
Abstract
Salinity stress is one of the abiotic factors that greatly affect agriculture by limiting plant growth and yield worldwide. A set of 138 barley accessions from different geographical regions was characterized with the aim to identify metabolite, biochemical, and morphological phenotypes under salinity stress. Salt stress resulted in significant increases in the phytochemicals, including the contents of total phenolic (TPC), total flavonoid (TFC), proline (ProC), and total antioxidant capacity (TAC) except soluble protein (SP). Positive relationships between proline content and the secondary metabolites or antioxidants, including total phenolics and flavonoids, were detected among barley accessions, indicating a critical adaptive strategy against free radicals under salt stress. Genome-wide association study (GWAS) revealed 122 significant quantitative trait nucleotides (QTNs) associated with the measured traits which resulted in the identification of 203 potential candidate genes. Interestingly, the QTN G:A was located inside the candidate gene HORVU.MOREX.r3.3HG0291720 at position 501,703,401 on chromosome 3H. This gene encodes Ca-binding protein and contributes to the signalling pathway that in turn triggers the expression of salt-stress responsive genes. The identified QTNs/ candidate genes provide information useful for the genetic improvement of barley genotypes under salt stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Salinity is one of the abiotic factors that harm agriculture by limiting plant growth and yield all over the world (Thabet et al. 2021a, b). The most fundamental physiological and biochemical mechanisms governing plant growth and yield were substantially hampered by soil salinity (Thabet et al. 2021a). High salt concentration in the form of sodium and chloride ions causes several undesirable effects on plant growth and performance, leading to disruption in the biochemical and molecular processes of the plant cell (Shahid et al. 2020). Na and Cl toxicity induces nutritional disorders by limiting the uptake of some nutrients (K+, Ca2+, P, N) from the soil (Rehman et al. 2019). Salt stress retards seed germination and seedling-related traits through osmotic and ionic imbalance (Tester and Davenport 2003). Salinity enhances reactive oxygen species (ROS) molecules in plant cells which causes toxic effects on lipid peroxidation, membrane deterioration, as well as DNA and protein damage, finally leading to causes oxidative damage. At the vegetative developmental stage, salt stress disturbs plant water relations, reduces cell turgor, and depresses plant biomass, which ultimately leads to osmotic stress (Munns and Tester 2008). This suggests that water uptake is the primary effect of environmental salinity. In general, salinity stress interferes with many physiological and biochemical activities, such as lowering water and nutrients uptake into plant tissues, changing metabolic processes, disturbing photosynthetic machinery, transpiration, and gaseous exchange by reducing the content of chlorophyll and carotenoids, and decreasing stomatal conductance, all of which impede plant growth and development.
Leaf protein content is one of the crucial indicators of the effects of salt stress (Isayenkov and Maathuis 2019). Therefore, plants respond to salinity stress conditions by accumulating small molecular mass proteins, which act as a source of storage nitrogen and also involve in osmotic adjustment (Ashraf and Harris 2004; Singh et al. 1987). On the other hand, it has been reported that a higher accumulation of osmolytes like proline could enhance salinity stress tolerance. Proline can modulate redox potential, thereby facilitating osmotic adjustment, safeguarding cellular membranes, and maintaining enzyme stability during abiotic stress (Raza et al. 2007). The most prevalent secondary metabolites, such as phenolic compounds, act as the most powerful antioxidants for scavenging the free radicals generated in response to different environmental stimuli. Another category of phenolic compounds known for having antioxidant effects is flavonoids (Tohidi et al. 2017). As a result, plants move toward cellular ion homeostasis, free radical scavenging, and stimulation of antioxidant machinery, polyamines biosynthesis, and mineral uptake leading to the maintenance of plant growth and productivity.
Barley (Hordeum vulgare L.), a cereal crop, is among the most well-known cereals in the world that can tolerate abiotic stresses (Thabet and Alqudah 2019). Since barley is one of the most saline-tolerant crops and has a higher tolerance to salt stress than other cereals like wheat, corn, and rice, it is commonly used as a model for studying plant resistance mechanisms (Thabet and Alqudah 2023; Thabet et al. 2021b). Identifying quantitative trait loci (QTLs) controlling salt tolerance is a promising strategy for enhancing salinity tolerance in barley. Thabet et al. (2021b) identified QTLs associated with salt tolerance at seed germination and seedling-related traits in barley. Thabet et al. (2021a) identified several QTLs for agronomic traits such as plant height, spikelet per spike, grains per spike, spike density, and thousand kernel weight under salt stress. Moreover, multiple QTLs were identified for several physiological traits of different plant species under salinity stress. For example, Rivandi et al. (2011) detected a QTL associated with the shoot Na+ exclusion trait that mapped to HvNax4. Shavrukov et al. (2010) identified a QTL mapped to HvNax3, a locus controlling shoot sodium content in the wild barley. Interestingly, Thabet et al. (2021a) identified several QTLs associated with enzymatic and non-enzymatic antioxidant components that cope with salt stress tolerance in barley using a genome-wide association study (GWAS).
GWAS is an effective approach to map QTLs more accurately and pinpoint the genes underlying several biochemical and agronomic parameters with relative ease in many crop species (Alqudah et al. 2020, 2021). In barley, GWASs have focused on complex traits in barley such as drought resistance, salt tolerance, and frost tolerance (Mwando et al. 2020; Thabet and Alqudah 2023; Thabet et al. 2018, 2020). In our recent work, we have explored several biochemical and agronomic traits as potential selection criteria for plant salt tolerance. Here, we measured bio-physiological and agronomic traits in a core set of 138 barley accessions to identify significant QTNs/candidate genes. We hypothesized that the studied components are controlled by multiple genes and would result in multiple QTNs for salt tolerance in barley. Our results shed light on the understanding of the genetic basis of plant architecture based on morpho-physiological attributes, provided QTNs and markers that can be used by breeders, and constructed a theoretical basis for fine mapping and for marker-assist-selected breeding.
Materials and Methods
Germplasm and Genotyping
A core set collection consisting of 138 worldwide spring barley accessions was selected out of 22,621 wild and domesticated barley accessions hosted in the German ex-situ IPK-Gatersleben Gene Bank as highly diverse accessions. The collection is originally from different geographical origins including 54 cultivars and 84 landraces. The core set/panel was split into 63 two-rowed and 75 six-rowed. All the accessions were genotyped using GBS that yielded 19,276 SNPs as reported by Milner et al. (2019). This collection was tested for the population structure that is considered in the GWAS analysis. Principal component analysis was used to check the structure within the population and plotted based on geographical regions, row-type, and biological status. Detailed information on barley accessions is provided in Table S1.
Experimental Setup
A core collection of 138 barley accessions was evaluated under field conditions at Fayoum University (29° 11′ 20.36″ N latitude, 30° 10′ 06.45″ E longitude) from November 2021 to April 2022. Five seeds from each accession were planted in plastic pots (40 cm × 25 cm × 25 cm) that were filled with silty loam soil and each pot was manually irrigated as required. Each accession was grown under two treatments, including control and salinity (200 mM NaCl), and was replicated three times for each treatment using a randomized complete block design (RCBD). At the early reproductive phase (25 days after sowing (DAS)), the plants were thinned into three plants per pot then exposed to both well-watered and salt treatments (200 mM NaCl). In this experiment, we used standard techniques for irrigation, fertilization, and pest and disease management. Salt treatment was conducted up to 50 DAS, after which distilled water irrigation was used until harvest. After 8 weeks of planting, leaf samples from each accession were collected to measure the physiological traits, including proline content (ProC), soluble protein content (SP), total phenolic (TPC), total flavonoid (TFC), and total antioxidant capacity (TAC). Five phenotypic traits such as spike length (SL), spikelet number per spike (SS), grain number per spike (GS), the weight of grains per spike (WGS) in gram (g), and thousand-grain weight (TGW) in gram (g) were measured as described in Thabet et al. (2021a). Additionally, salt tolerance indices (STI) for all traits were calculated according to the following equation: STI = trait value under salinity/trait value under control.
Estimation of Proline and Protein Content
Proline content (ProC) was determined and extracted as described in methods by Bates et al. (1973). Briefly, 400 ml of the mixture (1.25 g ninhydrin, 20 mL phosphoric acid, and 30 mL glacial acetic acid) was added to a 200 mL supernatant aliquot and heated for 1 h at 100 °C. The mixture was then allowed to cool, and 4 mL of toluene was added. At 520 nm, the absorbance was taken and the results were recorded as mmol g−1 DW. Soluble protein (SP) was determined by using a dye-binding method and bovine serum albumin as a standard. Then, the absorbance was measured spectrophotometrically at 595 nm as mg g DW−1.
Determination of Total Antioxidant Capacity (TAC)
The total antioxidant capacity (TAC) was estimated according to Prieto et al. (1999). Briefly, barley extract of 10 µL was mixed with 1 mL of 250 µM DPPH solution and 4 mL distilled water, then the extract was placed in the dark for 30 min. The absorbance was measured at 517 nm. The reference standard was Trolox, and also the results were recorded as Trolox equivalent mg g−1 DW. The inhibition (%) of free radical DPPH was calculated using the equation as follows:
Antioxidant activity (I %) = [(A0 − Ae)/A0] × 100, where A0 is the absorbance of the blank sample and Ae is the absorbance of the tested sample.
Determination of Metabolites
The content of total phenolic (TPC) was estimated and extracted according to Song et al. (2010). Briefly, 100 µL of leaf extract sample was added to 50 µL of 2N Folin–Ciocalteau reagent. After 5 min, 400 µL of 2N Na2CO3 and 1 mL of water were added and incubated for 90 min at room temperature and then removed to a microplate. The absorbance was measured at 740 nm, and the results were expressed as gallic acid (GAE) equivalent µg g−1 of DW.
Total flavonoid content (TFC) was measured and extracted using the method described in Zou et al. (2004). Briefly, 400 µL of barley extracts was mixed with 60 µL of 5% NaNO2, 500 µL of distilled water, and 140 µL of 10% AlCl3. After 10 min, 400 µL of 1 mM NaOH was added, and incubated for 10 min at room temperature, and then removed to a microplate. The absorbance was measured at 500 nm and the results were expressed as rutin (RE) equivalent µg g−1 of DW.
Data Analysis
Two-way ANOVA was determined using R software to compare the mean values of the different traits under both treatments at a P-value < 0.05. A mean value comparison of different traits was made with Student’s t-test. Measured trait relationships under each treatment were calculated using Pearson correlation analysis applying the corrplot function in the R package corrplot (Wei and Simko 2017). The best linear unbiased estimations (BLUEs) for the mean values of the measured traits were calculated by assuming the intercept and genotype effects using the lme4 package (Bates et al. 2015). The broad-sense heritability (H2) was performed according to Schmidt et al. (2019).
Genome-Wide Association Scan (GWAS)
GWAS was conducted using Genomic Association and Prediction Integrated Tool (GAPIT) in R software on a diverse set of 138 barley accessions (Lipka et al. 2012). The accessions were genotyped by sequencing (GBS) that yielded 19,276 SNPs. First, the GWAS analysis was computed using a mixed linear model (MLM) that took into account the variance–covariance kinship matrix and principal component analysis (PCA) and was conducted by incorporating the phenotypic and genotypic dataset. The kinship matrix was calculated using the VanRaden method (VanRaden 2008) to determine relative kinship among the sampled individuals. Both PCA and kinship matrix were used for population correction and stratification. We also applied a GAPIT model known as “fixed and random model circulating probability unification” (FarmCPU) to our data analysis. This was applied by considering the incorporation of multiple markers simultaneously as a covariate in a fixed-effects model and optimization on the associated covariate markers separately in a random effects model, which enabled us to avoid any false-negatives and to control the false-positive associations by preventing model over-fitting (Liu et al. 2016). Thus, FarmCPU is a powerful tool with less false-positives than MLM. The selection of an appropriate model and thresholds is important steps in identifying markers that are truly associated with specific traits and that might be located within or very close to genes that control the trait variation while also controlling both false-positive and false-negative associations. To determine which of the tested models best fitted the data, we plotted quantile–quantile (Q-Q) plots based on the observed and expected − log10(P) values. Based on the GWAS output of three models (GLM, MLM, and FarmCPU), the number of significant associations, and the resulting Q-Q plots, we then selected the FarmCPU model. A threshold P-value 0.001 equal to − log10(P) ≥ 4 was used to determine the significance of marker–trait associations. The region of interest was defined using the linkage disequilibrium (LD) decay around the most significantly associated SNP markers on each chromosome, which extended until a squared correlation (r2) decayed to < 0.5 within LD ± 1.2 Mbp. The physical locations of the SNPs were identified based on the barley database BARLEX used to mine the candidate genes (Mascher et al. 2021). Gene annotations, gene ontology (GO), and even gene annotations are all included in the web-based platform known as BARLEX. The annotation of the high-confidence (HC) candidate gene was detected using Morex v3 (Mascher et al. 2021).
Results
Molecular Markers, Population Structure, and Linkage Disequilibrium (LD)
The core collection of barley accessions belongs to ten geographical regions (Table S1 and Fig. 1). There are two main clusters among the accessions with biological status and row-type whereas most of the accessions were 2-rowed and landraces were 6-rowed (Table S1 and Fig. 2a and b). The accessions of the collection were originally from different geographical origins and could see a trend among the accessions that most of Northern Europe, Northern America, and South America were clustered around the center while the accessions from other origins were distributed (Table S1 and Fig. 2c). These results showed a wide diversity among the landraces coming from central Asia, the Near and the Far East, North Africa, and Ethiopia compared to accessions mostly coming from Europe and the Americas (Table S1 and Fig. 2).

Geographical distribution of barley population

Population structure based on a biological status, b row-type, and c geographical region
All of the barley accessions were genotyped using GBS, which yielded 19,276 SNPs. Information on the number of markers for each chromosome, with the map length and marker density for each chromosome, is presented in Figure S1. The largest number of markers was detected on chromosome 2H by 3,373 SNPs, while the highest marker density was detected on chromosome 7H. Only 2193 markers were noted on chromosome 1H with the lowest marker density among chromosomes. For the whole barley genome, the overall average values of r2 decreased as the distance between SNPs increased as well as the average LD decay distance was approximately 2.4 Mbp (Alqudah et al. 2020) (Figure S1).
Natural Variation of Biochemical Attributes
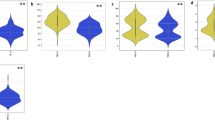
Natural variation was detected for metabolites and biochemical compounds that expressed normal distribution under salt stress (Fig. 3 and Figure S2). Proline content (ProC) accumulation remarkably increased by 47% under salt treatment (Fig. 3a), compared to the control condition (Table S2). Soluble protein (SP) significantly decreased under salt stress by 57% when compared to the control (Fig. 3b). The mean salt tolerance indices (STI) for SP and ProC were 43 and 190, respectively. A significant increase in TPC, TFC, and TAC was detected under salt treatment by 32%, 66%, and 72%, respectively, in comparison to the control across over genotypes (Table S2). Genotypic mean values for such traits STI ranged from 42.99 (SP) to 359.69 (TAC) (Fig. 4a and Table S2). H2 values ranged from 91.44 for TFC_C to 61.78 for TAC_C. Under salt treatment, moderate H2 was detected for TFs by 72.43. H2 values for STI were low for ProC_STI (34.70) and reached 89.31 in TFC_STI.

Natural phenotypic variation of physiological and agronomic traits: a proline content (ProC) mmol g−1 DW, b soluble protein (SP) mg g DW−1, c total antioxidant capacity (TAC) µg g−1 DW, d total phenolic content (TPC) µg g−1 DW, e total flavonoid content (TFC) µg g−1 DW, f spike length (SL) cm, g spikelet per spike (SS), h grains per spike (GS), i weight of grains per spike (WGS) gm, and j thousand kernel weight (TKW) gm in barley accessions under control and salinity

Natural phenotypic variation of all of the studied traits for salt tolerant indices (STI): a biochemical attributes including, proline content (ProC) mmol g−1 DW, soluble protein (SP) mg g DW−1, total antioxidant capacity (TAC) µg g−1 DW, total phenolic content (TPC) µg g−1 DW, and total flavonoid content (TFC) µg g−1 DW. b Agronomic traits including spike length (SL) cm, spikelet per spike (SS), grains per spike (GS), weight of grains per spike (WGS) gm, and thousand kernel weight (TKW) gm in barley accessions
Natural Variation of Morphological Attributes
Natural phenotypic variation was detected for all agronomic traits with normal distribution under both control and salt treatment (Fig. 3 and Figure S3–S4). Means for all of the studied morphological traits showed a significant decrease in response to salt treatment (Fig. 1f–j and Table S3). Yield traits, including WGS, showed a highly significant reduction under salt treatment by 48% compared to control treatment, indicating that salt stress has negative effects on barely yield grains (Fig. 3i and Table S3). The highest significant mean value was detected for GS_STI by 85.26% (Fig. 4b and Table S3). Moreover, high heritability was detected for GS under both treatments by 99.3% (Table S3).
Correlation
Pearson correlation was detected among all the studied traits under both treatments. Under control conditions, weight grains per spike showed a positive correlation with total phenolic content and soluble protein (Fig. 5a). In parallel, a positive correlation was detected under salinity treatment between proline content and the content of total antioxidant capacity, phenolics, and flavonoids; meanwhile, it showed negative correlation with all of the studied agronomic traits (Fig. 5b). This indicated that vigorous relationships between proline content and the secondary metabolites or antioxidants, including total phenolics and flavonoids, would imply a sophistication mechanism to scavenge the ROS molecules generated under salt stress.

Pearson correlation among all the studied traits under a control condition and b salinity condition. Soluble protein (SP) µg g−1, proline content (ProC) µg g−1, total phenolics (TPC) µg g−1, total flavonoid content (TFC) µg g−1, total antioxidant capacity (TAC), spike length (SL) cm, spikelet per spike (SS), grains per spike (GS), weight of grains per spike (WGS) gm, and thousand kernel weight (TKW) gm in barley accessions
GWAS Analysis and Candidate Genes
Based on the GWAS output of three models (GLM, MLM, and FarmCPU), the number of significant associations, and the resulting Q-Q plots, we then selected the FarmCPU model. A threshold P-value 0.001 equal to –log10(P) ≥ 4 was used to determine the significance of marker–trait associations (Figures S5–S6). On all seven chromosomes, 122 significant QTNs passed the FDR level of P ≤ 0.05 were found for ten measured traits using GWAS analysis (Table S4). Forty-one QTNs were identified for plants grown under control conditions and 53 QTNs were detected for plants exposed to salt treatment, while 28 QTNs were identified for STI of all of the measured traits (Table S4). The highest number of QTNs was found on chromosome 2H (31 QTNs), followed by chromosomes 3H and 7H (21 QTNs for each), 5H (16 QTNs), 1H (14 QTNs), 6H (10 QTNs), and 4H (9 QTNs) (Table S4).
Thirty-six QTNs were found for the physiological traits, including SP (12 QTNs), TFC (11 QTNs), TPC (7 QTNs), TAC (5 QTNs), and ProC (1 QTN) (Table S4 and Fig. 6). For TFC, most QTNs were localized on chromosome 3H (570,752,043–570,752,136 bp). For TPC_STI, the most significant QTN was associated for chr2H:607,230,828:G:C which positively influenced the trait by + 17.8 g (–log10(P) = 6.62748789570469E-07) resulting in regulating the antioxidant defense system in response to salt stress (Table S4 and Fig. 6).

a Manhattan plot of the traits under control (C): ProC_C—proline content control, SP_C—soluble protein control, TAC_C—total antioxidant capacity control, TFC_C—total flavonoid content control, and TPC_C—total phenolic content control in barley. b Manhattan plot of the traits under salt treatment (S): ProC_S—proline content salinity, and SP_S—soluble protein, TAC_S—total antioxidant capacity salinity, TFC_S—total Flavonoid content salinity, and TPC_S—total phenolic content salinity in barley. The x-axis shows the chromosomes and the SNP order. The y-axis shows the − Log10 (P-value) for each SNP marker. c Manhattan plot of all biochemical attributes based on salt tolerant indices (STI) in spring barley. The x-axis shows the chromosomes and the SNP order. The y-axis shows the − Log10 (P-value) for each SNP marker
Most QTNs for TFC_STI showed the highest positive effect (~ + 35.02 µg g−1 DW) and were influenced by G:A (at a position 570,7520,43 bp) on chromosome 3H, followed by TPC_STI which showed a positive effect of 17.77 µg g−1 DW by G:C (at position 607,230,828 bp) on chromosome 2H (Table S4 and Fig. 6).
The highest negative effect was detected for TAC_STI by − 27.02 mg g−1 DW and influenced by QTN T:A located at position 579,232,393 bp on chromosome 4H. In general, GWAS outputs revealed several QTNs that affected traits like TFC and TPC positively under salinity conditions. This information might be useful for the selection of salt-tolerant accessions and help to extend the understanding of genetic mechanisms of plants exposed to salt stress.
Eighty-six QTNs were found for the morphological-related traits (Table S4 and Fig. 7). Most QTNs were detected for SL (36) and TKW (19) for plants grown either under control or salinity. Highly significant QTNs (− log10(P) > 8.66E-08) were found to be associated with GS and TKW under both treatments (Table S4 and Fig. 7). The most significant QTN (− log10(P) = 2.81809401251597E-13) was detected for grain number per spike under control conditions and was associated with the marker chr1H:58,239,661:C:A located on chromosome 1H at 582,396,61 bp (Table S4 and Fig. 7). In response to salinity conditions, the most significant QTN (− log10(p) = 9.09430571441244E-13) was also found for grain number per spike on chromosome 2H at position 572,438,008 bp and was associated with the marker chr2H:572,438,008:C:A (Table S4 and Fig. 7).

a Manhattan plot of the morphological traits under control (C): SL_C—spike length control, SS_C—spikelets per spike control, grains per spike control, WGS_C—weight grains per spike control, and TKW_C—thousand kernel weight control in barley. b Manhattan plot of the traits under salt condition: SL_S—spike length salinity, SS_S—spikelet per spike salinity, GS_S—grains per spike salinity, WGS_S—weight grains per spike salinity, and TKW_S—thousand kernel weight salinity in barley. The x-axis shows the chromosomes and the SNP order. The y-axis shows the − Log10 (P-value) for each SNP marker. c Manhattan plot of all morphological traits based on salt tolerant indices (STI) in spring barley. The x-axis shows the chromosomes and the SNP order. The y-axis shows the − Log10 (P-value) for each SNP marker
According to the physical position of QTNs, 203 putative candidate genes were detected within the defined interval (Table 1 and Table S5). Out of the high number of candidate genes, three important putative genes were found to be associated with involved salt-stress tolerance. For example, the QTN G:C was found inside the candidate gene HORVU.MOREX.r3.2HG0193490 at position 607,230,828 on chromosome 2H that annotated as a growth-regulating factor, controlling the variation of SP_STI and TPC_STI. This candidate showed overexpression in response to salt stress in several plant species (Fig. 8a–c).

a Manhattan plot of SP_STI and TPC_STI, respectively, SP: soluble protein, TPC: total phenolics, and STI: salt tolerant index. The x-axis shows the chromosomes and the SNP order. The y-axis shows the –log10 (P-value) for each SNP marker. b Locus zoom in on regional plot colocalization of highly associated SNP (G:C). c Structure of the candidate gene (HORVU.MOREX.r3.2HG0193490) with sequence variation in the second exon inside LD heat map. d Allele effect on the accessions at the natural genetic variant (G:C) of single gene-based. e Quantile–quantile scale representing expected versus observed − log10 (P-value)
The allelic variation explained by this marker G:C showed that accessions carrying allele C had slightly lower soluble proteins and total phenolics compared to the accessions carrying G allele (Fig. 8d). This indicates that accessions carrying allele G showed more resistance to salinity stress, thereby scavenging the deleterious effects of the free radicals generated under salt stress conditions.
The HORVU.MOREX.r3.2HG0217090 annotates the C2 calcium/lipid-binding and GRAM domain protein which were found to have higher functional activity in the transmembrane regions and are involved in abiotic stress, including drought and salinity (Table S5 and S6). Interestingly, the QTN G:A was located inside the candidate gene HORVU.MOREX.r3.3HG0291720 at position 501,703,401 on chromosome 3H and annotated as Ca-binding protein that harbors the variation of thousand kernel weight under both control and salinity conditions (Fig. 9a–c). Ca-binding protein is a type of protein that binds to specific intracellular free Ca2+ and contributes to the signalling process that triggers the expression of salt-stress responsive genes. The allelic variation explained by this marker G:A showed that accessions carrying allele G had a lower thousand kernel weight compared to the accessions carrying A allele (Fig. 9d). This indicates that accessions carrying allele G are more sensitive to salinity stress and thereby produced fewer grains under such conditions.

a Manhattan plot of TKW_C and TKW_S, respectively, TKW: thousand kernel weight, C: control, and S: salt. The x-axis shows the chromosomes and the SNP order. The y-axis shows the –log10 (P-value) for each SNP marker. b Locus zoom in on regional plot colocalization of highly associated SNP (G:A). c Structure of the candidate gene (HORVU.MOREX.r3.3HG0291720) with sequence variation in the second exon inside LD heat map. d Allele effect on the accessions at the natural genetic variant (G:A) of single gene-based. e Quantile–quantile scale representing expected versus observed − log10 (P-value)
Discussion
Globally, salinity stress is one of the abiotic factors that greatly affect agriculture by limiting plant growth and production. Therefore, plants respond to the detrimental effects of salt stress through triggering antioxidant defense mechanisms (Thabet et al. 2021a, 2022). The leaf proteins have a major role in plant development, reproduction, and ultimately final grain yield. It is generally known that proteins in biological systems are the initial targets of ROS, and thus, decrease in protein content in plant leaves as a result of salt stress is expected. One of the main targets of ROS is the chloroplast, which modifies a wide range of proteins, such as stromal and thylakoid, significantly, including the degradation and inactivation of Rubiscom (Ishida et al. 1997). Therefore, leaf protein content is one of the crucial indicators of the effects of salt stress (Isayenkov and Maathuis 2019). Our results showed that soluble protein (SP) was significantly decreased under salinity conditions in comparison to control. Our results are in agreement with Khosravinejad et al. (2009), those who demonstrated a considerable decline in total protein content as a result of specific stress protein synthesis produced under salinity stress. Soluble protein content decreased as salinity increased, owing to a disruption in the protein synthesis mechanism or increased proteolytic activity, which eventually induced programmed cell death (Kovács 2017). Therefore, plants may accumulate small molecular mass proteins in response to salt stress, which may be a source of storage nitrogen and may also involve osmotic adjustment (Ashraf and Harris 2004; Singh et al. 1987).
On the other hand, it has been proposed that a higher accumulation of compatible solutes, such as proline, can improve salinity tolerance. Proline, along with other osmolytes, modulates redox potential in the end by facilitating osmotic adjustment, safeguarding cellular membranes, and maintaining enzyme stability during abiotic stress (Raza et al. 2007). Our study detected a significant increase in proline contents (ProC) for all barley accessions in response to salinity stress compared to the control condition. This finding is consistent with observations on wheat by Kumar et al. (2017). As a result, the osmoadaptive response involves the accumulation of proline, suggesting an imprecise involvement in the resistance to abiotic stress such as drought and salinity.
Moreover, plants respond to the detrimental effects of salt stress through the evolvement of a variety of metabolic mechanisms. Phenolic compounds are not only the most prevalent secondary metabolites in higher plants, but they also serve as the most powerful antioxidants for scavenging the excessive ROS generated in response to different environmental stimuli. Another category of phenolic compounds known for having antioxidant effects is the flavonoids (Tohidi et al. 2017). Our findings showed a significant increase in the accumulation of total phenolic and flavonoids compounds under salt treatment. This agrees with the observations made by Hichem et al. (2009), who reported the significant effect of salt stress on the total phenolic and flavonoids compounds in two maize (Zea mays L.) cultivars. The well-established link between polyphenols and abiotic stress tolerance, including salinity, is a strong indicator of tolerance and may therefore be utilized as a marker for the maintenance of the redox state in the cells (Sharma et al. 2019).
Under salt stress, our results detected a high positive correlation between proline content and the content of total antioxidant capacity, phenolics, and flavonoids; meanwhile, it showed a negative correlation with all of the studied agronomic traits, indicating that the vigorous relationships between proline content and the secondary metabolites, including total phenolics and flavonoids, would imply a sophistication adaptive mechanism to scavenge the free-radicals generated under salt stress (Hodaei et al. 2018).
Understanding the molecular basis of plant responses to salt stresses depends on identifying and characterizing salt stress-responsive genes and, ultimately, developing salt-tolerant crops. Here, we identify several candidate genes that might control the biological mechanisms of plant crop adaptation to salt stress, including barley. The most important candidate is located on chromosome 2H at position (607,227,256–607,231,009 bp), namely HORVU.MOREX.r3.2HG0193490, that encodes growth-regulating factors (GRFs). GRFs are transcription factors that are specific to plants and play significant roles in the plant’s response to abiotic stress (Omidbakhshfard et al. 2015). The GRFs have been discovered in a variety of plant species, including Arabidopsis, rice, rapeseed, Chinese cabbage, tobacco, and wheat (Li et al. 2021). Sakuma et al. (2006) reported that the overexpression of AtGRF7 in Arabidopsis results in improved tolerance to salt and drought stress. In cotton, Cao et al. (2020) reported that expression patterns of GhGRF1A, GhGRF1D, and GhGRF17D confer increased in response to salt stress. In strawberry, expression profile analysis detected that FvGRF3, FvGRF6, and FvGRF8 were up-regulated under salt stress (Li et al. 2021).
Moreover, the HORVU.MOREX.r3.2HG0217090 annotates the C2 calcium/lipid-binding and GRAM domain protein which were found to have higher functional activity in the transmembrane regions and are involved in abiotic stress, including drought and salinity. C2 domain-containing proteins (C2DPs), a type of Ca-dependent protein kinase, have higher functional activity in the transmembrane regions and are involved in abiotic stress, including drought and salinity (Zhang et al. 2022). In apple, overexpression of a gene encoding a C2-domain phospholipid-binding protein MdCAIP1 conferred tolerance to osmotic and salt stress (Liu et al. 2019). In transgenic Arabidopsis, a rice gene encoding a small C2 domain protein OsSMCP1 was up-regulated under biotic and abiotic stress responses (Yokotani et al. 2009). Based on RNA-seq data, the transcript profiles of GmC2-58, GmC2-88, and GmC2-148 in soybean showed overexpression in response to drought, salinity, and abscisic acid (ABA) treatments. Furthermore, the soybean plants with GmC2-148 transgenic hairy roots had delayed leaf rolling, increased the contents of proline, and reduced the contents of H2O2, O2−, and malondialdehyde under both salt and drought stress conditions (Sun et al. 2021). Altogether, these findings reported that C2 domain genes are implicated in the response to salt and drought conditions.
Interestingly, the QTN G:A was located inside the candidate gene HORVU.MOREX.r3.3HG0291720 at position 501,703,401 on chromosome 3H that harbors the variation of thousand kernel weight under both control and salinity conditions. Ca-binding protein is a type of protein that binds to specific intracellular free Ca2+ and contributes to the signaling process that triggers the expression of salt-stress responsive genes. Calcium ion (Ca2+) is an important second messenger for plant development as it serves as a signaling factor for plant transduction pathways regulating stress-induced gene expression (Kudla et al. 2010). Ca2+ maintains the steadiness of the cell wall, membrane, and membrane-binding protein and improves the plant responses to abiotic and biotic stresses, thereby triggering multiple signaling pathways (Cho et al. 2016). Ca-binding protein is a type of protein that binds to specific intracellular free Ca2+ and contributes to the signalling process that triggers the expression of stress response genes. Under salt stress, high levels of cytosolic Ca2+ can also be captured by three main Ca-binding proteins, namely calmodulins, Ca-dependent protein kinase, and calcineurin B-like proteins (Galon et al. 2010). Genetic evidence suggests that perception of salt stress leads to cytosolic Ca2+-mediated signal transduction that activates the Ca2+ sensor SOS3 (calcineurin-like protein [CBL4]). SOS3 senses the Na+-induced elevation of cytosolic Ca2+ via the interaction with SOS2 (calcineurin-interacting protein kinase [CIPK24]), which phosphorylates and activates of the plasma membrane Na+/H+ exchanger SOS1. The activation of SOS1 results in either the extrusion of Na+ out of cytosol or its compartmentation in the vacuole (El Mahi et al. 2019). Similar to SOS3, when calmodulins (CaMs) perceive a change in cytosolic Ca2+ associated with a particular stress response, they may modify the affinity of the Ca2+/CaM complex by controlling specific downstream receptors (such as phosphodiesterase) and targets (such as Cyclic Nucleotide-Gated Channel12) (DeFalco et al. 2016). Kaewneramit et al. (2019) reported that the overexpression of OsCaM1-1 enhanced salt stress tolerance in transgenic rice. Yoo et al. (2005) reported that overexpression of GmCaM4 in soybean conferred salt stress tolerance via modulating the expression levels of salt stress-responsive genes. Shen et al. (2020) reported that HvCaM1 (gene identifier HORVU0Hr1G001270) negatively regulates salt tolerance in barley via interaction with the transcriptional activator HvCAMTA4 to modulate the down-regulation of HvHKT1;5 and/or the up-regulation of HvHKT1;1, certain class I high-affinity potassium transporter (HvHKT1;5 and HvHKT1;1)-mediated Na+ translocation in roots, and ultimately reduce Na+ accumulation rates in shoots. Our findings detected the role of our candidate in the regulation of ion homeostasis was enhanced by Ca2+/CaM, thus providing a new strategy for increasing the tolerance of plants to salt stress.
Conclusion
To our knowledge, this is the first report that establishes the genetic factors determining the protective role of polyphenols (TPC and TFC) and antioxidants against salt stress-induced oxidative damage and the differential response of barley accessions, using GWAS. The vigorous activity of antioxidants would imply significant roles in the evolvement of defense-oriented strategies to scavenge the ROS molecules generated under salt stress. Highly significant QTN G:A was located inside the candidate HORVU.MOREX.r3.3HG0291720 gene on chromosome 3H. This gene annotates as Ca-binding protein that binds to specific intracellular free Ca2+ and contributes to the signalling process that in turn activates the expression of stress response genes. The identified QTNs/ candidate genes shed light on the genetic improvement of barley genotypes under salt stress.
Availability of Data and Materials
All data supporting the findings of this study are available within the paper and within its supplementary materials published online.
References
Alqudah AM, Sallam A, Baenziger PS, Börner AJJoar (2020) GWAS: Fast-forwarding gene identification and characterization in temperate Cereals: lessons from Barley–a review 22:119–135
Alqudah AM, Sharma R, Börner A (2021) Insight into the genetic contribution of maximum yield potential, spikelet development and abortion in barley 3:721–736
Ashraf M, Harris PJC (2004) Potential biochemical indicators of salinity tolerance in plants. Plant Sci 166:3–16
Bates LS, Waldren RP, Teare ID (1973) Rapid determination of free proline for water-stress studies. Plant Soil 39:205–207
Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48
Cao JF, Huang JQ, Liu X, Huang CC, Zheng ZS, Zhang XF, Shangguan XX, Wang LJ, Zhang YG, Wendel JF, Grover CE, Chen ZW (2020) Genome-wide characterization of the GRF family and their roles in response to salt stress in Gossypium. BMC Genomics 21:575
Cho K-M, Nguyen HTK, Kim SY, Shin JS, Cho DH, Hong SB, Shin JS, Ok SH (2016) CML10, a variant of calmodulin, modulates ascorbic acid synthesis 209:664–678
DeFalco TA, Marshall CB, Munro K, Kang HG, Moeder W, Ikura M, Snedden WA, Yoshioka K (2016) Multiple calmodulin-binding sites positively and negatively regulate Arabidopsis CYCLIC NUCLEOTIDE-GATED CHANNEL12. Plant Cell 28:1738–1751
El Mahi H, Pérez-Hormaeche J, De Luca A, Villalta I, Espartero J, Gámez-Arjona F, Fernández JL, Bundó M, Mendoza I, Mieulet D, Lalanne E, Lee SY, Yun DJ, Guiderdoni E, Aguilar M, Leidi EO, Pardo JM, Quintero FJ (2019) A critical role of sodium flux via the plasma membrane Na(+)/H(+) exchanger SOS1 in the salt tolerance of rice. Plant Physiol 180:1046–1065
Galon Y, Finkler A, Fromm H (2010) Calcium-Regulated Transcription in Plants Molecular Plant 3:653–669
Hichem H, Mounir D, Naceur EA (2009) Differential responses of two maize (Zea mays L.) varieties to salt stress: changes on polyphenols composition of foliage and oxidative damages. Ind Crops Prod 30:144–151
Hodaei M, Rahimmalek M, Arzani A, Talebi M (2018) The effect of water stress on phytochemical accumulation, bioactive compounds and expression of key genes involved in flavonoid biosynthesis in Chrysanthemum morifolium L. Ind Crops Prod 120:295–304
Isayenkov SV, Maathuis FJM (2019) Plant salinity stress: many unanswered questions remain. Front Plant Sci 10:80
Ishida H, Nishimori Y, Sugisawa M, Makino A, Mae T (1997) The large subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase is fragmented into 37-kDa and 16-kDa polypeptides by active oxygen in the lysates of chloroplasts from primary leaves of wheat. Plant Cell Physiol 38:471–479
Kaewneramit T, Buaboocha T, Sangchai P, Wutipraditkul NJBP (2019) OsCaM1–1 overexpression in the transgenic rice mitigated salt-induced oxidative damage 63:335
Khosravinejad F, Heydari R, Farboodnia T (2009) Effect of salinity on organic solutes contents in barley. Pakistan Journal of Biological Sciences : PJBS 12:158–162
Kovács J (2017) Comparative study of salt stress-induced physiological and molecular responses in tomato (Solanum lycopersicum L.). szte
Kudla J, Batistic O, Hashimoto K (2010) Calcium signals: the lead currency of plant information processing. Plant Cell 22:541–563
Kumar S, Beena AS, Awana M, Singh A (2017) Physiological, biochemical, epigenetic and molecular analyses of wheat (Triticum aestivum) genotypes with contrasting salt tolerance. Front Plant Sci 8:1151
Li Z, Xie Q, Yan J, Chen J, Chen Q (2021) Genome-Wide Identification and Characterization of the Abiotic-Stress-Responsive Grf Gene Family in Diploid Woodland Strawberry (fragaria Vesca) 10:1916
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang Z (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28:2397–2399
Liu X, Huang M, Fan B, Buckler ES, Zhang Z (2016) Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet 12:e1005767
Liu X-J, Dong Y-H, Liu X, You C-X, Hao Y-J (2019) A C2-domain phospholipid-binding protein MdCAIP1 positively regulates salt and osmotic stress tolerance in apple. Plant Cell. Tissue and Organ Culture (PCTOC) 138:29–39
Mascher M, Wicker T, Jenkins J, Plott C, Lux T, Koh CS, Ens J, Gundlach H, Boston LB, Tulpova Z, Holden S, Hernandez-Pinzon I, Scholz U, Mayer KFX, Spannagl M, Pozniak CJ, Sharpe AG, Simkova H, Moscou MJ, Grimwood J, Schmutz J, Stein N (2021) Long-read sequence assembly: a technical evaluation in barley. Plant Cell 33:1888–1906
Milner SG, Jost M, Taketa S, Mazon ER, Himmelbach A, Oppermann M, Weise S, Knupffer H, Basterrechea M, Konig P, Schuler D, Sharma R, Pasam RK, Rutten T, Guo G, Xu D, Zhang J, Herren G, Muller T, Krattinger SG, Keller B, Jiang Y, Gonzalez MY, Zhao Y, Habekuss A, Farber S, Ordon F, Lange M, Borner A, Graner A, Reif JC, Scholz U, Mascher M, Stein N (2019) Genebank genomics highlights the diversity of a global barley collection. Nat Genet 51:319–326
Munns R (2008) Tester MJARPB. Mechanisms of Salinity Tolerance 59:651–681
Mwando E, Han Y, Angessa TT, Zhou G, Hill CB, Zhang X-Q, Li C (2020) Genome-wide association study of salinity tolerance during germination in barley (Hordeum vulgare L.) 11
Omidbakhshfard MA, Proost S, Fujikura U, Mueller-Roeber B (2015) Growth-regulating factors (grfs): a small transcription factor family with important functions in plant biology. Mol Plant 8:998–1010
Prieto P, Pineda M, Aguilar M (1999) Spectrophotometric quantitation of antioxidant capacity through the formation of a phosphomolybdenum complex: specific application to the determination of vitamin E. Anal Biochem 269:337–341
Raza SH, Athar HR, Ashraf M, Hameed A (2007) Glycinebetaine-induced modulation of antioxidant enzymes activities and ion accumulation in two wheat cultivars differing in salt tolerance. Environ Exp Bot 60:368–376
Rehman S, Abbas G, Shahid M, Saqib M, Umer Farooq AB, Hussain M, Murtaza B, Amjad M, Naeem MA, Farooq A (2019) Effect of salinity on cadmium tolerance, ionic homeostasis and oxidative stress responses in conocarpus exposed to cadmium stress: implications for phytoremediation. Ecotoxicol Environ Saf 171:146–153
Rivandi J, Miyazaki J, Hrmova M, Pallotta M, Tester M, Collins NC (2011) A SOS3 homologue maps to HvNax4, a barley locus controlling an environmentally sensitive Na+ exclusion trait. J Exp Bot 62:1201–1216
Sakuma Y, Maruyama K, Osakabe Y, Qin F, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2006) Functional analysis of an arabidopsis transcription factor, DREB2A, involved in drought-responsive gene expression. Plant Cell 18:1292–1309
Schmidt P, Hartung J, Rath J, Piepho H-P (2019) Estimating Broad-Sense Heritability with Unbalanced Data from Agricultural Cultivar Trials 59:525–536
Shahid MA, Sarkhosh A, Khan N, Balal RM, Ali S, Rossi L, Gómez C, Mattson N, Nasim W, Garcia-Sanchez F (2020) Insights into the Physiological and Biochemical Impacts of Salt Stress on Plant Growth and Development 10:938
Sharma A, Shahzad B, Rehman A, Bhardwaj R, Landi M, Zheng B (2019) Response of phenylpropanoid pathway and the role of polyphenols in plants under abiotic stress. Molecules 24
Shavrukov Y, Gupta NK, Miyazaki J, Baho MN, Chalmers KJ, Tester M, Langridge P, Collins NC (2010) HvNax3–a locus controlling shoot sodium exclusion derived from wild barley (Hordeum vulgare ssp. spontaneum). Funct Integr Genomics 10:277–291
Shen Q, Fu L, Su T, Ye L, Huang L, Kuang L, Wu L, Wu D, Chen ZH, Zhang G (2020) Calmodulin HvCaM1 negatively regulates salt tolerance via modulation of HvHKT1s and HvCAMTA4. Plant Physiol 183:1650–1662
Singh NK, Bracker CA, Hasegawa PM, Handa AK, Buckel S, Hermodson MA, Pfankoch E, Regnier FE, Bressan RA (1987) Characterization of osmotin : a thaumatin-like protein associated with osmotic adaptation in plant cells. Plant Physiol 85:529–536
Song FL, Gan RY, Zhang Y, Xiao Q, Kuang L, Li HB (2010) Total phenolic contents and antioxidant capacities of selected chinese medicinal plants. Int J Mol Sci 11:2362–2372
Sun Y, Zhao J-Y, Li Y-T, Zhang P-G, Wang S-P, Guo J, Chen J, Zhou Y-B, Chen M, Ma Y-Z, Fang Z-W, Xu Z-S (2021) Genome-wide analysis of the C2 domain family in soybean and identification of a putative abiotic stress response gene GmC2–148 12
Tester M, Davenport RJAob (2003) Na+ tolerance and Na+ transport in higher plants 91:503–527
Thabet SG, Alqudah AM (2019) Crops and Drought. eLS, pp 1–8
Thabet SG, Alqudah AM (2023) New genetic insights into improving barley cope with salt stress via regulating mineral accumulation, cellular ion homeostasis, and membrane trafficking. Environ Exp Bot 208:105252. https://doi.org/10.1016/j.envexpbot.2023.105252
Thabet SG, Moursi YS, Karam MA, Graner A, Alqudah AM (2018) Genetic basis of drought tolerance during seed germination in barley. PLoS ONE 13:e0206682
Thabet SG, Moursi YS, Karam MA, Börner A, Alqudah AM (2020) Natural Variation Uncovers Candidate Genes for Barley Spikelet Number and Grain Yield under Drought Stress 11:533
Thabet SG, Alomari DZ, Alqudah AM (2021a) Exploring natural diversity reveals alleles to enhance antioxidant system in barley under salt stress. Plant Physiol Biochem 166:789–798
Thabet SG, Moursi YS, Sallam A, Karam MA, Alqudah AM (2021b) Genetic associations uncover candidate SNP markers and genes associated with salt tolerance during seedling developmental phase in barley. Environ Exp Bot 188:104499
Thabet SG, Alomari DZ, Börner A, Brinch-Pedersen H, Alqudah AM (2022) Elucidating the genetic architecture controlling antioxidant status and ionic balance in barley under salt stress. Plant molecular biology
Tohidi B, Rahimmalek M, Arzani A (2017) Essential oil composition, total phenolic, flavonoid contents, and antioxidant activity of Thymus species collected from different regions of Iran. Food Chem 220:153–161
VanRaden PMJJods (2008) Efficient methods to compute genomic predictions 91:4414–4423
Wei T, Simko V (2017) R package “corrplot”: visualization of a Correlation Matrix (Version 0.84). Vienna
Yokotani N, Ichikawa T, Kondou Y, Maeda S, Iwabuchi M, Mori M, Hirochika H, Matsui M, Oda K (2009) Overexpression of a rice gene encoding a small C2 domain protein OsSMCP1 increases tolerance to abiotic and biotic stresses in transgenic Arabidopsis. Plant Mol Biol 71:391–402
Yoo JH, Park CY, Kim JC, Heo WD, Cheong MS, Park HC, Kim MC, Moon BC, Choi MS, Kang YH, Lee JH, Kim HS, Lee SM, Yoon HW, Lim CO, Yun DJ, Lee SY, Chung WS, Cho MJ (2005) Direct interaction of a divergent CaM isoform and the transcription factor, MYB2, enhances salt tolerance in arabidopsis. J Biol Chem 280:3697–3706
Zhang H, Zeng Y, Seo J, Kim YJ, Kim ST, Kwon SW (2022) Global identification and characterization of C2 domain-containing proteins associated with abiotic stress response in rice (Oryza sativa L.). Int J Mol Sci 23
Zou Y, Lu Y, Wei D (2004) Antioxidant activity of a flavonoid-rich extract of Hypericum perforatum L. in vitro. J Agric Food Chem 52:5032–5039
Acknowledgements
The author (M.D.A) would like to acknowledge Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R355), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
SGT and AMA designed the experiment and analyzed the data. SGT, M.D.A, A.A.J, and AMA wrote and edited the manuscript. SGT conceived the idea and participated in the interpretation of the results.
Corresponding authors
Ethics declarations
Ethical Approval
This article does not contain any studies involving animals or human participants performed by any of the authors.
Competing interests
The authors have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• This study focuses on characterizing a metabolite-based genome-wide association study (mGWAS) that is involved in salt adaptation in barley.
• The identified QTN/candidate genes provide information useful for the genetic improvement of barley genotypes under salt stress.
• However, the genetic control of metabolomes underlying crop environmental stress adaptation remains elusive.
Supplementary Information
Below is the link to the electronic supplementary material.
11105_2023_1408_MOESM1_ESM.pptx
Fig. S1: The number of SNPs within 1 MB window size for barley; Fig. S2: Distribution of accessions for antioxidants under control and salt stress in barley. Proline content (ProC), Soluble protein (SP), Total antioxidant capacity (TAC), Total phenolic content (TPC), and Total flavonoid content (TFC); Fig. S3: Phenotypic distribution of accessions for all agronomic traits under control and salt stress in barley. Spike Length (SL), spikelet per spike (SS), grains per spike (GS), Weight of grains per spike (WGS), and Thousand kernel weight (TKW); Fig. S4: Phenotypic distribution of accessions for all of the studied traits underlie salt-tolerant indices (STI) in barley; Fig. S5: Comparison association analysis for FarmCPU, GLM, MLM models indicates Manhattan plot for; ProC—Proline content, SP—Soluble protein, TAC—Total antioxidant capacity, TFC— Total Flavonoid content, and TPC— Total phenolic content, SL— Spike Length, SS—Spikelets per Spike, GS — Grains per Spike, WGS—Weight Grains per Spike, and TKW—Thousand Kernel Weight in barley. The x-axis shows the chromosomes and the SNP order. The y-axis shows the − Log10 (P-value) for each SNP marker. The x-axis shows the chromosomes and the SNP order. The y-axis shows the − Log10 (P-value) for each SNP marker; Fig. S6: Quantile–quantile scale representing expected versus observed -log10 (p-value) underlying FarmCPU, GLM, MLM models for traits, including ProC—Proline content, SP—Soluble protein, TAC—Total antioxidant capacity, TFC— Total Flavonoid content, and TPC— Total phenolic content, SL— Spike Length, SS—Spikelets per Spike, GS — Grains per Spike, WGS—Weight Grains per Spike, and TKW—Thousand Kernel Weight in barley (PPTX 17788 KB)
11105_2023_1408_MOESM2_ESM.xlsx
Table S1: The detailed information of barley accessions; Table S2: Heritability and analysis of variance for all biochemical traits; Table S3: Heritability and analysis of variance for all morphological traits; Table S4: Marker trait association with the studied traits. Physical position of markers which are passing -log10(4) and R2 10%; Table S5: Marker trait association with the multi traits. Physical position of markers which are passing -log10(4) and R2 10%; Table S6: The list of candidate genes based on the linkage disequilibrium of multi-traits associated marker. (XLSX 54 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Thabet, S.G., Alqahtani, M.D., Jabbour, A.A. et al. Genetic Associations Underpinning the Metabolite-Mediated Salt Stress Tolerance in Barley. Plant Mol Biol Rep (2023). https://doi.org/10.1007/s11105-023-01408-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11105-023-01408-3