ga3 was down-regulated the combination of which would result in a decrease in the accumulation of growth-active GAs (Kurepin et al. 2013). In addition, transcripts involved in GA signalling [GAI (down); gasa4 (down) and gasa5 (down)] were also captured in the analyses of genes responding to HEP. The gene, GAI, encodes DELLA proteins which normally repress growth in the absence of growth-active GAs (Hussain and Peng 2003). Also, the plastidic RNA-polymerase subunits, known as sigE and sigC were induced, while other sigma-factors remained unaltered. In addition, a multitude of ROS scavenging enzymes were affected by HEP [i.e. tAPx (down), APx4 (down), csd1 (down), gpx5 (down), gpx6 (up), gr1 (up), gpx1 (down), gpx3 (down), prxIIF (down) in addition to the known zinc finger ROS scavenging regulators zat10 (up) and zat6 (up) (Mittler 2006; Perez and Brown 2014)].
Effects of DCMU and DBMIB on global transcript abundance
In order to distinguish which genes are regulated by the PQ-pool and which ones were altered due to the infiltration process or to possible nonspecific effects of the inhibitors, plants were infiltrated with either DCMU, DBMIB or water, respectively, and then incubated for 1 h in either control growth light conditions (50 µmol photons m−2 s−1) or in the darkness. An ANOVA was performed in order to create gene lists comparing the inhibitor and water infiltration effects in the light compared to dark controls. The resulting lists of genes altered only by infiltration with either inhibitor in the light were compared to the list of genes altered by H2O infiltration only in the light and duplicates were removed in order to create lists of genes that were truly regulated by either DCMU or DBMIB. Both inhibitors were used at the minimal concentrations necessary to inhibit the PETC on the one hand (Fig. 2) but, on the other hand, avoid potential pleiotropic effects at higher concentrations of these inhibitors.
Infiltration with either DCMU or DBMIB resulted in a more specific change in the gene expression profiles. While about 9.3 % of all genes (1057 up-regulated and 1071 down-regulated) were differentially expressed after treatment with DBMIB (Supplemental Fig. S4A, B), only about 3.1 % of the monitored transcripts (285 up-regulated and 420 down-regulated) were altered in response to the DCMU inhibition (Supplemental Fig. S4C, D).
Contribution of PQH2 and PSII(Q −A ) to changes in global transcript abundance
The site for DCMU inhibition of PETC is the QB-binding site of the D1 polypeptide of PSII (Ke 2001; Kurepin et al. 2013). Since the PQ-pool was already fairly oxidized (1 − qP ≈ 0.15) (Fig. 2) during the control, LEP growth conditions (Fig. 1, diamonds), the addition of DCMU on the redox state of PQ would be expected to be minimal since DCMU inhibits electron transfer from PSII into the PQ pool but allows PQH2 to be oxidized by PSI via the Cyt b 6 /f complex (Ke 2001; Kurepin et al. 2013). The major effect of the addition of this inhibitor was to close PSII reaction centres (1 − qP = 1.0; Fig. 2) which indicates that QA is fully reduced. However, the site for DBMIB inhibition of the PETC is the Qo-binding site of the Cyt b 6 /f complex (Ke 2001). In the presence of DBMIB, PSII reduces the PQ pool but electron transfer from PQH2 to PSI is inhibited. Thus, DBMIB not only closed all PSII reaction centres (Fig. 2) but concomitantly, results in the accumulation of PQH2 (Ke 2001; Kurepin et al. 2013; Petrillo et al. 2014). Hence, in order to distinguish the list of genes that was purely regulated by PQH2 from those regulated by PSII(Q −A ), the list of DCMU-regulated genes, that is, genes regulated solely by PSII(Q −A ), was subtracted from the list of DBMIB-regulated genes, that is genes regulated by PSII(Q −A ) plus PQH2 (Fig. 4). The number of genes regulated by PQH2 was still about 8.6 % of the total with 970 up-regulated and 1000 down-regulated transcripts (Fig. 4, Supplemental Fig. S4). Many photosynthetic transcripts were up-regulated, such as lhca1; lhca3; lhca5; lhca6; lhcb2.1; lhcb2.3; lhcb4.2 and lhcb6, while none of them appeared to be down-regulated by PQH2. Genes that have been previously reported to be involved in plastid retrograde signalling, such as gun5 (Koussevitzky et al. 2007) and stn7 (Pesaresi et al. 2011) were up-regulated by PQH2 about 1.8 fold and 2.0 fold, respectively, but not up-regulated by any of the other treatments. In addition, the transcripts of cbf1 and cbf3 normally associated with cold stress were up-regulated by PQH2, along with the GA related transcript kao2. Other transcripts associated with GA biosynthesis and signalling were down-regulated by PQH2 (kao1 and gasa4) as were the phytochrome transcripts phyB and phyD.

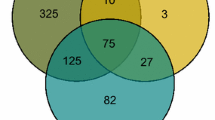
Comparison of differentially expressed genes in different groups. Number of genes altered in each treatment compared to its respective control condition and comparisons of different treatments. HEP = genes that are equally regulated by high light and low temperature; PQH2 = genes regulated by plastoquinol; PSII = genes regulated by photosystem II; ∩ = intersection between two gene lists. The black part of the bars represents down-regulated genes and the white part represents the up-regulated genes. Genes were considered altered at a fold-change level of 1.5x and P = 0.01
In order to identify those genes strictly regulated by excitation pressure, that is by PSII(Q −A ), the gene expression data from DCMU-treated plants were used since DCMU closes all PSII reaction centres (Fig. 2), but keeps the PQ pool in the oxidized state (Kurepin et al. 2013; Petrillo et al. 2014). Thus, this list of genes was inferred to be regulated by PSII(Q −A ) (Fig. 4). Genes up-regulated directly by PSII(Q −A ) included the following: the PSII core antennae transcript psbB; another smaller subunit of PSII, psbTN; and two nuclear-encoded subunits of the plastidic RNA polymerase complex, namely sigC and sig4.
By definition, excitation pressure is the a measure of the proportion of closed PSII reaction centres (P680+ Pheo Q −)A which reflects the relative reduction state of QA (Q −A /QA + QA) (Dietz et al. 1985; Hüner et al. 1998; Ensminger et al. 2006; Hüner et al. 2013). Theoretically, the list of genes regulated by HEP should be the same as the list of genes regulated by PSII(Q −A ). However, when we compared the set of genes regulated by HEP with that regulated solely by PSII(Q −A ) (HEP ∩ PSII), there was an overlap of only 5.5 % (51 up- and 86 down-regulated genes) between the two groups (Fig. 4). This included the cor15A gene which was up-regulated and the sigC subunit of the plastidic RNA polymerase. Similarly, a comparison of the list of genes regulated by HEP with that regulated by PQH2 (HEP ∩ PQH2) indicated an overlap of only 5.7 % (47 up-regulated and 95 down-regulated), (Fig. 4) which included the up-regulated cbf3 transcript, as well as var1 which encodes the protease, FtsH5, involved in PS II reaction centre repair (Yu et al. 2007; Miura et al. 2007).
Functional analysis of the differentially regulated genes in response to HEP stress
The genes that were differentially regulated as a result of HEP stress (HL ∩ LT) appeared to be ubiquitously distributed over all of the most important metabolic processes, regardless of their cellular localization (Fig. 5a; see also Table S2). Similarly, that set of genes regulated by PQH2 (Fig. 5b; see also Table S3) also appeared to be distributed amongst all major metabolic pathways. However, although genes regulated by PQH2 accounted for 79 % of the HEP genes, HEP appeared to inhibit gene expression, while PQH2 increased transcript abundance, sometimes for the same genes (e.g. lhcb6, lpa2, lhcb4.2, lhca6, atpd, ted4). Only aoat2 seemed to be up-regulated by both regulators (see Tables S2, S3).

MapMan display of major metabolic pathways affected by short-term photosynthetic stress. MapMan software (Thimm et al. 2004) was used to display significant changes in transcript abundance of genes associated with major metabolic pathways. a The differentially expressed genes that were affected by HEP. b The genes that were regulated by PQH2 and c the genes that were differentially expressed by PSII. Red squares represent up-regulated transcripts, blue squares represent down-regulated transcripts, while white squares represent transcripts that remained unaltered in comparison to the control
The impact of PSII(Q −A ) on regulating gene expression in the major metabolic pathways remained distinctly smaller than the impact of either HEP or PQH2. However, there appeared to be no apparent concentration of regulated transcripts in the photosynthesis related pathways of genes controlled by PSII(Q −A ) (Fig. 5c; see also Table S4).
The additional MapMan analysis of the effects of short-term stress on cellular responses showed that in spite of the different total number of regulated genes, all three redox regulators (HEP, PQH2 and PSII(Q −A ) modulated gene expression involved with most general cellular response processes (Fig. 6). However, ROS signalling was the predominant process affected by HEP stress followed by cell cycle, cell division, abiotic and cell development processes regardless of the chloroplast redox regulator (Fig. 6). PQH2 appeared to contribute to a similar extent to the modulation of genes associated with abiotic stress as HEP, whereas processes associated with biotic stress were the least affected by HEP stress (Fig. 6).

Display of cellular responses affected by short-term photosynthetic stress. MapMan software (Thimm et al. 2004) was used to display an overview of significant changes in transcript abundance of genes associated with cellular responses, showing the percentage of genes altered in each category. Shown are the differentially expressed genes that were regulated by HEP, PQH2 and closed PSII reaction centres
Long-term acclimation
Acclimation entails long-term growth and development to establish a new, homeostatic state. To ensure comparison of acclimated plants at comparable developmental states (Krol et al. 1984; Claeys et al. 2014), comparative growth curves were used to estimate the time required to achieve a comparable vegetative growth stage under the various growth regimes (Fig. 1a, b). Based on the growth curves (Fig. 1) and the exponential growth rates (Table 1), an irradiance of 450 μmol photons m−2 s−1 was sufficient to saturate the growth response of Arabidopsis thaliana at either 25 °C (Fig. 1a) or 12 °C (Fig. 1b). In contrast, the growth response at 50 μmol photons m−2 s−1 exhibited an extensive lag time and an exponential rate that was approximately 50 % lower than that observed at either 150 or 450 μmol photons m−2 s−1 at 25 °C (Fig. 1a). Based on the growth curves, Arabidopsis grown at 25/50 for 45–50 days were at a comparable vegetative growth state as plants grown for 18–20 days at either 25/150 or 25/450 (Fig. 1a). Similar trends were observed for growth at 12 °C. Plants grown for 70–75 days at 12/50 were at a comparable vegetative growth stage as those grown between 42 and 48 days at either 12/150 or 12/450 (Fig. 1b). Thus, our growth analyses allowed us to choose an appropriate time for leaf sampling which minimized differences in vegetative growth state regardless of the growth condition.
The phenotypes of plants acclimated to different levels of excitation pressure induced by growth under varying levels of light and temperature indicated that with increasing excitation pressure the petioles tended to be shorter and the leaves exhibited decreased Chl (Fig. 7a) consistent with previous results for Arabidopsis (Rosso et al. 2006). All of these growth effects appear more pronounced at 12 °C than at 25 °C and with increased growth irradiance.

a Representative photographs of Arabidopsis acclimated to various light and temperature regimes. Plants were grown to mid-log phase of vegetative growth at their respective growth regime in order to have them at a comparable developmental age. Photos display representative plants from each growth regime. b The effects of growth irradiance and temperature on excitation pressure and photoacclimation. Excitation pressure (1 − qP) light response curves were performed for Arabidopsis leaves grown at 25 °C under varying growth light regimes [25/50 (squares), 25/450 (diamonds)]. Measurements were performed on attached leaves at the respective growth temperature of the plant, with increasing irradiance from 0 to 1550 µmol photons m−2 s−1. Plants were grown with an 8/16-h day/night cycle, and attached leaves were measured 4 h into the photoperiod. Data represent the mean ± SE calculated from 2 to 4 measurements per plant in 3 to 5 different plants per treatment
Previous results have shown that photoacclimation typically results in an increase in the quantum requirement for PSII closure as indicated by an increase in the number of photons required to close 50 % of the PSII reaction centres (Rosso et al. 2009; Dahal et al. 2012, 2014). Consequently, to assess the extent of photosynthetic acclimation under our different growth regimes, we compared the light response curves for excitation pressure in plants developed under our various growth regimes (Fig. 7b, Supplemental Fig. S5). Plants grown under HL conditions (450 μmol photons m−2 s−1) at 25 °C required an irradiance of about 1200 μmol photons m−2 s−1, whereas those grown at low light (50 μmol photons m−2 s−1) at the same growth temperature required an irradiance of only 650 μmol photons m−2 s−1 to close 50 % of PSII reaction centres. Thus, as expected, the quantum requirement for PSII closure for plants acclimated to HL was almost double that of plants acclimated to LL (Fig. 7b). Similar trends in quantum requirement for PSII closure were observed for plants grown at either HL or LL but 12 °C (Supplemental Fig. S5). In addition, the maximum photochemical efficiencies of PSII (F v/F m) of all the plants grown at the various light and temperature regimes were comparable at a value of about 0.8 (Table 1), demonstrating that neither of the growth regimes induced chronic photodamage. These results are consistent with the designation of plants grown at either 12/50 or 25/50 as acclimated to LEP, whereas those plants grown at either 12/450 or 25/450 were acclimated to HEP.
Acclimation to either LT or HL versus acclimation to HEP
In order to distinguish genes that were differentially expressed as a result of acclimation to HEP from differential gene expressions due to acclimation to either light or temperature, we used plants grown at 25/50 as our LEP control group. As illustrated in Fig. 8a, we then compared this control group to plants that were grown at the same irradiance but LT (12/50) to assess changes in transcript abundance due to acclimation to LT. To assess the effects of acclimation to HL on transcript abundance, we compared control plants (25/50) with plants grown at the same temperature but at high irradiance (25/450). Finally, to assess the combined effects of HL and LT (HL + LT) on transcript abundance, we compared control plants (20/50) with plants grown at 12/450 (Fig. 8a). Two-way ANOVAs were used in all comparisons. Only genes that increased or decreased their expression levels with a fold-change value ≥1.5 and P ≤ 0.01 were considered differentially expressed. This resulted in three separate gene lists that were differentially expressed due to acclimation to either low growth temperature (LT), high growth light (HL) or both (HL + LT) (Fig. 8a). Differentially expressed genes that overlapped both HL and LT were considered to be altered due to acclimation to HEP (Fig. 8b, c). These HEP acclimation genes were compared to HL + LT as another test to verify whether the genes associated with acclimation to HEP were also found to be regulated in the plants acclimated to 12/450 (Fig. 8b).

Determination of differential gene expressions as a result of acclimation to HEP. a A two-way ANOVA was performed between the microarray gene expression data from the plants grown at 12 °C and 50 µmol photons m−2 s−1 (12/50) and the control plants grown under 25 °C and 450 µmol photons m−2 s−1 (25/50); the resulting gene list was considered to be regulated by acclimation to low temperature (LT). The same process was applied to plants grown under 25 °C and 450 µmol photons m−2 s−1 (hight light; HL) and plants grown under 12 °C and 450 µmol photons m−2 s−1 (high light and low temperature; HL/LT). Transcripts were considered changed at a fold-change value of 1.5x and P ≤ 0.01. b Transcripts that were altered in the same manner due to both lower growth temperature LT and higher growth irradiance (HL) were considered to be regulated by high excitation pressure (HEP). As a control, the HEP list was compared to those of the genes regulated by high light and low temperature at the same time (HL/LT). c Heatmap of and 735 acclimation altered transcripts. Blue colour represents down-regulated genes and red colour represents up-regulated genes. Genes were considered changed at a fold-change value ≥1.5x and P ≤ 0.01
Acclimation to HL resulted in 12 % of the transcriptome being differentially expressed with 1282 up- and 1547 down-regulated genes (Figs. 8c, 9), including, as expected, various light harvesting genes (i.e. lhcb2.3: down; lhcb6: down; lhcb4.3: up; lhca6: down; lhcb4.2: down), transcripts for subunits of both, PSII (psbP2: down; psbH: up; psbY: down; psbK: down; psbQ: down) and PSI (psaD2: down; psaE2: down; psaH1: down; psaH2: down), Calvin-Benson cycle enzymes (rbcL: up; gapcp1: up; rpe: down) and many more photosynthesis-associated genes (petG: down; atpI: up; atpA: down; fnr2: up; ndhO: down).

Comparison of differentially expressed genes in plants acclimated to varying light and temperature regimes. Number of genes altered in plants grown at hight light (HL), in plants grown at low temperature (LT), in plants grown at high light and low temperature at the same time (HL + LT), the list of common genes (HEP) between HL and LT and the intersection of genes regulated by HL + LT and HEP. The black part of the bars represents down-regulated genes and the white part represents the up-regulated genes. Genes were considered changed at a fold-change value ≥1.5x and P ≤ 0.01
Acclimation to LT (Fig. 8c) resulted in 10 % of all genes being regulated differentially, with 1162 up- and 1127 down-regulated genes (Fig. 9). Apart from a range of photosynthetic genes (lhcb4.3: up; lhca6: down; psbE: down; psaD2: down; atpH: up), the expression of several genes associated with LT acclimation were up-regulated and included cor47, cor15A, cor15B, cor414-tm1, cbf1-3, and ice1. cbf-b was down-regulated. As expected, acclimation to HL + LT resulted in the largest change in gene expression in comparison to the control (Fig. 8a) with approximately 16 % of the genome altered (1764 up- and 1931 down-regulated) (Fig. 9). Transcripts related to photoacclimation (lhcb4.2, psbN, lhca6, psaA, petE) were generally down-regulated in response to HL + LT except for ndhA which was up-regulated. In contrast, transcript abundance of genes associated with LT acclimation (cor47, cor15A, cor414-TM1, cor15B, cor413-PM1, cor314-TM2, cbf1, and cbf3) were all up-regulated in response to HL + LT.
The overlap between HL and LT (HL ∩ LT) (Fig. 8b) was designated as the source of genes regulated by acclimation to HEP (Table 1) which constituted approximately 3 % of all differentially expressed mRNAs (344 up- and 391 down-regulated) (Fig. 9). About 79 % of these HEP acclimation genes were also found modulated in the plants grown at 12/450 (HEP ∩ 12/450) corroborating the validity of designating this list of genes as associated with acclimation to HEP (Fig. 9). Interestingly, the list of transcripts associated with acclimation to HEP included several transcripts not only associated with the PETC such as subunits of PS II (lhcb4.3: up; psbp2: down), PS I (lhca6: down; psad2: down, ycf3: up) and the Cyt b 6/ f complex (petg: down), but also components of the thylakoid ATP synthase (atpa: down; atph: up). In addition to these photosynthetic transcripts, some of the transcripts associated with cold acclimation were also present in this HEP acclimation list such as cor15A: up, cor15B: down in addition to two phytochrome associated transcripts, hfr1 (down), epr1 (down). This indicates that many transcripts that have previously been ascribed to either light or temperature regulation, are indeed regulated by HEP rather than either HL or LT per se. According to the MapMan software (Thimm et al. 2004) ca 11 % of the transcripts in the HEP acclimation gene list encode transcription factors.
In plants exposed to the short-term (1 h) HEP stress, we determined that about 11 % of the entire Arabidopsis transcriptome was changed (Fig. 4). Hence, we asked what proportion of the list of short-term, HEP stress genes was still present in the gene list for plants acclimated to long-term growth under HEP. To answer this question, we made a comparison between the lists of genes that were involved in HEP stress with the gene list associated with acclimation to HEP (Fig. 10). Out of the 735 genes that were differentially expressed as a result of acclimation to HEP, almost one third (224 transcripts) were transcripts that were induced by the 1-h HEP stress (Fig. 10). Amongst those genes were the following down-regulated photosynthetic transcripts, lhca6, psad2 and psbp2, and the up-regulated, cold-induced transcripts, cor15A, cor15b, as well as a variety of transcription factor families including the ABI3/VPI-related B3-domain family, the APETALA2/ETHYLENE-RESPONSIVE-BINDING PROTEIN family (AP2/EREBP), MYB domain and MYB-related transcription factor family as well as the GRAS transcription factor family important in GA signalling (Table 2).

Common gene expression in HEP stress and acclimation. a Venn Diagramm comparison of genes differentially regulated by short-term (1 h) HEP stress and long-term HEP acclimation. Genes were considered changed at a fold-change value ≥1.5x and P ≤ 0.01. b 2991 transcripts are displayed in this figure. Grey colour represents unaltered genes, while blue colour represents down-regulated genes and red colour represents up-regulated genes. Green arrows indicate co-regulated genes, while the purple arrow indicates genes that display a negative correlation between the stress response and the acclimation response. Genes were considered changed at a fold-change value ≥1.5x and P ≤ 0.01
Functional analysis of the acclimation response to HEP
The set of genes that were responsible for the acclimation to HEP displayed very little accumulation of altered transcripts related to any one specific metabolic pathway when it was plotted using MapMan. Interestingly, it seems that particularly processes associated with energy metabolism such as photosynthesis, photorespiration, and respiration appeared mostly unchanged in the acclimated plants (Fig. 11a). Of the 136 genes associated with PETC, only eight were affected by acclimation to HEP; lhcb4.3 and atph were up-regulated, while the remainder (psbp-2, lhca6, psad2, petg and atpa) were all down-regulated. Furthermore, two oxygen scavenging transcripts were down-regulated (gpx1 and apx3). Unlike lipid metabolism, cell wall biogenesis and the secondary metabolism pathways where both up- and down-regulation of transcripts were observed, only up-regulated transcripts in starch and sucrose metabolism were detected (Fig. 11a).

MapMan display of gene expression data as a result of long-term acclimation to HEP. MapMan software (Thimm et al. 2004) was used to display significant changes in transcript abundance of genes associated with major metabolic pathways. a The differentially expressed genes which represent the acclimation to HEP. b The genes that were regulated by both HEP acclimation and short-term HEP stress. Red squares represent up-regulated transcripts, blue squares represent down-regulated transcripts, while white squares represent transcripts that remained unaltered
Figure 11b illustrates that out of the 224 genes regulated by both stress and acclimation to HEP (Fig. 10), there was a paucity of genes (12 %) on this list associated with any major metabolic pathway. Only three of the genes involved in photosynthetic processes (lhca6, psbp-2, psad-2; all down-regulated) appeared on this list of genes common to both stress and acclimation to HEP (Fig. 11b). While this list contained a few genes in pathways involved in cell wall biogenesis and lipid metabolism, genes associated with other pathways, such as amino acid anabolism and catabolism, nucleotide metabolism, the Krebs cycle, N-, S- and C1-metabolism were noticeably absent from it. In contrast, genes involved in sucrose and starch metabolism (bam3, pwd & sus1), glycolysis and the OPP cycle (pfk7 & g6pd2) were present in this common list and were all up-regulated by HEP.
Consequently, we assessed the gene lists associated with acclimation to HEP with respect to known transcription factors. Within this list, 79 genes (11 %) were differentially regulated transcription factors (31 up- and 48 down-regulated) from various families (Table 2). Apart from 15 putative and unclassified transcription factors, the families with the most altered transcripts were the “Basic Helix–Loop–Helix family” (8), “C2H2 zinc finger family” (7), “MYB-related transcription factor family” (5), “MYB domain transcription factor family” (4), “Heat-shock transcription factor family” (4), “Homeobox transcription factor family” (4), “C2C2(Zn) CO-like, Constans-like zinc finger family” (3) and the “Pseudo ARR transcription factor family” (3). A comparison of this list with the list of 224 genes common to both stress and acclimation to HEP indicated a common pool of 28 differentially expressed transcription factors: 17 down- and 11 up-regulated (Table 3). While “MYB-related transcription factor family” had already 4 altered transcripts 1 h into the HEP stress, none of the actual “MYB domain transcription factor family” transcripts were activated or repressed during this early stage. The only two other notable families with >2 members altered were “C2C2(Zn) CO-like, Constans-like zinc finger family” (3) and “Pseudo ARR transcription factor family” (3).
Discussion
Our recent model (Kurepin et al. 2013) predicted that the redox state of the PQ pool would represent the major sensor/regulator within the PETC involved in the regulation of genes in response to HEP stress. This was assessed by examining changes in gene expression in the presence of either DCMU or DBMIB (Kurepin et al. 2013; Petrillo et al. 2014). The most striking result of this investigation was the huge impact of both HL stress and LT stress on overall gene expression (Figs. 3, 4) and the large overlap of genes that are regulated in the same way, by either factor. Based on the present experimental design and our previous research on the role of excitation pressure on cold acclimation and photoacclimation in green algae (Maxwell et al. 1994, 1995; Wilson and Hüner 2000, 2003), cyanobacteria (Miskiewicz et al. 2000; 2002) and winter cereals (Gray et al. 1997; Hüner et al. 1998; Dahal et al. 2012), we suggest that these overlapping genes should be considered to be regulated by HEP stress rather than on either HL or LT per se. However, our results show clearly that, in spite of an almost equally large number of PQH2-regulated genes, there is only a 7 % overlap between the gene complement regulated by PQH2 and the total gene complement regulated by HEP stress. One explanation for this apparent incongruence might be the differential degree of reduction applied through our experimental design. While exposure to either HL and LT stress effectively cause the reduction of the PETC, they only close ca. 60 % of all PSII reaction centres (1 − qP ≈ 0.6), whereas the chemical inhibitors, DCMU and DBMIB, cause complete closure of PSII reaction centres (1 − qP = 1.0; Fig. 2a). Thus, the chemical inhibitors presumably create a more severe, and therefore, a slightly different stress condition than either LT or HL stress alone. Alternatively, exposure to either HL or LT stress induces myriad interconnected, signal transduction pathways (Penfield 2008; Yamaguchi-Shinozaki and Shinozaki 2006; Medina et al. 2011; Dietz 2015) that might override signalling from the chloroplast PQH2 pool. In comparison with the PQH2 pool, it is interesting to note how few genes appear to be regulated by PSII(Q −A ). This may be partially explained by the fact that the life time of redox state of the PQH2 pool (ms) is much longer than that of PSII(Q −A ) (100 μs) (Ke 2001). Consequently, on the time scale of transcription and signal transduction, the contributions of the redox state of QA may be inseparable from those of the PQH2 pool. Nevertheless, in contrast to the report of Piippo et al. (2006), our results confirm that the redox state of the PQ pool makes a significant contribution, and therefore, is an important component of redox signalling in Arabidopsis.
Although previous experiments have reported that lhcb1 and rbcS respond to photosynthetic redox signalling (Piippo et al. 2006; Kindgren et al. 2012; Ruckle et al. 2012), our experimental design for the induction of short-term stress did not result in a differential accumulation of either one of these transcripts, even though many other light harvesting complex transcripts of both PSII and PSI were affected (HEP: lhca6, lhcb4, lhcb6 are down-regulated; PQH2: lhca1, lhca3, lhca5, lhca6, lhcb2.1, lhcb4, lhcb2.3; are up-regulated; PSII: no lhc’s were affected). This may be due to the fact that we used a comparatively short exposure time (1 h) of the plants to the respective stress condition, in order to detect potentially transient, primary transcript targets of redox signalling. This suggests that in Arabidopsis, both rbcS and lhcb1 are not primary targets of redox regulation within the first hour of a shift in temperature or irradiance. Our results for changes in transcript abundance under a 1-h stress condition are consistent with published functional and biochemical data which indicate that, in the time frame of the short-term exposure to temperature or light stress, cold-tolerant plants such as rye, wheat, Arabidopsis and Brassica exhibit minimal changes in total Chl content, Chl a/b ratio, Lhcb1 and Rubisco contents (Hüner et al. 1984; Pocock et al. 2001; Savitch et al. 2002; Rosso et al. 2006; Dahal et al. 2012).
We conclude that short-term redox sensing/signalling through chloroplast excitation pressure is part of a complex network of sensors and signal transducers that are integrated and partially redundant (Yamaguchi-Shinozaki and Shinozaki 2006; Penfield 2008; Medina et al. 2011; Koscy et al. 2013; Kurepin et al. 2013; Holmstrom and Finkel 2014; Dietz 2015). However, there appear to be at least three sensors within the PETC that affect distinct target genes. We suggest that both PQH2 and PSII (Q −A ) are important components of this sensing/signalling machinery in contrast to the conclusions of Piippo et al. (2006). However, together these two sensors regulate only up to 11.7 % (PSII: 3.1 %; PQH2: 8.6 %) of the entire Arabidopsis genome which indicates that there must be additional major player(s) in chloroplast redox sensing/signalling. We suggest that a prime candidate that can fulfil the role of a major redox sensing/signalling component within the chloroplast is PSI. Thus, we predict that the remaining 89 % of the HEP- induced gene regulation is probably regulated by PSI and/or the myriad acceptor-side redox components of PSI (Dietz 2008; Brautigam et al. 2009). Since there are no specific inhibitors of PSI, the contribution of PSI to sensing/signalling in response to short-term HEP was difficult to assess in our experimental design.
Khandelwal et al. (2008) examined the regulation of redox homeostasis in Arabidopsis thaliana by comparing exposure of plants to either short-term, photoinhibitory HL conditions or to the inhibition of PSII by DCMU. They concluded that 20 % of the Arabidopsis transcriptome is regulated by HL, whereas 8 % is regulated by DCMU, that is, regulated by PSII(Q −A ). Although we also report that approximately 20 % of the Arabidopsis transcriptome is regulated by HL, our results show that PSII(Q −A ) regulates only 3.1 % of the Arabidopsis transcriptome. We suggest that Khandelwal et al. (2008) overestimated the contribution made by PSII to redox sensing/signalling due to the fact that they did not correct their PSII contribution to redox signalling by correcting for the contribution made by the PQ pool since these authors did not complement the effects of inhibition of the PETC by DCMU with that of DBMIB. Both inhibitors must be used to quantify redox signalling by the PETC accurately (Escoubas et al. 1995; Kurepin et al. 2013; Petrillo et al. 2014).
In contrast to short-term stress, acclimation typically requires growth and development under the stress regime which results in system-wide reprogramming of plant development and metabolism through modulation of complex interacting signal transduction pathways (Gray et al. 1997; Gray and Heath 2005; Penfield 2008; Wingler 2015; Dietz 2015). We hypothesized that the family of genes whose expression is modulated by short-term stress to HEP are distinct and not associated with changes in gene expression associated with long-term growth and acclimation to HEP. However, we found that about 10 % of all genes regulated by short-term HEP stress were still captured in the gene expression profile associated with long-term acclimation to HEP. This indicates that a significant overlap exits between gene expression regulated by short-term stress versus long-term acclimation to HEP. The AP2/EREPB transcription factors represent a large gene family implicated not only in abiotic and biotic stress responses but they also govern plant growth and development (Licausi et al. 2013). Vogel et al. (2014) convincingly showed that several AP2/EREPBs respond extremely rapidly within 10 min upon a shift of low-light grown Arabidopsis to HL. We note that AP2/EREPBs were modulated both by short-term (1 h) HEP stress as well as long-term acclimation to HEP (Tables 2, 3). We suggest that this is consistent with the fact that AP2/EREPBs are known to be important not only in regulating short-term stress response (Vogel et al. 2014) but also the regulation of plant growth and development which is required for long-term plant acclimation and the establishment of photostasis. From the onset of a short-term stress event, plants integrate information generated by the modulation of these interacting signal transduction pathways over time to establish a new, homeostatic acclimated state. Acclimation is a stochastic response which involves a complex network(s) of genes over developmental time induced by an initial stress event. Thus, our global transcriptome analyses are consistent with the thesis that plant acclimation to HEP stress is a time-nested phenomenon (Falkowski and Chen 2003; Brautigam et al. 2009; Dietz 2015).
However, the global transcriptome response associated with HEP stress differed significantly from that associated with acclimation to HEP. First, the major cellular responses associated with HEP stress were associated with ROS signalling (27 %) followed by genes involved in the regulation of the cell cycle (18 %), cell division (14 %) and abiotic stress (13 %) (Fig. 6). In contrast, the global transcriptome response to acclimation to HEP resulted in the modulation of genes primarily associated with cell cycle (8 %) and developmental processes (7 %) rather than ROS signalling (4 %) and abiotic stress (2 %) (Supplemental Fig. S8). The greater modulation of various transcripts involved in ROS signalling in response to short-term HEP stress compared to long-term acclimation to HEP is corroborating evidence that sudden exposure to HEP results in a significant perturbation of the cellular redox poise, which can eventually be re-adjusted and dampened through acclimation to the respective growth condition. Second, HEP stress appeared to result in the regulation of 2489 genes (Fig. 1) which generally reflected a down-regulation of most of the major metabolic pathways (Fig. 5a), whereas acclimation to HEP resulted in the modulation in 70 % fewer genes (735) than the global transcriptome response to HEP stress (Fig. 10). We interpret these differences in the quality and quantity of the global transcriptome response for stress and acclimation to HEP to reflect the fact that the short-term stress causes a significant negative, perturbation of the homeostatic state, whereas long-term acclimation to the same stress reflects the establishment of a new, homeostatic state, that is, the establishment of photostasis. These results are consistent with the functional results illustrated for growth (Fig. 1) and the light response curves for excitation pressure (Fig. 7b, Supplemental Fig. S5) as well as previously published physiological and biochemical responses to stress versus acclimation (Krol et al. 1984; Hüner et al. 1993, 1998, 2003; Savitch et al. 2001; Gray and Heath 2005; Ensminger et al. 2006; Hüner et al. 2012). Furthermore, a detailed nontargeted, metabolomic analysis of cold-stressed versus cold-acclimated Arabidopsis indicated that plants shifted to LT exhibit dynamic changes in their metabolomes, whereas plants grown and developed at LT exhibit a distinct and more stable complement of metabolites than cold-stressed plants (Gray and Heath 2005), which is consistent with our data for the global modulation of the Arabidopsis transcriptome in response to stress and acclimation to either LT or HL.
One objective of this study was to test our proposed model (Kurepin et al. 2013) linking chloroplast redox regulation to the dwarf phenotype associated with growth at either HL or LT (Gray et al. 1997; Dahal et al. 2012, 2014). Consistent with this proposed model, we observed that cbf3, cor15A and cor15b, genes which had previously been reported to be regulated primarily by LT (Chinnusamy et al. 2007; Penfield 2008; Yamaguchi-Shinozaki and Shinozaki 2006; Thomashow 2010; Medina et al. 2011) are also regulated by short-term HEP stress (Fig. 3). These results are consistent with the previous reports that WCS19, a cold-regulated gene in winter wheat, is also regulated by excitation pressure rather than by LT as previously assumed (Gray et al. 1997; Ndong et al. 2001). As expected, the list of genes regulated by LT stress included cbf1-4. In contrast, only cfb3 of the cbf family was detected in the list of genes regulated under short-term HEP. Furthermore, we show for the first time that PQH2 is the chloroplast redox sensor/regulator that governs the expression of cbf3. Concomitantly, PQH2 up-regulates gun5 and stn7, genes involved in plastid retrograde signalling (Koussevitzky et al. 2007; Pesaresi et al. 2011). The up-regulation of cbf3, gun5 and stn7 by PQH2 is coupled to the up-regulation by HEP of GA2ox, an important enzyme in GA catabolism resulting in a shift in the biosynthesis of growth- active GAs to growth- inactive GAs (Hedden 2003; Kurepin et al. 2013). Thus, these results are consistent with our model linking chloroplast redox signalling to CBF expression and GA biosynthesis in Arabidopsis. Our global transcriptome analyses provide further evidence that the dwarf phenotype associated with cold acclimation is regulated by a complex signal transduction pathway which involves chloroplast redox status (Gray et al. 1997; Kurepin et al. 2013; Hüner et al. 2014). Although plant responses to LT stress and cold acclimation have been thoroughly reviewed (Levitt 1980; Steponkus 1984; Guy 1990; Guy et al. 2006; Yamaguchi-Shinozaki and Shinozaki 2006; Penfield 2008; Patel and Franklin 2009; Thomashow 2010; Medina et al. 2011; Wingler 2015), these reviews fail to acknowledge the important role that the chloroplast plays in redox sensing/signalling to establish the cold-acclimated state.
Photosynthetic acclimation to chloroplast redox imbalance results in increased resistance to photoinhibition in overwintering annuals which is accounted for by an enhanced photosynthetic capacity (Hüner et al. 1993, 1998; Ensminger et al. 2006; Hüner et al. 2013). The apparent up-regulation of genes associated with starch/sucrose metabolism (bam3, pwd, and sus1), glycolysis and the OPP cycle (pfk7 and g6pdh) upon establishment of photostasis after acclimation to HEP (Fig. 11) is consistent with the reported enhanced sink capacity required to maintain a balance in cellular energy budget during cold acclimation in Arabidopsis (Stitt and Hurry 2002; Strand et al. 2003) as well as winter rye and wheat (Gray et al. 1996; Dahal et al. 2012, 2014). This is consistent with the data in Fig. 7b and Supplemental Fig. S5, which indicate that plants acclimated to HEP exhibit significant resistance to closure of PSII reaction centres compared to control plants. The ability to re-establish photostasis in response to an initial, stress-induced chloroplast redox imbalance is reflected in a 70 % decrease in the perturbation of global transcript abundance during long-term growth and development under the stress condition compared to the energy imbalance induced by the short-term (1 h) shift (Fig. 10). Failure to acclimate and compensate for such an energy imbalance has dire consequences since it affects plant survival and fitness (Kulheim et al. 2002).
Excitation pressure is defined as the relative redox state of QA, the first stable quinone electron acceptor in the PSII reaction centre (Dietz et al. 1985; Hüner et al. 2003, 2013). However, based on our global transcriptome analyses, Q −A contributes minimally to the overall chloroplast redox signalling process (Fig. 4). Thus, to suggest that photosynthetic performance and phenotypic plasticity associated with chloroplast redox signalling is governed by excitation pressure per se is misleading. Although plants do respond to environmental modulation of chloroplast redox imbalance the extent of which can be estimated by excitation pressure measured as 1 − qP, this reflects the overall reduction state of the entire PETC, not just the redox state of QA (Hüner et al. 2003, 2012; Ensminger et al. 2006; Rosso et al. 2009). Thus, the term excessive excitation energy (EEE), first suggested by Karpinski et al. (1999), is probably a more appropriate term to describe the driving force for environmental modulation of chloroplast redox status rather than excitation pressure per se.
How are the signals generated by chloroplast redox imbalance transmitted over long distances between the leaf chloroplast and meristematic tissue to affect plant growth habit during long-term acclimation? Karpinski et al. (1999) were the first to report that the signal generated by chloroplast redox imbalance measured as excitation pressure (1 − qP) in an Arabidopsis leaf stressed by HL can be transmitted systemically to nonstressed leaves of the same plant to induce a HL-acclimation response. Such a response has been called systemic acquired acclimation (SAA) with ROS playing a major role in signalling associated with SAA (Karpinski et al. 1999; Baxter et al. 2014). Our global transcriptome analyses indicate that modulation of ROS processes dominated the short-term stress response to HEP (Fig. 6), whereas this ROS dependence on gene regulation appeared to have been dampened due to long-term acclimation to HEP (Supplemental Fig. S8). We suggest that this is consistent with the conversion of a physiological stressed state, initially characterized by chloroplast redox imbalance quantified by HEP measured as 1 − qP (Fig. 2), to the establishment of photostasis upon growth and development under the stress condition (Fig. 7B, Supplemental Fig. S5). ROS signalling is a complex cellular communication process (Apel and Hirt 2004; Miller et al. 2008; Galvez-Valdivieso and Mullineaux 2010; Kim and Apel 2013) that is spatially propagated systemically throughout the plant as a ‘ROS wave’ to affect gene expression to short-term stress as well as long-term acclimation to abiotic stress (Mittler et al. 2004, 2011; Baxter et al. 2014). Our global transcriptome data are consistent with this suggestion. Consequently, we suggest that chloroplast redox imbalance is one important component involved in the regulation of photosynthetic performance as well as plant phenotype through a combination of intracellular retrograde control of CBF expression combined with long distance, systemic pathways involved in ROS and GA signalling. Thus, the chloroplast exhibits a dual function: not only is it the primary energy transformer but it is also a primary redox sensor modulated by environmentally induced changes in EEE (Anderson et al. 1995; Hüner et al. 1998, 2012, 2014; Pfannschmidt 2003; Wilson et al. 2006; Murchie et al. 2009; Dietz 2015). However, further research is required to confirm the proposed mechanistic links between changes in EEE, CBF expression, GA biosynthesis and phenotypic plasticity through long-distance intercellular ROS signal transduction.
Abbreviations
- Chl:
-
Chlorophyll
- Cyt b6/f :
-
Cytochrome b6/f
- DBMIB:
-
2,5-Dibromo-3-methyl-6-isopropyl-p-benzoquinone
- DCMU:
-
3-(3,4-Dichlorophenyl)-1,1-dimethylurea
- Fo :
-
Minimum yield of chlorophyll fluorescence in open PSII centres
- F v/F m :
-
Maximum photochemical efficiency of PSII in the dark-adapted state
- GA:
-
Gibberellic acid
- LHCII a/b :
-
Light-harvesting chlorophyll a/b-protein complex of PSII
- P700:
-
Reaction centre chlorophyll of PSI
- PQ:
-
Plastoquinone
- PSI:
-
Photosystem I
- PSII:
-
Photosystem II
- qN:
-
Non-photochemical quenching
- qP:
-
Photochemical quenching parameter
- QA :
-
Primary electron-accepting quinone in PSII
- Q B :
-
Secondary electron-accepting quinone in PSII
- ROS:
-
Reactive oxygen species
References
Allen J (1993) Control of gene expression by redox potential and requirement for chloroplast and mitochondrial genomes. J Theor Biol 165:609–631
Anderson J (1986) Photoregulation of the composition, function and structure of thylakoid membranes. Annu Rev Plant Physiol 37:93–136
Anderson J, Chow WS, Park Y-I (1995) The grand design of photosynthesis: acclimation of the photosynthetic apparatus to environmental cues. Photosynth Res 46:129–139
Apel K, Hirt H (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55:373–399
Aro E-M, Virgin I, Andersson B (1993) Photoinhibition of photosystem II. Inactivation, protein damage and turnover. Biochim Biophys Acta 1143:113–134
Asada K, Heber U, Schreiber U (1993) Electron flow to the intersystem chain from stromal and cyclic electron flow in maize chloroplasts as detected in intact leaves by monitoring redox change of P700 and chlorophyll fluorescence. Plant Cell Physiol 34:39–50
Baker NR (1991) A possible role for photosystem II in environmental perturbations of photosynthesis. Physiol Plant 81:563–570
Baker NR (2008) Chlorophyll fluorescence: a probe of photosynthesis in vivo. Annu Rev Plant Biol 59:89–113
Baxter A, Mittler R, Suzuki N (2014) ROS as key players in plant stress signalling. J Exp Bot 65:1229–1240
Brautigam K, Dietzel L, Kleine T, Stroher E, Wormuth D, Dietz K-J, Radke D, Wirtz M, Hell R, Dormann P, Nunes-Nesi A, Schauer N, Fernie AR, Oliver SN, Geigenberger P, Leister D, Pfannschmidt T (2009) Dynamic plastid redox signals integrate gene expression and metabolism to induce distinct metabolic states in photosynthetic acclimation in Arabidopsis. Plant Cell 21:2715–2732
Chi W, Sun X, Zhang L (2013) Intracellular signaling from plastid to nucleus. Annu Rev Plant Biol 64:559–582
Chinnusamy V, Zhu J, Zhu J-K (2007) Cold stress regulation of gene expression in plants. Trends Plant Sci 12:444–451
Chory J (1997) Light modulation of vegetative development. Plant Cell 9:1225–1234
Claeys H, Van Landeghem S, Dubois M, Maleux K, Inze D (2014) What Is Stress? Dose-response effects in commonly used in vitro stress assays. Plant Physiol 165:519–527
Dahal K, Gadapati W, Savitch L, Singh J, Hüner NPA (2012) Cold acclimation and BnCBF17-over-expression enhance photosynthetic performance and energy conversion efficiency during long-term growth of Brassica napus under elevated CO2 conditions. Planta 236:1639–1652
Dahal K, Knowles VL, Plaxton WC, Hüner NPA (2014) Enhancement of photosynthetic performance, water use efficiency and grain yield during long-term growth under elevated CO2 in wheat and rye is growth temperature and cultivar dependent. Environ Exp Bot 106:207–220
Demmig-Adams B, Adams WW III (1992) Photoprotection and other responses of plants to high light stress. Annu Rev Plant Physiol Plant Mol Biol 43:599–626
Derks A, Shaven K, Bruce D (2015) Diverse mechanisms for photoprotection in photosynthesis. Dynamic regulation of photosystem II excitation in response to rapid environmental change. Biochim Biophys Acta 1847:468–485
Dietz K-J (2003) Plant peroxiredoxins. Annu Rev Plant Biol 54:93–107
Dietz K-J (2008) Redox signal integration: from stimulus to networks and genes. Physiol Plant 133:459–468
Dietz K-J (2015) Efficient high light acclimation involves rapid processes at multiple mechanistic levels. J Exp Bot 66:2401–2414
Dietz K-J, Pfannschmidt T (2011) Novel regulators in photosynthetic redox control of plant metabolism and gene expression. Plant Physiol 155:1477–1485
Dietz K-J, Schreiber U, Heber U (1985) The relationship between the redox state of QA and photosynthesis in leaves at various carbon-dioxide, oxygen and light regimes. Planta 166:219–226
Durnford DG, Falkowski PG (1997) Chloroplast redox regulation of nuclear gene transcription during photoacclimation. Photosynth Res 53:229–241
Ensminger I, Busch FA, Hüner NPA (2006) Photostasis and cold acclimation: sensing low temperature through photosynthesis. Physiol Plant 126:28–44
Escoubas J-M, Lomas M, LaRoche J, Falkowski PG (1995) Light intensity regulates cab gene transcription via the redox state of the plastoquinone pool in the green alga, Dunaliella tertiolecta. Proc Natl Acad Sci USA 92:10237–10241
Falkowski PG, Chen Y-B (2003) Photoacclimation of light harvesting systems in eukaryotic algae. In: Green BR, Parson WW (eds) Advances in photosynthesis and respiration. Light harvesting systems in photosynthesis, vol 13. Kluwer, Dordrecht, pp 423–427
Falkowski PG, LaRoche J (1991) Acclimation to spectral irradiance in algae. J Phycol 27:8–14
Feng S, Martinez C, Gusmaroli G, Wang Y, Zhou J, Wang F, Chen L, Yu L, Iglesias-Pedraz JM, Kircher S, Schafer E, Fu X, Fan L-M, Deng XW (2008) Coordinated regulation of Arabidopsis thaliana development by light and gibberellins. Nature 451:475–479
Fernandez AP, Strand A (2008) Retrograde signaling and plant stress: plastid signals initiate cellular stress responses. Curr Opin Plant Biol 11:509–513
Foyer CH, Noctor G (2009) Redox regulation in photosynthetic organisms: signaling, acclimation, and practical implications. Antiox Redox Signal 11:861–905
Foyer CH, Neukermans J, Queval G, Noctor G, Harbinson J (2012) Photosynthetic control of electron transport and the regulation of gene expression. J Exp Bot 63:1637–1661
Fujita Y, Murakami A, Aizawa K, Ohki K (1994) Short-term and long-term adaptation of the photosynthetic apparatus: homeostatic properties of thylakoids. In: Bryant DA (ed) The Molecular Biology of Cyanobacteria. Advances in Photosynthesis, vol 1. Kluwer, Dordrecht, pp 667–692
Galvez-Valdivieso G, Mullineaux PM (2010) The role of reactive oxygen species in signalling from chloroplasts to the nucleus. Physiol Plant 138:430–439
Gilmour SJ, Sebolt AM, Salazar MP, Everard JD, Thomashow MF (2000) Overexpression of the Arabidopsis CBF3 transcriptional activator mimics multiple biochemical changes associated with cold acclimation. Plant Physiol 124:1854–1865
Gray GR, Heath D (2005) A global reorganization of the metabolome in Arabidopsis during cold acclimation is revealed by metabolic fingerprinting. Physiol Plant 124:236–248
Gray GR, Savitch LV, Ivanov AG, Hüner NPA (1996) Photosystem II excitation pressure and development of resistance to photoinhibition. II. Adjustment of photosynthetic capacity in winter wheat and winter rye. Plant Physiol 110:61–71
Gray GR, Chauvin L-P, Sarhan F, Hüner NPA (1997) Cold acclimation and freezing tolerance. A complex interaction of light and temperature. Plant Physiol 114:467–474
Guy CL (1990) Cold acclimation and freezing tolerance: role of protein metabolism. Annu Rev Plant Physiol Plant Mol Biol 41:187–223
Guy C, Porat R, Hurry V (2006) Plant cold and abiotic stress gets hot. Physiol Plant 126:1–4
Hedden P (2003) The genes of the green revolution. Trends Genet 19:5–9
Holmstrom KM, Finkel T (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15:411–421
Hopkins WG, Hüner NPA (2009) Introduction to plant physiology, 4th edn. Wiley, New York
Horton P, Ruban AV, Walters RG (1996) Regulation of light harvesting in green plants. Annu Rev Plant Physiol Plant Mol Biol 47:655–684
Horton P, Johnson MP, Perez-Bueno ML, Kiss AZ, Ruban AV (2008) Photosynthetic acclimation: does the dynamic structure and macro-organisation of photosystem II in higher plant grana membranes regulate light harvesting states? FEBS J 275:1069–1079
Hüner NPA, Grodzinski B (2011) Photosynthesis and photoautotrophy. In Moo-Young M (ed) Comprehensive Biotechnology, vol 1, 2nd edn. Elsevier, Amsterdam, pp 315-322
Hüner NPA, Elfman B, Krol M, MacIntosh A (1984) Growth and development at cold hardening temperatures. Chloroplast ultrastructure, pigment content and composition. Can J Bot 62:53–60
Hüner NPA, Öquist G, Hurry VM, Krol M, Falk S, Griffith M (1993) Photosynthesis, photoinhibition and low temperature acclimation in cold tolerant plants. Photosynth Res 37:19–39
Hüner NPA, Öquist G, Sarhan F (1998) Energy balance and acclimation to light and cold. Trends Plant Sci 3:224–230
Hüner NPA, Öquist G, Melis A (2003) Photostasis in plants, green algae and cyanobacteria: the role of light harvesting antenna complexes. In: Green BR, Parson WW (eds) Advances in photosynthesis and respiration. Light harvesting antennas in photosynthesis, vol 13. Kluwer, Dordrecht, pp 401–421
Hüner NPA, Dahal K, Hollis L, Bode R, Rosso D, Krol M, Ivanov AG (2012) Chloroplast redox imbalance governs phenotypic plasticity: the “grand design of photosynthesis” revisited. Front Plant Physiol 3:Article 255
Hüner NPA, Bode R, Dahal K, Busch FA, Possmayer M, Szyszka B, Rosso D, Ensminger I, Krol M, Ivanov AG, Maxwell DP (2013) Shedding some light on cold acclimation, cold adaptation, and phenotypic plasticity. Botany 91:127–136
Hüner NPA, Dahal K, Kurepin LV, Savitch L, Singh J, Ivanov AG, Kane K, Sarhan F (2014) Potential for increased photosynthetic performance and crop productivity in response to climate change: role of CBFs and gibberellic acid. Front Chem 2:Artcle 18
Hussain A, Peng J (2003) Della proteins and GA signalling in Arabidopsis. J Plant Growth Regul 22:134–140
Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31:e15. doi:10.1093/nar/gng015
Ivanov AG, Morgan RM, Gray GR, Velitchkova MY, Hüner NPA (1998) Temperature/light dependent development of selective resistance to photoinhibition of photosystem I. FEBS Lett 430:288–292
Ivanov AG, Hendrickson L, Krol M, Selstam E, Öquist G, Hurry V, Hüner NPA (2006) Digalactosyl-diacylglycerol deficiency impairs the capacity for photosynthetic intersystem electron transport and state transitions in Arabidopsis thaliana due to photosystem I acceptor-side limitations. Plant Cell Physiol 47:1146–1157
Ivanov AG, Morgan-Kiss RM, Krol M, Allakhverdiev SI, Zanev Yu, Sane PV, Hüner NPA (2015) Photoinhibition of photosystem I in a pea mutant with altered LHCII organization. J Photochem Photobiol B 152:335–346
Jaglo-Ottosen KR, Gilmour SJ, Zarka DG, Schabenberger O, Thomashow MF (1998) Arabidopsis CBF1 overexpression induces COR genes and enhances freezing tolerance. Science 280:104–106
Jahns P, Holzwarth AJ (2012) The role of the xanthophyll cycle and of lutein in photoprotection of photosystem II. Biochim Biophys Acta 1817:182–193
Karpinski S, Reynolds H, Karpinska B, Wingsle G, Creissen G, Mullineaux P (1999) Systemic signaling and acclimation in response to excess excitation energy in Arabidopsis. Science 284:654–657
Ke B (2001) Photosynthesis. Photobiochemistry and photobiophysics. In: Advances in photosynthesis, vol 10. Kluwer, Dordrecht
Khandelwal A, Elvitigala T, Ghosh B, Quatrano RS (2008) Arabidopsis transcriptome reveals control circuits regulating redox homeostasis and the role of an AP2 transcription factor. Plant Physiol 148:2050–2058
Kim C, Apel K (2013) Singlet oxygen-mediated signaling in plants: moving from flu to wild type reveals an increasing complexity. Photosynth Res 116:455–464
Kindgren P, Noren L, Barajas Lopez J-D, Shaikhali J, Strand Å (2012) Interplay between heat shock protein 90 and HY5 controls PhANG expression in response to the GUN5 plastid signal. Mol Plant 5:901–913
Koscy G, Tari I, Vankova R, Zechmann B, Gulyas Z, Poor P, Galiba G (2013) Redox control of plant growth and development. Plant Sci 211:77–91
Koussevitzky S, Nott A, Mockler TC, Hong F, Sachetto-Martins G, Surpin M, Lim J, Mittler R, Chory J (2007) Signals from chloroplasts converge to regulate nuclear gene expression. Science 316:715–719
Kramer DM, Johnson G, Kiirats O, Edwards GE (2004) New fluorescence parameters for the determination of QA redox state and excitation energy fluxes. Photosynth Res 79:209–218
Krause GH (1988) Photoinhibition of photosynthesis. An evaluation of damaging and protective mechanisms. Physiol Plant 74:566–574
Krol M, Griffith M, Hüner NPA (1984) An appropriate physiological control for environmental temperature studies: comparative growth kinetics for winter rye. Can J Bot 62:1062–1068
Kulheim C, Agren J, Jansson S (2002) Rapid regulation of light harvesting and plant fitness in the field. Science 297:91–93
Kurepin L, Dahal K, Savitch L, Singh J, Bode R, Ivanov AG, Hurry V, Hüner NPA (2013) Role of CBFs as integrators of chloroplast redox, phytochrome and plant hormone signaling during cold acclimation. Int J Mol Sci 14:12729–12763
Leonardos ED, Savitch LV, Hüner NPA, Öquist G, Grodzinski B (2003) Daily photosynthetic and C-export patterns in winter wheat leaves during cold stress and acclimation. Physiol Plant 117:521–531
Levitt J (1980) Responses of plants to environmental stresses. In: Chilling, freezing, and high temperature stresses, vol 1. Academic Press, New York
Li Z, Wakao S, Fischer JBB, Niyogi KK (2009) Sensing and responding to excess light. Annu Rev Plant Biol 60:239–260
Licausi F, Ohme-Takagi M, Perata P (2013) APETALA2/Ethylene Responsive Factor (AP2/ERF) transcription factors: mediators of stress responses and developmental programs. New Phytol 199:639–649
Long SP, Humphries S, Falkowski PG (1994) Photoinhibtion of photosynthesis in nature. Annu Rev Plant Physiol Plant Mol Biol 45:633–662
Machalek KM, Davison IR, Falkowski PG (1996) Thermal acclimation and photoacclimation of photosynthesis in the brown alga Laminaria saccharina. Plant Cell Environ 19:1005–1016
Maxwell DP, Falk S, Trick CG, Hüner NPA (1994) Growth at low temperature mimics high-light acclimation in Chlorella vulgaris. Plant Physiol 105:535–543
Maxwell DP, Laudenbach DE, Hüner NPA (1995) Redox regulation of light-harvesting complex II and cab mRNA abundance in Dunaliella salina. Plant Physiol 109:787–795
Medina J, Catalá R, Salinas J (2011) The CBFs: three Arabidopsis transcription factors to cold acclimate. Plant Sci 180:3–11
Melis A (1991) Dynamics of photosynthetic membrane composition and function. Biochim Biophys Acta 1058:87–106
Melis A (1998) Photostasis in Plants. In: Williams G, Thistle T (eds) Photostasis and related Phenomena. Plenum Press, New York, pp 207–220
Melis A (1999) Photosystem-II damage and repair cycle in chloroplasts: what modulates the rate of photodamage in vivo ? Trends Plant Sci 4:130–135
Miller G, Shulaev V, Mittler R (2008) Reactive oxygen signaling and abiotic stress. Physiol Plant 133:481–489
Miskiewicz E, Ivanov AG, Williams JP, Khan MU, Falk S, Hüner NPA (2000) Photosynthetic acclimation of the filamentous cyanobacterium, Plectonema boryanum UTEX 485, to temperature and light. Plant Cell Physiol 41:767–775
Miskiewicz E, Ivanov AG, Hüner NPA (2002) Stoichiometry of the photosynthetic apparatus and phycobilisome structure of the cyanobacterium Plectonema boryanum UTEX 485 are regulated by both light and temperature. Plant Physiol 130:1414–1425
Mittler R (2006) Abiotic stress, the field environment and stress combination. Trends Plant Sci 11:15–19
Mittler R, Vanderauwera S, Gollery M, Van Breusegem F (2004) Reactive oxygen gene network of plants. Trends Plant Sci 9:490–498
Mittler R, Vanderauwera S, Suzuki N, Miller G, Tognetti VB, Vandepoele K, Gollery M, Shulaev V, van Breusegem F (2011) ROS signalling: the new wave? Trends Plant Sci 16:300–308
Miura E, Kato Y, Matsushima R, Albrech V, Laalami S, Sakamoto W (2007) The balance between protein synthesis and degradation in chloroplasts determines leaf variegation in Arabidopsis yellow variegated mutants. Plant Cell 19:1313–1328
Murata N, Allakhverdiev SI, Nishiyama Y (2012) The mechanism of photoinhibition in vivo: re-evaluation of the roles of catalase α-tocopherol, non-photochemical quenching, and electron transport. Biochim Biophys Acta 1817:1127–1133
Murchie EH, Pinto M, Horton P (2009) Agriculture and the new challenges for photosynthesis research. New Phytol 181:532–552
Ndong C, Danyluk J, Hüner NPA, Sarhan F (2001) Survey of gene expression in winter rye during changes in either growth temperature, irradiance or excitation pressure. Plant Mol Biol 45:691–703
Niyogi KK (1999) Photophotoprotection revisited: genetic and molecular approaches. Annu Rev Plant Physiol Plant Mol Biol 50:333–359
Osmond CB (1994) What is photoinhibition? Some insights from comparison of shade and sun plants. In: Bowyer JR, Baker NR (eds) Photoinhibition of photosynthesis—from molecular mechanisms to the field. Bios Scientific Publishers, Oxford UK, pp 1–24
Patel D, Franklin KA (2009) Temperature-regulation of plant architecture. Plant Signal Behav 4:577–579
Penfield S (2008) Temperature perception and signal transduction in plants. New Phytol 179:615–628
Peng J, Harberd NP (1997) Giberellin deficiency and response mutations suppress stem elongation phenotype of phytochrome-deficient mutants of Arabidopsis. Plant Physiol 113:1051–1058
Peng J, Carol P, Richards DE, King KE, Cowling RJ, Murphy GP, Harberd NP (1997) The Arabidopsis GAI gene defines a signalling pathway that negatively regulates gibberellin responses. Genes Dev 11:3194–3205
Peng J, Richards DE, Hartley NM, Murphy GP, Devos KM, Flintham JE, Beales J, Fish LJ, Worland AJ, Pelica F, Sudhakar D, Christou P, Snape JW, Gale MD, Harberd NP (1999) ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 400:256–261
Perez IB, Brown PJ (2014) The role of ROS signalling in cross-tolerance: from model to crop. Front Plant Physiol 5:article 754
Pesaresi P, Pribil M, Wunder T, Leister D (2011) Dynamics of reversible protein phosphorylation in thylakoids of flowering plants: the roles of STN7, STN8 and TAP38. Biochim Biophys Acta 1807:887–896
Petrillo E, Godoy Herz MA, Fuchs A, Reifer D, Fuller J, Yanovsky MJ, Simpson C, Brown JWS, Barta A, Kalyna M, Kornblihtt AR (2014) A chloroplast retrograde signal regulates nuclear alternative splicing. Science 344:427–430
Pfannschmidt T (2003) Chloroplast redox signals: how photosynthesis controls its own genes. Trends Plant Sci 8:33–41
Piippo M, Allahverdiyeva Y, Paakkarinen V, Suoranta U-M, Battchikova N, Aro E-M (2006) Chloroplast-mediated regulation of nuclear genes in Arabidopsis thaliana in the absence of light stress. Physiol Genomics 25:142–152
Pocock T, Hurry V, Savitch LV, Hüner NPA (2001) Susceptibility to low-temperature photoinhibition and the acquisition of freezing tolerance in winter and spring wheat: the role of growth temperature and irradiance. Physiol Plant 113:499–506
Pogson BJ, Woo NS, Förster B, Small ID (2008) Plastid signalling to the nucleus and beyond. Trends Plant Sci 13:602–609
Queval G, Foyer CH (2012) Redox regulation of photosynthetic gene expression. Philos Trans R Soc B 367:3475–3485
Rosso D, Ivanov AG, Fu A, Geisler-Lee J, Hendrickson L, Geisler M, Stewart G, Krol M, Hurry V, Rodermel SR, Maxwell DP, Hüner NPA (2006) IMMUTANS does not act as a stress-induced safety valve in the protection of the photosynthetic apparatus of Arabidopsis during steady-state photosynthesis. Plant Physiol 142:574–585
Rosso D, Bode R, Li W, Krol M, Saccon D, Wang S, Schillaci LA, Rodermel SR, Maxwell DP, Hüner NPA (2009) Photosynthetic redox imbalance governs leaf sectoring in the Arabidopsis thaliana variegation mutants immutans, spotty, var1, and var2. Plant Cell 21:3473–3492
Ruckle ME, Burgoon Lyle D, Lawrence Lauren A, Sinkler CA, Larkin RM (2012) Plastids are major regulators of light signaling in Arabidopsis. Plant Physiol 159:366–390
Savitch LV, Gray GR, Hüner NPA (1997) Feedback-limited photosynthesis and regulation of sucrose-starch accumulation during cold acclimation and low-temperature stress in a spring and winter wheat. Planta 201:18–26
Savitch LV, Barker-Astrom J, Ivanov AG, Hurry V, Oquist G, Huner NPA, Gardestrom P (2001) Cold acclimation of Arabidopsis thaliana results in incomplete recovery of photosynthetic capacity which is associated with an increased reduction of the chloroplast stroma. Planta 217:295–301
Savitch LV, Leonardos ED, Krol M, Jansson S, Grodzinski B, Hüner NPA, Öquist G (2002) Two different strategies for light utilization in photosynthesis in relation to growth and cold acclimation. Plant Cell Environ 25:761–771
Schreiber U, Bilger W, Neubauer C (1994) Chlorophyll fluorescence as a nonintrusive indicator for rapid assessment of in vivo photosynthesis. In: Schulze ED, Caldwell MM (eds) Ecophysiology of photosynthesis. Springer, Berlin, pp 49–70
Steponkus PL (1984) Role of the plasma membrane in freezing injury and cold acclimation. Annu Rev Plant Physiol 35:543–584
Stitt M, Hurry V (2002) A plant for all seasons: alterations in photosynthetic carbon metabolism during cold acclimation in Arabidopsis. Curr Opin Plant Biol 5:199–206
Stoher E, Dietz K-J (2008) The dynamic thiol-disulphide redox proteome of the Arabidopsis thaliana chloroplast as revealed by differential electrophoretic mobility. Physiol Plant 133:566–583
Strand A, Foyer CH, Gustafsson P, Gardeström P, Hurry V (2003) Altering flux through the sucrose biosynthesis pathway in transgenic Arabidopsis thaliana modifies photosynthetic acclimation at low temperatures and the development of freezing tolerance. Plant Cell Environ 26:523–535
Sukenik A, Wyman KD, Bennett J, Falkowski PG (1987) A novel mechanism for regulating the excitation of photosystem II in a green alga. Nature 327:704–707
Terashima I, Noguchi K, Itohnemoto T, Park YM, Kubo A, Tanaka K (1998) The cause of PSI photoinhibition at low temperatures in leaves of Cucumis sativus, a chilling-sensitive plant. Physiol Plant 103:295–303
Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kriuger P, Selbig J, Muller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37:914–939
Thomashow MF (2010) Molecular basis of plant cold acclimation: insights gained from studying the CBF cold response pathway. Plant Physiol 154:571–577
Verhoeven A (2014) Sustained energy dissipation in winter evergreens. New Phytol 201:57–65
Vogel MO, Moore M, König K, Pecher P, Alsharafa K, Lee J, Dietz K-J (2014) Fast retrograde signaling in response to high light involves metabolite export, MITOGEN-ACTIVATED PROTEIN KINASE6, and AP2/ERF transcription factors in Arabidopsis. Plant Cell 26:1151–1165
Wilson KE, Hüner NPA (2000) The role of growth rate, redox-state of the plastoquinone pool and the trans-thylakoid Delta pH in photoacclimation of Chlorella vulgaris to growth irradiance and temperature. Planta 212:93–102
Wilson KE, Krol M, Hüner NPA (2003) Temperature-induced greening of Chlorella vulgaris. The role of the cellular energy balance and zeaxanthin-dependent nonphotochemical quenching. Planta 217:616–627
Wilson KE, Ivanov AG, Öquist G, Grodzinski B, Sarhan F, Hüner NPA (2006) Energy balance, organellar redox status and acclimation to environmental stress. Can J Bot 84:1355–1370
Wingler A (2015) Comparison of signalling interactions determining annual and perennial plant growth in response to low temperature. Front Plant Physiol 5:794–814
Woodson JD, Chory J (2008) Coordination of gene expression between organellar and nuclear genomes. Nat Rev Genet 9:383–395
Yamaguchi-Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57:781–803
Yu F, Fu A, Aluru M, Park S, Xu Y, Liu H, Liu X, Foudree A, Nambogga M, Rodermel S (2007) Variegation mutants and mechanisms of chloroplast biogenesis. Plant Cell Environ 30:350–365
Acknowledgments
We are grateful to David Carter at the London Regional Genomics Center (Robarts Research Institute, London, Ontario, Canada) for processing all the microarray chips and providing the license and training for the use of Partek. This research was supported by grants to NPAH from NSERC, CFI and the CRC programmes.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bode, R., Ivanov, A.G. & Hüner, N.P.A. Global transcriptome analyses provide evidence that chloroplast redox state contributes to intracellular as well as long-distance signalling in response to stress and acclimation in Arabidopsis . Photosynth Res 128, 287–312 (2016). https://doi.org/10.1007/s11120-016-0245-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-016-0245-y