
Abstract
The optimal balance between sugar and acidity is an essential criterion to elaborate equilibrated and stable wines. The aim of this study was to locate quantitative trait loci (QTLs) for these traits using an F1 population derived from Monastrell and Syrah wine cultivars. Several parameters related to acidity were evaluated during six consecutive years by measuring total soluble solids, total acidity, malic acid, and tartaric acid. Three genetic maps were developed using 104 SSR (simple sequence repeat) and 146 SNP (single-nucleotide polymorphism) markers. The consensus map covered 1174 cM with 238 markers assembled in 19 linkage groups (LGs). Significant QTLs at the genome-wide level were detected, and, although they exhibited a large degree of instability from year to year, QTLs for the ratio of soluble solids to acidity (LG2) and malic acid (LG8) and the ratio of tartaric to malic acid (LG8) were stable in at least 2 years. Several annotated genes involved in sugar and acidity pathways co-located with the confidence intervals of these QTLs and are proposed as putative candidate genes for future studies of these traits.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acidity is one of the main enological parameters involved in wine quality. The level and composition of the acid fraction in the must influence the development of balanced and stable wines, since acidity affects the growth of the microorganisms that are needed in the fermentation process of wine. Moreover, the role of the acids in maintaining a low pH is crucial for the color stability of red wines. Excessive acidity produces wines that are too tart, and poor acidity produces wines that have a flat and uninteresting taste (Conde et al. 2007; Jackson 2008a). The total acidity is the combination of both volatile (readily removed by steam distillation) and fixed (weakly volatile) acidity. Tartaric and malic acids account for over 70–90% of the total acids present in juice and wines and represent the most significant influences on the acidity of the juice (Kliewer 1966; Conde et al. 2007).
The concentration of both acids increases in the fruit during the early period of berry growth until the onset of ripening or veraison (Coombe 1992). Berry tartaric acid levels subsequently remain unchanged until maturation, whereas malic acid levels decrease during the ripening period—making it available as a potential source of carbon for other pathways, such as energy supply. The berries contain more tartaric than malic acid at maturity because, generally, the loss of malic acid is greater. This loss of malic acid reduces the grape titratable acidity and influences the sugar/acid balance.
The organic acid composition depends on the cultivar, cultural practices, environment, and interaction between genotype and environment. In particular, many studies show that organic acids content is significantly influenced by temperature. Generally, in ripe grapes, high temperatures reduce the concentration of organic acids and, therefore, the acid levels are lower in a warmer climatic region than in a cooler one (Kliewer 1973; Gladstones 1992; Jones et al. 2005; Jackson 2008b). Regarding the inheritance of sugars and acids in grape berries, Sato et al. (2000) and Wei et al. (2002) studied the inheritance of total soluble solids content and titratable acidity of grape berries. Later, Liu et al. (2007) studied the inheritance patterns of glucose, fructose, total sugar, malic acid, tartaric acid, and total acid for table grape.
Acidity in grapevine, like most characters of agricultural interest, is a complex quantitative trait, likely controlled by a large number of genes. Quantitative trait locus (QTL) analysis has been widely used to investigate the genetic determinism of traits of interest in viticulture and enology, based on linkage maps and phenotypic evaluation of segregating progenies (reviewed by Chialva et al. 2016). In spite of the relevance of acidity in wine grapes, the current understanding of its genetic bases remains very limited. Viana et al. (2013, 2016) identified some QTLs that explain a small amount of phenotypic variation in soluble solid concentrations, pH, and titratable acidity in table grape, but their position and stability are quite approximate due to the low chromosome coverage by molecular markers and the lack of phenotypic data over multiple seasons. More recently, Chen et al. (2015), Zhao et al. (2015, 2016), Ban et al. (2016), and Yang et al. (2016) reported QTLs controlling the production of individual or total sugars and acids in grape berries, as well as some candidate genes related to these characters. However, the QTL analysis performed by Yang et al. (2016) is based on the phenotyping of a small progeny in a single year. Moreover, all the studies refer to interspecific crosses. The only berry acidity QTLs identified in a Vitis vinifera intra-specific cross were reported so far by Houel et al. (2015). Finally, association studies by Laucou et al. (2018) allowed finding SNPs associated with the acidity of the wine. In light of the above considerations, it would be important to acquire new insights about the genetic determination of acidity in Vitis vinifera with the dual objective of assisting breeding programs and of identifying candidate genes for further functional studies.
In this work, an intra-vinifera Monastrell x Syrah progeny was used for the genetic dissection of total acidity and individual acids in grape berries. Molecular maps were combined with phenotypic data collected over six seasons to identify stable QTLs related to berry acidity. Additionally, some putative candidate genes were identified based on their functional annotation and co-location with the detected QTLs.
Materials and Methods
Plant Material
This study is based on an F1 progeny of 229 seedlings (one plant per genotype) planted in a pre-selection frame in the experimental field of the IMIDA in Murcia (southeast Spain) in the year 2000. The progeny was generated by controlled crosses between the wine cultivars Monastrell (female) and Syrah (male). Monastrell constitutes the main cultivar in the Jumilla, Yecla, and Bullas Denomination of Origin (D.O.) areas in the Region of Murcia. It is a cultivar spread across the whole Mediterranean basin and is well adapted to the dry conditions of this area; Syrah blends very well with Monastrell contributing quality characters. A plant of each genotype and representative plants of both progenitors were grown on their own roots under standard conditions of irrigation, fertilization, and pest and disease control (Bayo-Canha et al. 2012). Three plants of each parent were included in the experiment to assess the environmental variance.
Assessment of Parameters Related to Acidity
A set of fruiting genotypes and the two parents were used in each of the 6 years of the study (2008–2013). The number of individuals analyzed for parameters related to acidity varied from year to year depending on the environmental conditions, disease incidence, and fruit setting. In total, 96 genotypes were evaluated in 2008, 97 in 2009, 119 in 2010, 76 in 2011, 67 in 2012, and 148 in 2013 for acidity parameters. One hundred thirty-four of the total plants analyzed got data for more than 1 year.
The physicochemical analyses of each sample were performed in triplicate from at least 3–9 representative bunches at harvest, considered as the date when three berries picked randomly from the top, medium, and bottom regions of at least three representative clusters located in different parts of the canopy reached approximately 13.5 °Baumé (colored grapes) or 12.5 °Baumé (uncolored grapes). About 100 g of berries from different positions within each cluster, considered as one replication, was mixed and squeezed to determine the physicochemical parameters of the juice. Total soluble solids (TSS) were measured as °Baumé (°Bé), using an Atago RX-5000 digital refractometer (Atago, Tokyo, Japan). The juice titratable acidity was determined by titration with 0.1 N NaOH, using a Metrohm 686 automatic titrator (Metrohm, Herisau, Switzerland), and was expressed as grams per liter of tartaric acid equivalents. Tartaric and malic acids were measured using enzymatic kits from Boehringer Mannheim GmbH (Mannheim, Germany). Given the link between acid and sugar metabolisms (Sweetman et al. 2009) and the higher robustness of ratios of compound concentrations compared with individual metabolite levels (Morreel et al. 2006; Yang et al. 2016), also the ratio of soluble solids to total acidity and the ratio of tartaric acid to malic acid were calculated.
Statistical Analysis
All the statistical analyses were performed using the program SPSS 13.0. Normality of trait distribution was tested with the Kolmogorov-Smirnov test. Year effect of the data and the correlation between traits were determined with the Kruskal-Wallis and the Spearman tests, respectively. The broad-sense heritabilities were calculated as the proportion of the phenotypic variance that can be attributed to the variance of genotypic values using the following equation: H2 = SG2/SP2. Here, the phenotypic (P) variance is expressed as the sum of genotypic (G) variance and environmental variance. The total phenotypic variance was calculated within the F1 population, while the environmental variance was estimated with an analysis of variance on the parental controls.
Marker Analysis
Total DNA was isolated from 50 mg of young frozen leaves, using the DNeasy Plant Mini Kit (Qiagen, Valencia, CA) with extraction buffer supplemented with 1% polyvinylpyrrolidone to reduce polyphenols (Lodhi et al. 1995). The mapping population (229 F1 individuals) and the parents were genotyped using SSRs, SNPs, and one CAPS (cleaved amplified polymorphic sequence).
The selection of suitable SSR markers was based on their presence over the 19 linkage groups in previous genetic linkage maps of Vitis vinifera (Adam-Blondon et al. 2004; Riaz et al. 2004; Doligez et al. 2006). The mapping population was genotyped for 104 SSR markers (http://www.ncbi.nlm.nih.gov; Supplementary Table 1). The PCR amplifications were performed in 20-μl reactions containing 10–30 ng of template DNA, 0.2 μM of each primer (Applied Biosystems), 0.2 mM of each dNTP, 1× PCR buffer (Ecogen, Barcelona, Spain), 1.9 mM MgCl2, and 0.25 units of Taq DNA polymerase (Ecogen, Barcelona, Spain). Amplification reactions were carried out in a 96-well block Thermal cycler (Eppendorf, Barcelona, Spain), using the following program: 5-min initial denaturation step at 94 °C followed by 35 cycles (1-min denaturation at 94 °C, 45 s at the annealing temperature for the primer (Supplementary Table 1) and 1-min extension at 72 °C), followed by 10-min final extension at 72 °C. Primers failing to amplify were further tested using a touchdown PCR amplification program (Don et al. 1991), in which the initial annealing temperature (Ta) was reduced by 0.2 °C per cycle for the following 14 cycles, followed by 20 cycles with an annealing temperature of Ta-3 °C. All forward primers were labeled at their 5′-ends with fluorescent dyes (6-FAM, NED, VIC, or PET), and the PCR products were separated by capillary electrophoresis using the ABI Prism 3730 Genetic Analyzer sequencer (Applied Biosystems, Carlsbad, CA) in an external platform (Unidad de Genómica-Campus Moncloa del Parque Científico de Madrid [http://www.fpcm.es]). Alleles were identified using GeneMapper software v3.7 (Applied Biosystems), and their sizes were determined using the internal size standard GS500LIZ (Applied Biosystems).
The mapping population was also genotyped for 238 SNP markers (Lijavetzky et al. 2007; Cabezas et al. 2011) using the Applied Biosystems SNPlexTM Genotyping System 48-plex platform (De la Vega et al. 2005; Tobler et al. 2005) in the Centro Nacional de Genotipado (http://www.cegen.org).
In addition, new SNP-based markers were analyzed in the mapping progeny by applying the candidate gene approach (Pflieger et al. 2001). These markers were identified and developed at Fondazione Edmund Mach (FEM) from the cultivars Monastrell, Syrah, and Pinot Noir, in collaboration with the research team of Dr. Stella Grando. The candidate genes were selected within the confidence intervals of common QTLs for phenological and productivity traits in two different progenies sharing Syrah as a parent. Thirty-five pairs of primers were designed using the NCBI tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) and were tested on the parents. Three of them produced multiple bands and were discarded. The amplicons obtained with the remaining primers were sequenced as described in Battilana et al. (2009), and the sequences were analyzed with the software GAP 4 (http://www.gap-system.org) to find informative SNPs in the three cultivars under study. Then, the segregation was confirmed in a few seedlings. For suitable polymorphisms, new primers were developed and a mini-sequencing was performed, employing the SNaPshot Multiplex Kit protocol reported at http://docs.appliedbiosystems.com/search.taf. Subsequently, the polymorphisms were scored by GeneMapper v3.7 (Applied Biosystems, Carlsbad, CA).
Finally, the mapping population was also genotyped with the CAPS marker 20D18CB9, linked to berry color (Walker et al. 2007).
Genetic Linkage Maps
Genetic maps were constructed using the JoinMap 3.0 software (Van Ooijen and Voorrips 2001), following the double pseudo-testcross strategy (Grattapaglia and Sederoff 1994) and applying the Kosambi mapping function (Kosambi 1944) to convert recombination rates into genetic distances. Both parental maps and consensus map for the cross were constructed using double-haploid and cross-pollinated population types, respectively. The segregation of each locus was tested for goodness-of-fit to the expected ratio using the chi-square test. Most markers showing distorted segregation were originally included in the map calculation unless they significantly affected the order of neighboring markers. Linkage groups were determined using threshold values of 4.0 (3.0 for LG13 and 2.0 for LGs 8 and 12) for LOD (logarithm of odds) and 0.35 for recombination rate. We kept the marker order obtained at round 2. Female, male, and consensus genetic maps were aligned using MapChart 2.2 software (Voorrips 2002).
The observed genome size (Gob) for each linkage map was estimated using the sum of all LG sizes. The estimated genome length (Ge) was determined according to method 3 of Chakravarti (Hulbert et al. 1988; Chakravarti et al. 1991). The confidence interval was computed according to Gerber and Rodolphe (1994), for an α of 5%. The observed map coverage was defined as the ratio between Gob and Ge (Gob/Ge). The expected genome map coverage (Gcl) was calculated by the formula given by Lange and Boehnke (1982), adjusted for chromosomal ends.
QTL Analysis
The QTL detection was carried out separately for the parental and consensus maps, using MapQTL 4.0 software (Van Ooijen et al. 2002), and the phenotypic raw data from each season since the normality of the raw data distribution was not achieved by applying different transformations (squared values, square root, or Naperian logarithm). Indeed, we have confirmed that the QTLs obtained with transformed but non-normal data are equivalent to those obtained with non-transformed data (data not shown). The analysis was based on three different methods: Kruskal-Wallis non-parametric test (KW; Lechmann 1975), simple interval mapping (SIM, Lander and Botstein 1989), and multiple QTL mapping (MQM, Jansen and Stam 1994). First, KW rank-sum test was applied to verify the global segregation of each locus and to detect putative QTLs. Second, a simple interval mapping was performed to find regions with potential QTL effects; then, scored markers in these regions were used as co-factor (no more than five) in restricted MQM mapping. All the cofactors were selected with the “automatic cofactor selection” test implemented in the MapQTL software. Both linkage group–wide (LGW) and genome-wide (GW) LOD (logarithm of odds) thresholds corresponding to α = 0.05 were used for SIM and restricted MQM detection of QTLs. The LOD thresholds were established through 1000 permutations of the phenotypic data. The QTLs with a LOD score higher than the GW threshold were considered as significant, while the QTLs with a LOD score higher than the LGW threshold were considered as putative. Putative QTLs were considered worth retaining since several relevant agronomical traits are controlled by multiple genes each making a small contribution to the overall outcome. The QTLs detected with GW and LGW thresholds were analyzed separately. The percentage of variance explained by each QTL and by all QTLs detected in the same season, as well the QTL location, was estimated in the final MQM model. The QTL position was estimated from the location of the maximum LOD value and was indicated by the 1-LOD confidence interval and the cofactor. The 1-LOD support intervals were calculated using restricted MQM mapping. A QTL was considered stable when it was detected in at least two growing seasons. Stable genetic determinants are the most promising to be used in marker-assisted selection and in the search for candidate genes, and they are especially relevant when dealing with traits with a strong environmental influence. In order to avoid statistical artifacts, unstable QTLs were considered of interest here only when they were consistent with evidence from other works.
Candidate Genes
Based on the annotated molecular function available at NCBI (http://www.ncbi.nlm.nih.gov/projects/mapview/map_search.cgi?taxid=29760), candidate genes for the traits under study were looked for within each confidence interval of the QTLs detected in more than 1 year, including together significant and putative QTLs whose confidence intervals overlap. The most proximal marker (SSR or SNP) was selected to delimit the confidence interval, and the physical position of the marker was identified in the NCBI database.
Results
Genetic Maps
One hundred and seventy-seven SSR loci were initially analyzed in eight progeny individuals and both genitors. This allowed the identification of 104 SSRs (59%) polymorphic for at least one parent (Supplementary Table 1). Out of the 238 SNPs previously identified by Lijavetzky et al. (2007), 138 were polymorphic (58%) for at least one parent (Supplementary Table 2). In addition, starting from the 35 pairs of primers designed and tested on the parents, eight new SNPs (representing six candidate genes) were developed for the genotyping of the mapping progeny (Supplementary Table 2). The total number of molecular markers useful for linkage analysis in the Monastrell x Syrah mapping progeny was 251 (104 SSRs, 146 SNPs, and 1 CAPS), of which 84% allowed discrimination between maternal and paternal inherited alleles (markers type abxcd, abxac, abxbc, abxaa, aaxab). One SSR marker (vmc5h11-200) showed a segregation pattern consistent with the presence of a null allele in Syrah <aaxa0> and was re-coded as described by Doligez et al. (2002).
Finally, 238 markers (100 SSRs, 137 SNPs, and 1 CAPS) were assembled in the consensus map (Table 1; Supplementary Fig. 1) over the expected 19 LGs (the haploid number of chromosomes in V. vinifera) with an average distance of 5.23 cM. The total number of positioned markers per LG ranged from 7 (LG10 and 16) to 18 (LG7 and 18). The average size of the LGs was 61.84 cM, ranging from 42.20 (LG13) to 90.70 (LG7) cM.
Chi-square analysis revealed a distorted segregation ratio (P < 0.05) for 18% of the polymorphic markers in Monastrell and for 15% of the polymorphic markers in Syrah. Most of them were randomly distributed throughout the genome, but some were located in clusters with a high distortion level (Table 1).
In this work, the LGs 7, 14, 18, and 19 were the most distorted regions of the map containing at least three adjacent distorted markers (Supplementary Fig. 1). The marker order was generally consistent between parental and consensus homologous linkage groups, with the local inversions of closely linked markers and proximal markers (Supplementary Fig. 1). A general consistency in marker order was found in the manual comparison with other published maps (Doligez et al. 2006; Cabezas et al. 2011), with only minor discrepancies.
The heterozygosity level of the parents was similar, irrespective of the markers used (45% in Monastrell and 52% in Syrah). When comparing the level of heterozygosity of the 177 SSRs with that of the 273 SNPs, a great difference was observed: 79–81% of the SSRs were heterozygous in Monastrell and Syrah versus only 28–38% of the SNPs.
Phenotypic Segregation
The phenotypic segregation data of the evaluated parameters for each year of study are shown in Supplementary Table 3. Continuous variation and transgressive segregation were observed for the evaluated parameters related to acidity (Supplementary Fig. 2). The Kolmogorov-Smirnov test indicated that only the total acidity in 2009, 2010, and 2013, malic acid in 2008 and 2013, and tartaric acid in 2010 showed a normal distribution. The Kruskal-Wallis test revealed a significant year effect (P < 0.05) for all the parameters evaluated.
The data obtained during the 6 years were averaged, and minimum and maximum values were identified (Table 2). Although the mean tartaric acid content of the progeny was positioned between the values of the two progenitors, the mean total acidity content of the progeny was slightly higher than that of both progenitors while the mean malic acid content and total soluble solids were slightly lower (Table 2). The percentage of seedlings that had values higher than that of both progenitors ranged between 2 and 30% for total soluble solids, 25 and 80% for total acidity content, 6 and 56% for malic acid content, and 12 and 82% for tartaric acid content (Supplementary Fig. 2).
Spearman’s correlation coefficients (r) among variables were calculated within each year (P < 0.01). The values of the coefficients corresponding to all the years are shown in Supplementary Table 4. Total acidity and malic acid were positively correlated in 5 years. In contrast, total acidity and tartaric acid were positively correlated only in 2 years. As expected, total acidity correlated negatively in all years with the ratio of total soluble solids to total acidity and with the ratio of tartaric acid to malic acid, except in 2009. Likewise, the ratio of tartaric acid to malic acid correlated positively with the ratio of total soluble solids to total acidity in all years except 2009. Finally, the ratio of total soluble solids to total acidity correlated negatively with malic acid in 5 years (Supplementary Table 4).
Broad-sense heritabilities of the traits analyzed in this study are shown in Supplementary Table 5. In our experimental conditions, the heritability of TSS ranged from 0.54 to 0.84, the total acid content from 0.77 to 0.98, the malic acid from 0.51 to 0.69, and the tartaric acid from 0.49 to 0.56.
QTL Analysis
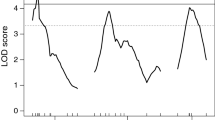
Five variables were analyzed for the study of this trait: total acidity, malic and tartaric acids, and the ratios of total soluble solids to total acidity and of tartaric acid to malic acid. Several QTLs were identified including significant (at the genome-wide level, Table 3) and putative (at the linkage group level, Supplementary Table 6) QTLs, some of which were stable over time (detected in more than one season). First, we present the stable and significant QTLs, which are mostly represented in Fig. 1. For the ratio of total soluble solids to total acidity, one significant QTL was detected on LG2 (TSS/Ac2), which was stable in the Monastrell and consensus maps, explaining up to 19.7% of the total phenotypic variance (Fig. 1 and Table 3). Malic acid was under the control of several genomic regions but only the Ma8 QTL was stable at the GW level in the Syrah map, explaining up to 19.5% of the total phenotypic variance (Fig. 1 and Table 3). It was also stable at the LGW level in Syrah and consensus maps (Supplementary Table 6). For the ratio of tartaric acid to malic acid, a significant QTL was found on LG8 (Tar/Ma8), which was stable in Syrah map and explained up to 21.1% of the total variance (Fig. 1 and Table 3) and it was also stable at the LGW level in Syrah and consensus map (Supplementary Table 6).

One-LOD support confidence intervals of the main QTLs for acidity parameters detected with restricted MQM test. The QTLs are shown on the right of each linkage group. C, consensus; Mn, Monastrell; Sy, Syrah. Ac, total acidity; TSS_Ac, ratio of total soluble solids to total acidity; Ma, malic acid; Tar_Ma, ratio of tartaric acid to malic acid
Several QTLs were detected only during 1 year (unstable) at the GW level whose confidence intervals overlapped with other stable QTLs (Fig. 1 and Table 3): Ac2 for total acidity, Ma5 for malic acid, and Tar/Ma5 for the ratio of tartaric acid to malic acid.
Finally, several putative and stable QTLs were found (Supplementary Table 6): Ac2, Ac5, and Ac18 for total acidity; TSS/Ac5 and TSS/Ac11 for the ratio of total soluble solids to total acidity; Tar16 for tartaric acid; Ma7, Ma11, and Ma17 for malic acid; and Tar/Ma17 for the ratio of tartaric acid to malic acid (Supplementary Table 6).
Most of the QTLs on LG2 mapped around the berry color locus. Given the differential harvesting procedure used for colored and uncolored genotypes, we cannot exclude a priori that these QTLs are an artifact.
Candidate Gene Identification
Functional candidate genes for acidity were retrieved from the literature (Davies and Robinson 2000; Or et al. 2000; Terrier et al. 2005; DeBolt et al. 2006; Sweetman et al. 2009; Weinl and Kudla 2009; Cuéllar et al. 2013). The location of these genes within the 1-LOD support intervals of the detected QTLs was investigated by determining their position in the grape genome (NCBI Map Viewer, http://www.ncbi.nlm.nih.gov/projects/mapview/map_search.cgi?taxid=29760). Novel candidate genes were also looked for within the confidence intervals of significant QTLs whose confidence intervals overlap (Fig. 1). As a result, several candidate genes for total acidity, total soluble solids/acidity ratio, malic acid, tartaric acid, and tartaric/malic acid ratio were located on LGs 2, 5, 8, 16, and 17 (Table 4). Based on their annotation and on the literature, these genes may participate in acid metabolism or transport, and in the sugar and tricarboxylic acid (TCA) cycle.
Discussion
Genetic Maps
Complete parental and consensus genetic maps were developed using 238 informative molecular markers. The parental maps have a similar length (1035.4 cM in Monastrell and 1038.9 cM in Syrah), with an observed coverage of 61% and 60%, respectively (Table 1). In general, if the map is unsaturated, a greater number of positioned markers produce a larger map and greater coverage, increasing the power of QTL detection. As a consequence, the incomplete coverage of our maps could theoretically limit the number of stable and significant QTLs detected. However, the observed sizes of the parental and consensus maps are within the range set by other published maps (reviewed by Cipriani et al. 2011), although the presence of highly distorted markers in our maps could inflate the observed sizes. There were four and three marker-free regions of between 20 and 30 cM on the Monastrell map and the Syrah map, as well as two marker-free regions longer than 30 cM on the Monastrell map, indicating that the maps reported here are at a good level of saturation for the proposed objective. The comparison of common markers among the three maps showed strong conservation of marker order with only minor changes detected (Supplementary Fig. 1). The changes of position in these maps are localized in regions of the genome in which they are most likely; for example, regions harboring markers with distorted segregation, partially informative markers (abxab), and clusters of markers in a very narrow strip. A general consistency in marker order was also found, compared with other published maps (Doligez et al. 2006; Cabezas et al. 2011). Regions containing at least two adjacent distorted markers were observed in the maps on several LGs (Table 1). Distorted segregation may arise mainly from misclassification of some genotypes or from differential survival of different genotypes due to gamete/zygote lethality, meiotic drive/preferential segregation, or sampling/selection during population development (Kearsey and Pooni 1996; Boopathi 2013). Segregation distortion might theoretically affect the local marker order and the power of QTL detection; however, drastic consequences are not expected here. Distortion due to misclassification is normally restricted to the locus concerned and will normally only affect recombination frequency, while distortion due to selection does not generally create problems for mapping and will affect all linked loci in inverse proportion to their distance from the affected locus (Kearsey and Pooni 1996). Moreover, the independence LOD score calculated by JoinMap for the recombination frequency is not affected by segregation distortion (Van Ooijen and Voorrips 2001). As concerns QTL mapping, segregation distortion does not produce more false QTLs nor does it have a significant impact on the position and effect estimations of QTLs, especially in large-size mapping populations. Because segregation distortion loci (SDL) can decrease as well as increase the statistical power of QTL mapping, the presence of SDL is not necessarily detrimental to QTL mapping (Xu 2008; Zhang et al. 2010). Wen et al. (2013) proposed a multi-QTL mapping approach that uses distorted markers to detect SDL. The results from their studies show that the power of QTL detection is higher under the dominant genetic model of SDL than under the additive genetic model. Consistently, several authors have found QTLs for important traits, such as seedlessness, terpenol content, and powdery mildew resistance, in distorted areas (Cabezas et al. 2006; Welter et al. 2007; Costantini et al. 2008; Duchêne et al. 2009). In the present work, LG18 in the consensus map and LG19 in the Syrah map were the LGs with the highest number of markers having distorted segregation (8 in each of them), in agreement with other authors in the case of the LG18 (Cabezas et al. 2006; Troggio et al. 2007; Costantini et al. 2008). Markers in these distorted regions could be linked to genes subject to gametic or zygotic selection, which control the viability of individuals bearing different genotypes of the locus (Xu 2008; Xu and Hu 2009).
Phenotypic Segregation
The parameters evaluated in this study showed continuous variation within the progeny (Supplementary Fig. 2), suggesting a polygenic inheritance—in agreement with previous studies (Liu et al. 2007; Viana et al. 2013; Duchêne et al. 2014; Chen et al. 2015). Transgressive segregation was apparent in the total soluble solids, total acidity, tartaric acid, and malic acid (Supplementary Fig. 2). Among the genotypes with transgressive segregation, those with high acidity content may be useful to overcome the negative effect of high temperatures and to develop balanced and stable wines in warm areas. In this sense, three seedlings of the Monastrell x Syrah progeny had malic acid content higher than 3.5 g/L (higher than that of both progenitors) during 5 of the 6 years studied and therefore could be selected to improve the acidity trait in warm areas.
As expected, total acidity and malic acid were positively correlated in 5 years. In contrast, tartaric acid only correlated positively in 2 years with total acidity (Supplementary Table 4). These results indicate that in our conditions the correlation of total acidity with malic acid is more stable than with tartaric acid. Moreover, the negative correlation found between malic acid and the total soluble solids/acid balance is consistent with the fact that the malic acid loss reduces the grape titratable acidity and influences the sugar/acid balance (Supplementary Table 4).
Broad-sense heritabilities of TSS (0.54–0.84) and total acid (0.77–0.98) obtained in this study (Supplementary Table 5) are consistent with those (0.61–0.74 and 0.53–0.90, respectively) reported by Liu et al. (2007), whereas in our experimental conditions heritabilities of malic acid (0.51–0.69) and tartaric acid (0.49–0.56) are lower than those obtained by the same authors (0.68–0.91 and 0.47–0.75, respectively). In any case, our results indicate that a significant portion of the variance for all the investigated traits is genetic.
Supplementary Fig. 2 additionally highlights that the total soluble solids content (TSS) estimated from a small number of berries in the field does not always coincide with the more precise TSS measure obtained by squeezing 100 g of harvested berries in the laboratory, which is quite variable. We cannot exclude that such variability in sugar content might have affected malic acid content and in turn QTL results. However, this is the most widely used approach in genetic studies of berry composition in grapevine since it is presently impossible to obtain detailed kinetics of berry development during maturation for each individual genotype in a large population. Moreover, there is no significant correlation between TSS and malic acid as shown in Supplementary Table 4. Therefore, we consider that the variability in sugar content has not affected malic acid content and in turn QTL results.
QTLs and Candidate Genes for Acidity
Even taking into account that the limited size of the Monastrell x Syrah population and the departure from a normal distribution for some phenotypic data might have reduced the power of QTL detection and influenced the number and stability of significant QTLs, we were able to draw a picture of the most reliable QTLs for acidity by integrating data from different seasons, maps, and significance levels (Fig. 1, Table 3, and Supplementary Table 6). We also report some candidate genes for this trait, most of which are supported by the literature while the rest represent novel findings (Table 4).
It is known that the principal synthesis of malic acid is linked with sugar metabolism (Sweetman et al. 2009), and Yang et al. (2016) highlighted the power of the analysis of the TSS/Ac ratio to detect branch-points in the regulation of specific metabolites involved in the same pathway. For that reason, we looked for QTLs related to the total soluble solids/acidity ratio. A significant and stable QTL for the TSS/Ac ratio was found on LG2; it co-located with Ac2, a significant but unstable QTL that was stable only at the LGW level in Monastrell map. In this region, the CBL01 (calcineurin B-like protein 01) gene was found (Table 4). This protein acts as a complex with a CBL-interacting protein kinase (CIPK) in the activation of a K+ channel of the Shaker family, VvK1.2. Cuéllar et al. (2013) found that the expression of the VvK1.2 gene is induced at veraison and during ripening and that the CBL/CIPK complex is involved in this activity. The CBL and CIPK families develop several functions as stress responses: H+ transport, K+ homeostasis, or nitrate responses (Weinl and Kudla 2009). Additional genes in this region could play a role like those linked to the seven SNPs found significantly associated to wine acidity by Laucou et al. (2018).
The significant but unstable Ma5 and Tar/Ma5 QTLs co-located on LG5 of the Syrah map, and several genes related with the sugar and tricarboxylic acid (TCA) cycle were found within these QTL intervals. One gene encoded a CMP-sialic acid transporter 2 (VIT_0001358001) and another one a fructokinase-2-like activity (VIT_00010790001), both linked with sugar metabolism (Pego and Smeekens 2000). Another candidate gene encoded a 2,3-dimethylmalate lyase-like activity (VIT_00013539001), involved in the TCA cycle and related to citric acid (Schnarrenberger and Martin 2002). All of these genes could be associated with malic acid because it is a source of energy when sugar has been accumulated in the vacuoles (Table 4).
Significant and stable Ma8 and Tar/Ma8 QTLs were detected, and five candidate genes were found within these QTL intervals (pyruvate kinase, ripening-related protein-like (GRIP32), pyruvate decarboxylase 2 (PDC1), and CBL-interacting protein kinase 07 and 08 (CIPK07 and CIPK08)) (Table 4). GRIP32 is a grape ripening-induced protein involved in abiotic stresses that increases during ripening (Davies and Robinson 2000; Zenoni et al. 2010). PDC1 was cloned in order to clarify the ethanol formation in berries (Or et al. 2000). The increase in ethanol is linked with the pyruvate production derived from malic decarboxylation. PDC is the key enzyme in the fermentative metabolism and in the ethanol synthesis. Additionally, a malate synthase gene (VIT_00008494001) and a malate dehydrogenase (VIT_00007997001) were located within the Ma17 and Tar/Ma17 QTL intervals (Table 4). Malate synthase is upregulated in the first phase of berry development, when malic acid is synthesized (Terrier et al. 2005).
Additionally, in the present work, the putative Tar16 was stable on the Syrah map in 2 years. In this QTL interval, the gene encoding L-idonate 5-dehydrogenase was found (Table 4). This is the key enzyme in the conversion of L-idonate to 5-keto D-gluconic acid—that produces oxalic acid (DeBolt et al. 2006). It is highly expressed in the first phase of berry development, pre-veraison, when tartaric acid is accumulating. In summary, several annotated genes involved in sugar and acidity pathways co-located with the confidence intervals of the QTLs found on LGs 2, 5, 8, 16, and 17.
Finally, several QTLs identified in this work co-localized with QTLs previously reported. Viana et al. (2013) found a QTL for pH on LG1 in a similar region as the significant and unstable Ac1 QTL detected here. However, the LG19 on which the same authors discovered a QTL for tartaric acid comprised only two markers, so we could not establish if the significant and unstable Tar19 QTL that we found is in the same region. Moreover, Cholet et al. (2016) located a gene related to tartaric acid synthesis (GDP-L-galactose phosphorylase 2) on LG19; this gene is close to our QTL region. This is the last enzyme in the Smirnoff-Wheeler pathway for production of ascorbic acid, the precursor of tartaric acid. Finally, Chen et al. (2015) and Houel et al. (2015) detected QTLs in a similar region as the putative Ac18 and Ma7 QTLs found in this work, respectively. Additional QTLs related with acidity were identified in other regions as LGs 6, 7, 13, and 18 by other authors (Chen et al. 2015; Houel et al. 2015; Ban et al. 2016; Yang et al. 2016). The QTLs found on LGs 5, 8, and 17 are reported in this work for the first time, suggesting new and interesting regions for the study of characters related to sugar and acidity.
Conclusions
The genetic maps developed in this work have allowed us to identify new QTLs related with acidity—one of the main enological parameters involved in wine quality—and to confirm some QTLs previously identified for this trait by other authors in other progenies. Significant QTLs were detected across 6 years of evaluation and, although they exhibited a large degree of instability from year to year, several of them were stable in at least 2 years. This proves the importance of collecting data from multiple seasons, especially for traits with a strong environmental influence. The present work also provides some candidate genes that are potentially involved in sugar and acidity pathways. These results open new perspectives for future studies to clarify the molecular determinants of vine acidity. Likewise, these data will support the selection of new wine grape cultivars derived from Monastrell and Syrah with increased acidity, a trait that is of great interest in the production of balanced wines in warm zones.
References
Adam-Blondon AF, Roux C, Claux D, Butterlin G, Merdinoglu D, This P (2004) Mapping 254 SSR markers on the Vitis vinifera genome: a tool for grape genetics. Theor Appl Genet 109:1017–1027. https://doi.org/10.1007/s00122-004-1704-y
Ban Y, Mitani N, Sato A, Kono A, Hayashi T (2016) Genetic dissection of quantitative trait loci for berry traits in interspecific hybrid grape (Vitis labruscana × Vitis vinifera). Euphytica 211:295–310. https://doi.org/10.1007/s10681-016-1737-8
Battilana J, Costantini L, Emanuelli F, Sevini F, Segala F, Moser S, Velasco R, Versini G, Grando MS (2009) The 1-deoxy-D-xylulose 5-phosphate synthase gene co-localizes with a major QTL affecting monoterpene content in grapevine. Theor Appl Genet 108:653–669. https://doi.org/10.1007/s0122-008-0927-8
Bayo-Canha A, Fernández-Fernández JI, Martínez-Cutillas A, Ruiz-García L (2012) Phenotypic segregation and relationships of agronomic traits in Monastrell x Syrah wine grape progeny. Euphytica 186:393–407. https://doi.org/10.1007/s10681-012-0622-3
Boopathi NM (2013) Genotyping of mapping population. In: Boopathi NM (ed) Genetic mapping and marker assisted selection. Basics, practice and benefits. Springer, India. ISBN 978-81-322-0957-7, pp 39–80
Cabezas JA, Cervera MT, Ruiz-García L, Carreño J, Martínez-Zapater JM (2006) A genetic analysis of seed and berry weight in grapevine. Genome 49:1572–1585. https://doi.org/10.1139/g06-122
Cabezas JA, Ibáñez J, Lijavetzky D, Vélez D, Bravo G, Rodríguez V, Carreño I, Jermakow AM, Carreño J, Ruiz-García L, Thomas MR, Martínez-Zapater JM (2011) A 48 SNP set for grapevine cultivar identification. BMC Plant Biol 11:153. https://doi.org/10.1186/1471-2229-11-153
Chakravarti A, Lasher LK, Reefer JE (1991) A maximum likelihood method for estimating genome length using genetic linkage data. Genetics 128:175–182
Chen J, Wang N, Fang L-C, Liang Z-C, Li S-H, Wu B-H (2015) Construction of a high-density genetic map and QTLs mapping for sugars and acids in grape berries. BMC Plant Biol 15:28. https://doi.org/10.1187/s12870-015-0428-2
Chialva C, Eichler E, Muñoz C, Lijavetzky D (2016) Development and use of biotechnology tools for grape functional analysis. In: Morata A, Loira I (eds) Grape and wine biotechnology. InTech, pp 75–101. https://doi.org/10.5772/64915
Cholet C, Claverol S, Claisse O, Rabot A, Osowsky A, Dumot V, Gerrari G, Gény L (2016) Tartaric acid pathways in Vitis vinifera L. (cv. Ugni blanc): a comparative study of two vintages with contrasted climatic conditions. BMC Plant Biol 16:144. https://doi.org/10.1186/s12870-016-0833-1
Cipriani G, Di Gaspero G, Canaguier A, Jusseaume J, Tassin J, Lemainque A, Thareau V, Adam-Blondon A-F, Testolin R (2011) Molecular linkage maps: strategies, resources and achievements. In: Adam-Blondon A-F, Martínez-Zapater JM, Kole C (eds) Genetics, genomics and breeding of grapes. Science Publishers, Enfield, pp 111–136
Conde C, Silva P, Fontes N, Dias ACP, Tavares RM, Sousa MJ, Agasse A, Delrot S, Gerós H (2007) Biochemical changes throughout grape berry development and fruit and wine quality. Food (Global Science Books) 1:1–22
Coombe BG (1992) Research on development and ripening of the grape berry. Am J Enol Vitic 43:101–110
Costantini L, Battilana J, Lamaj F, Fanizza G, Grando MS (2008) Berry and phenology-related traits in grapevine (Vitis vinifera L.): from quantitative trait loci to underlying genes. BMC Plant Biol 8:38
Cuéllar T, Azeem F, Andrianteranagna M, Pascaud F, Verdeil JL, Sentenac H, Zimmermann S, Gaillard I (2013) Potassium transport in developing fleshy fruits: the grapevine inward K+ channel Vvk1.2 is activated by CIPK-CBL complexes and induced in ripening berry flesh cells. Plant J 73:1006–1018. https://doi.org/10.1111/tpj.12092
Davies C, Robinson SP (2000) Differential screening indicates a dramatic change in mRNA profiles during grape berry ripening. Cloning and characterization of cDNAs encoding putative cell wall and stress response proteins. Plant Physiol 122:803–812. https://doi.org/10.1104/pp.122.3.803
De la Vega F, Lazaruk KD, Rhodes MD, Wenz MH (2005) Assessment of two flexible and compatible SNP genotyping platforms: TaqMan® SNP genotyping assays and the SNPlex™ genotyping system. Mutat Res 573:111–135. https://doi.org/10.1016/j.mrfmmm.2005.01.008
DeBolt S, Cook DR, Ford CM (2006) L-tartaric acid synthesis form vitamin C in higher plants. PNAS 103:5608–5613. https://doi.org/10.1073/pnas.0510864103
Doligez A, Bouquet A, Danglot Y, Lahogue F, Riaz S, Meredith CP, Edwards KJ, This P (2002) Genetic mapping of grapevine (Vitis vinifera L.) applied to the detection of QTLs for seedlessness and berry weight. Theor Appl Genet 105:780–795. https://doi.org/10.1007/s00122-002-0951-z
Doligez A, Adam-Blondon AF, Cipriani G, Di Gaspero G, Laucou V, Merdinoglu D, Meredith CP, Riaz S, Roux C, This P (2006) An integrated SSR map of grapevine based on five mapping populations. Theor Appl Genet 113:369–382. https://doi.org/10.1007/s00122-006-0295-1
Don RH, Cox PT, Wainwrigth BJ, Baker K, Mattick JS (1991) ‘Touchdown’ PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res 19:4008
Duchêne E, Butterlin G, Claudel P, Dumas V, Jaegli N, Merdinoglu D (2009) A grapevine (Vitis vinifera L.) deoxy-D-xylulose synthase gene colocates with a major quantitative trait loci for terpenol content. Theor Appl Genet 118:541–552. https://doi.org/10.1007/s00122-008-0919-8
Duchêne E, Dumas V, Jaegli N, Merdinoglu D (2014) Genetic variability of descriptors for grapevine berry acidity in Riesling, Gewürztraminer and their progeny. Aust J Grape Wine Res 20:91–99. https://doi.org/10.1111/ajgw.12051
Gerber S, Rodolphe F (1994) An estimation of the genome length of maritime pine (Pinus pinaster Ati). Theor Appl Genet 88:289–292. https://doi.org/10.1007/BF00223634
Gladstones J (1992) Viticulture and environment. Winetitles, Adelaide
Grattapaglia D, Sederoff R (1994) Genetic linkage map of Eucalyptus grandis and Eucalyptus urophylla using pseudo-testcross: mapping strategy and RAPD markers. Genetics 137:1121–1137
Houel C, Chatbanyong R, Doligez A, Rienth M, Foria S, Luchaire N, Roux C, Adivèze A, Lopez G, Farnos M, Pellegrino A, This P, Romieu C, Torregrosa L (2015) Identification of stable QTLs for vegetative and reproductive traits in the microvine (Vitis vinifera L.) using a 18K Infinium chip. BMC Plant Biol 15:205. https://doi.org/10.1186/s12870-015-0588-0
Hulbert SH, Ilott TW, Legg EJ, Lincoln SE, Lander ES, Michelmore RW (1988) Genetic analysis of the fungus Bremia lactucae, using restriction fragment length polymorphisms. Genetics 120:947–958
Jackson RS (2008a) Chemical constituents of grapes and wine. In: Jackson RS (ed) Wine science: principles and applications, 3rd edn. Elsevier, Burlington, pp 270–331
Jackson RS (2008b) Site selection and climate. In: Jackson RS (ed) Wine science: principles and applications, 3rd edn. Elsevier, Burlington, pp 239–369
Jansen RC, Stam P (1994) High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136:1447–1455
Jones GV, White MA, Cooper OR, Storchmann K (2005) Climate change and global wine quality. Clim Chang 73:319–343. https://doi.org/10.1007/s10584-005-4704-2
Kearsey MJ, Pooni HS (1996) Genes, genetic markers and maps. In: Kearsey MJ, Pooni HS (eds) The genetical analysis of quantitative traits. Stanley Thornes, UK. ISBN 0-7487-4082-1, pp 101–133
Kliewer WM (1966) Sugar and organic acids of Vitis vinifera. Plant Physiol 41:923–931. https://doi.org/10.1104/pp.41.6.923
Kliewer WM (1973) Berry composition of Vitis vinifera cultivars as influenced by photo- and nycto- temperatures during maturation. Amer Soc Hort Sci J 98:153–159
Kosambi DD (1944) The estimation of map distance from recombination values. Ann Eugenics 12:172–175
Lander E, Botstein D (1989) Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199
Lange K, Boehnke M (1982) How many polymorphic genes will it take to spam the human genome? Am J Hum Genet 34:842–845
Laucou V, Launay A, Bacilieri R, Lacombe T, Adam-Blondon AF, Bérard A, Chauveau A, de Andrés MT, Hausmann L, Ibáñez J, Le Paslier M-C, Maghradze D, Martinez-Zapater JM, Maul E, Ponnaiah M, Töpfer R, Péros J-P, Boursiquot J-M (2018) Extended diversity analysis of cultivated grapevine Vitis vinifera with 10K genome-wine SNPs. PLoS One 13:e0192540. https://doi.org/10.1371/journal.pone.0192540
Lechmann EL (1975) Nonparametrics. McGraw-Hill, New York
Lijavetzky D, Cabezas JA, Ibáñez A, Rodríguez V, Martínez-Zapater JM (2007) High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology. BMC Genomics 8:424. https://doi.org/10.1186/1471-2164-8-424
Liu HF, Wu BH, Fan PG, Xu HY, Li SH (2007) Inheritance of sugars and acids in berries of grape (Vitis vinifera L.). Euphytica 153:99–107. https://doi.org/10.1007/s10681-006-9246-9
Lodhi MA, Daly MJ, Ye GN, Weeden NF, Reisch B (1995) A molecular marker based linkage map of Vitis. Genome 38:786–794. https://doi.org/10.1139/g95-100
Morreel K, Goeminne G, Storme V, Sterck L, Ralph J, Coppieters W, Breyne P, Steenackers M, Georges M, Messens E, Boerjan W (2006) Genetical metabolomics of flavonoid biosynthesis in Populus: a case study. Plant J 47:224–237
Or E, Baybik J, Sadka A, Saks Y (2000) Isolation of mitochondrial malate dehydrogenase and phosphoenolpyruvate carboxylase cDNA clones from grape berries and analysis of their expression pattern throughout berry development. J Plant Physiol 157:527–534. https://doi.org/10.1016/S0176-1617(00)8018-X
Pego JV, Smeekens SCM (2000) Plant fructokinases: a sweet family get-together. Trends Plant Sci 5:531–536. https://doi.org/10.1016/S1360-1385(00)01783-0
Pflieger S, Lefebvre V, Causse M (2001) The candidate gene approach in plant genetics: a review. Mol Breed 7:275–291. https://doi.org/10.1023/A:1011605013259
Riaz S, Dangl GS, Edwards KJ, Meredith CP (2004) A microsatellite marker based framework linkage map of Vitis vinifera L. Theor Appl Genet 108:864–872. https://doi.org/10.1007/s00122-003-1488-5
Sato A, Yamada M, Iwanami H, Hirakawa N (2000) Optimal spatial and temporal measurement repetition for reducing environmental variation of berry traits in grape breeding. Sci Hortic 85:75–83
Schnarrenberger C, Martin W (2002) Evolution of the enzymes of the citric acid cycle and the glyoxylate cycle of higher plants. Eur J Biochem 269:868–883. https://doi.org/10.1046/j.0014-2956.2001.02722.x
Sweetman C, Deluc LG, Cramer GR, Ford CM, Soole KL (2009) Regulation of malate metabolism in grape berry and other developing fruits. Phytochemistry 70:1329–1344. https://doi.org/10.1016/j.phytochem.2009.08.006
Terrier N, Glissant D, Grimplet J, Barrieu F, Abbal P, Couture C, Ageorges A, Atanassova R, Léon C, Renaudin JP, Dédadéchamp F, Romieu C, Delrot S, Hamdi S (2005) Isogene specific oligo array reveal multifaceted changes in gene expression during grape berry (Vitis vinifera L.) development. Planta 222:832. https://doi.org/10.1007/s00425-005-0017-y
Tobler AR, Short S, Andersen MR, Paner TM, Briggs JC, Lambert SM, Wu PP, Wang Y, Spoonde AY, Koehler RT, Peyret N, Chen C, Leong LN, Ma CN, Rosenblum BB, Day JP, Ziegle JS, De La Vega RM, Rhodes MD, Hennessy KM, Wenz HM (2005) The SNPlex genotyping system: a flexible and scalable platform for SNP genotyping. J Biomol Tech 16:398–406
Troggio M, Malacarne G, Coppola G, Segala C, Cartwright DA, Pindo M, Stefanini M, Mank R, Moroldo M, Morgante M, Grando MS, Velasco R (2007) A dense single-nucleotide polymorphism-based genetic linkage map of grapevine (Vitis vinifera L.) anchoring pinot noir bacterial artificial chromosome contigs. Genetics 176:2636–2650. https://doi.org/10.1534/genetics.106.067462
Van Ooijen JW, Voorrips RE (2001) JoinMap® 3.0: software for the calculation of genetic linkage maps. Plant Research International, Wageningen, The Netherlands
Van Ooijen JV, Boer MP, Jansen RC, Maliepaard C (2002) MapQTL® 4.0, software for the calculation of QTL position on genetic maps. Plant Research International, Wageningen, The Netherland
Viana AP, Riaz S, Walker MA (2013) Genetic dissection of agronomic traits within a segregating population of breeding table grapes. Genet Mol Res 12:951–964
Viana AP, Resende MDV, Riaz S, Walker MA (2016) Genome selection in fruit breeding: application to table grapes. Sci Agric 73:142–149. https://doi.org/10.1590/0103-9016-2014-0323
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78. https://doi.org/10.1093/jhered/93.1.77
Walker AR, Lee E, Bogs J, McDavid DAJ, Thomas MR, Robinson SP (2007) White grapes arose through the mutation of two similar and adjacent regulatory genes. Plant J 49:772–785. https://doi.org/10.1111/j.1365-313X.2006.02997.x
Wei X, Sykes SR, Clingeleffer PR (2002) An investigation to estimate genetic parameters in CSIRO’s table grape breeding program. 2. Quality characteristics. Euphytica 128:343–351
Weinl S, Kudla J (2009) The CBL-CIPK Ca2+-decoding signaling network: function and perspectives. New Phytol 184:517–528. https://doi.org/10.1111/j.1469-8137.2009.02938.x
Welter LJ, Baydar NG, Akkurt M, Maul E, Eibach R, Töpfer R, Zyprian EM (2007) Genetic mapping and localization of quantitative trait loci affecting fungal disease resistance and leaf morphology in grapevine (Vitis vinifera L.). Mol Breed 20:359. https://doi.org/10.1007/s11032-007-9097-7
Wen J, Can VT, Zhang Y-M (2013) Multi-QTL mapping for quantitative traits using distorted markers. Mol Breed 31:395–404. https://doi.org/10.1007/s11032-012-9797-5
Xu S (2008) Quantitative trait locus mapping can benefit from segregation distortion. Genetics 180:2201–2208
Xu S, Hu Z (2009) Mapping quantitative trait loci using distorted markers. Int J Plant Genomics 2009:410825. https://doi.org/10.1155/2009/410825
Yang S, Fresnado-Ramírez J, Sun W, Manns DC, Sacks GL, Mansfield AK, Luby JJ, Londo JP, Reisch BI, Cadle-Davidson LE, Fennell AY (2016) Next generation mapping of enological traits in an F2 interspecific grapevine hybrid family. PLoS One 11:e0149560. https://doi.org/10.1371/journal.pine.0149560
Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, Malerba G, Bellin D, Pezzotti M, Delledonne M (2010) Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol 152:1787–1795. https://doi.org/10.1104/pp.109.149716
Zhang L, Wang S, Li H, Deng Q, Zheng A, Li S, Li P, Li Z, Wang J (2010) Effects of missing marker and segregation distortion on QTL mapping in F2 populations. Theor Appl Genet 121:1071–1082
Zhao YH, Guo YS, Lin H, Liu ZD, Ma HF, Guo XW, Li K, Yang XX, Niu ZZ, Shi GG (2015) Quantitative trait locus analysis of grape weight and soluble solid content. Genet Mol Res 14:9872–9881. https://doi.org/10.4238/2015
Zhao YH, Su K, Guo YH, Ma HF, Guo XW (2016) Molecular genetic map construction and QTL analysis of fruit maturation period in grapevine. Genet Mol Res 15(2). https://doi.org/10.4238/gmr.15028040
Acknowledgments
The authors wish to thank A. Fuentes-Denia for technical assistance and J.A. Martínez-Jiménez for plant management in the field. We are also grateful to the research team of Dr. Stella Grando at the Centre for Research and Innovation, Fondazione Edmund Mach (FEM), in San Michele all’Adige (Italy), for their help in the development of new SNP-based markers. A. Bayo-Canha was a pre-doctoral fellow of the Instituto Murciano de Investigación y Desarrollo Agrario y Alimentario.
Funding
This study was financially supported by projects RTA2007-00043 and RTA2011-00029-C02-02 from the Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, co-financed by the European Regional Development Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bayo-Canha, A., Costantini, L., Fernández-Fernández, J.I. et al. QTLs Related to Berry Acidity Identified in a Wine Grapevine Population Grown in Warm Weather. Plant Mol Biol Rep 37, 157–169 (2019). https://doi.org/10.1007/s11105-019-01145-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-019-01145-6