
Abstract
A novel genetic map of pomegranate (Punica granatum L.) enriched with quantitative trait loci (QTLs) for seven traits was constructed using an F2 population. The population was generated from a cross between two varieties of P. granatum: “Nana” and “Black.” Phenotyping of 76 F2 plants was conducted over two seasons. The map includes 1092 SNP markers which were newly developed from de novo transcriptome assembly and a comparison of the sequences of the two varieties or accessions. The map covers 1141 centimorgans (cM) with an average of 1.17 cM between markers over 11 linkage groups. Twenty-five QTLs were identified for fruit traits and plant size. The map includes QTLs for total soluble solids (TSS), fruit weight and perimeter, seed hardness, aril color and weight, and plant height. In an effort to explore the potential of the Agricultural Research Organization (ARO) pomegranate germplasm collection to associate traits with gene markers, an association study was conducted using a set of 346 SNPs described in an earlier study. Of these SNPs, 233 were mapped on the genetic map and found to be distributed in the different linkage groups (LGs). The associated traits were anchored to the map by these common markers. The F2 population described here and the corresponding genetic map provide a useful resource for further genomics and genetic studies of pomegranate, as well as a reservoir of markers for fruit improvement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pomegranate (Punica granatum L.) belongs to the Lythraceae family and is grown worldwide. According to Masoud et al. (2005), pomegranate is a diploid plant (2n = 16). The pomegranate fruit has a pleasant taste and an attractive appearance, and many parts of the plant are used for commercial purposes. Traditional and modern medicine indicate that the pomegranate tree, its flowers, and its fruit have a high content of important metabolites with health-promoting properties. The agricultural value of the pomegranate tree and its products is constantly increasing as its various benefits are being explored by modern science. Numerous scientific studies (Reviewed in Seeram et al. (2006) and Holland and Bar-Ya’akov (2014)), with human patients, animals, and cell cultures suggest that pomegranate-derived substances have antioxidant, anti-carcinogenic, anti-inflammatory, antimicrobial, and estrogenic properties. The prominent bioactive phytochemicals present in pomegranate are various polyphenolic compounds, including ellagitannins and flavonoids.
Worldwide genetic and molecular studies of pomegranate have so far been done mainly on local collections of accessions (Currò et al. 2010; Hasnaoui et al. 2010, 2012; Pirseyedi et al. 2010; Basaki et al. 2011; Sarkhosh et al. 2011; Soriano et al. 2011; Jian et al. 2012; Noormohammadi et al. 2012; Zhang et al. 2012; Hajiahmadi et al. 2013; Singh et al. 2013; Ferrara et al. 2014; Orhan et al. 2014). These studies involved the usage of various DNA markers in order to distinguish between accessions within local collections and in an effort to associate genetic markers with traits. The main molecular markers that were used to screen pomegranates were simple sequence repeats (SSRs) (Currò et al. 2010; Hasnaoui et al. 2010, 2012; Pirseyedi et al. 2010; Basaki et al. 2011; Soriano et al. 2011; Jian et al. 2012; Noormohammadi et al. 2012; Ferrara et al. 2014). However, studies with other molecular markers such as random amplification of polymorphic DNA (RAPD) (Noormohammadi et al. 2012; Zhang et al. 2012; Singh et al. 2013; Orhan et al. 2014), amplified fragment length polymorphisms (AFLP) (Sarkhosh et al. 2011), or internal transcribed spacers (Noormohammadi et al. 2012; Hajiahmadi et al. 2013; Singh et al. 2013) were reported in the literature. In most cases, no overlap was found between the classification by genotype and by the phenotype.
Recently, a wide molecular genetics study, which includes de novo establishment of the pomegranate transcriptome and the identification of several thousand SNP markers, was reported (Ophir et al. 2014). A subset of 346 SNPs was used to investigate the genetic structure of the Agricultural Research Organization (ARO) germplasm collection. The ARO collection includes more than 100 accessions, local accessions, and accessions introduced from foreign countries such as, India, China, Central Asia, the USA, Spain, Turkey, and the Mediterranean region (Ophir et al. 2014). The selected SNPs are highly diverse in the ARO pomegranate germplasm (half of the SNPs had PIC ≥0.43). Moreover, this set of SNPs was highly polymorphic, with only 10 % loci with minor allele frequencies of (MAF <0.05). These SNPs were successfully used to classify the ARO pomegranate collection by hierarchical clustering to three statistically significant groups (bootstrap value >90 %) (Online Resource Table 1). The STRUCTURE program was used suggesting that there are two major subpopulations.
Few genetic studies on pomegranate associated markers with traits (Basaki et al. 2011, 2013; Singh et al. 2015). Basaki et al. (2011, 2013) screened seven SSRs on 202 pomegranate accessions representing 22 provinces of Iran. Despite the limited number of markers, the associated SSRs explained up to 29.6 % of the variation for 20 individual traits, including fruit and flower traits, leaf size, and shoot production. The Indian pomegranate collection was genotyped with 44 microsatellites (Singh et al. 2015); three traits were found associated to three different markers.
Genetic maps based on segregating populations are powerful tools for linking genes and characters. A saturated linkage map should discover possible links between genotype and phenotype and also identify efficient markers for use in marker-assisted breeding. In melon, markers were mapped close to the trait of fruit acidity (Danin-Poleg et al. 2002; Harel-Beja et al. 2010). Another example is the genetic map of Artemisia annua L. that identified loci affecting yield that co-located with quantitative trait loci (QTLs) for artemisinin concentration, thus representing targets for a breeding program (Graham et al. 2010). In trees such as apple, QTL analysis and the candidate gene approach led to two haplotypes that were characterized and associated with fruit flesh browning (Di Guardo et al. 2013). In apricot and peach, QTL for chilling requirements was detected with a major locus at LG1 (Olukolu et al. 2009; Fan et al. 2010).
In the present study, an enriched pomegranate map was constructed based on an F2 population developed from a cross between two varieties, “Nana” and “Black.” These varieties differ in fruit quality traits, including aril characters such as sweetness, acidity, and color, seed hardness, external peel characters, and fruit size. In addition, the Nana parental variety is a dwarf pomegranate while the Black variety has a normal pomegranate tree size. Thus, the F2 population generated from a cross between Nana and Black is an excellent tool for mapping fruit quality traits along with tree growth characteristics such as tree size. The pomegranate genetic map was established with ~1100 SNP markers that were derived from transcripts. QTLs for total soluble solids (TSS), fruit size, aril color, seed hardness, and plant size were mapped. Finally, association mapping was done for fruit traits in the ARO collection.
Materials and methods
Plant material
A segregating F2 population (n = 76) was constructed from a cross between Black (P.G.127-28, according to the nomenclature of the ARO collection at Newe Ya’ar) cultivar and Nana seedling selection (P.G.232-243, P. granatum var. Nana). Nana seedlings are characterized as dwarf, bushy plants that have very small and sour fruits with hard seeds and a green to red peel. Black is a domesticated cultivar characterized as a deciduous normal-sized tree with a very distinct deep-purple peel. The Black accession has a fruit of medium size, with sweet taste and soft seeds. The parents are part of the ARO collection (Holland et al. 2009). The F1 plant, which was the progenitor of the F2 population, was planted in 2008 and was self-pollinated at 2009 (Nana pomegranate flower and fruit few months after germination). The F2 population was planted in 2010 at the Newe Ya’ar Research Center in northern Israel.
The ARO’s pomegranate germplasm collection is located at the Newe Ya’ar Research Center (http://igb.agri.gov.il/main/index.pl?page=22). It consists of more than 150 accessions originating from different geographical locations all over the world and in particular from Israel (Holland et al. 2009). For the association study, 94 accessions were analyzed (Online Resource Table 1).
Phenotype evaluation of a segregating F2 population
The two parents, the F1 plant and the F2 population, were grown in the orchard at Newe Ya’ar, Israel. Three mature fruits from each plant of the F2 population were harvested, from different parts of the tree in two successive fruiting seasons (2 and 3 years after planting). Maturity was determined by the combination of aril and peel color and by tasting for astringency and sourness. Each fruit was examined for the following traits: fruit perimeter, fruit and aril weight, TSS, peel and aril color, aril taste, and seed hardness. Peel and aril color, aril taste, and seed hardness were evaluated by visual screening and by tasting the fruit. These traits were graded from 1 to 5, where 1 is the least favored and 5 is the most favored. Fruit perimeter was measured by measuring tape; fruit weight was measured by scale (Merav 2000, Shekel Scales Ltd. Israel) and aril weight was measured by a digital scale (BBA-1200, M.R.C. Israel). TSS was measured by a hand refractometer (ATC-1 Atago Japan). Plant height was measured in the second, third, and fifth year after planting by measuring tape. Plant growth was calculated by subtracting the height in the second year from the height in the third year.
Fruits from the parental lines were harvested and evaluated like the fruits of the segregating population in the 2011 and 2014 fruit seasons; in addition, they were characterized as part of the ARO collection for at least 5 years. The F1 plant was examined during 2011, 2012, and 2014. The heights of mature plants of the maternal parent and of F1 plant were measured by measuring tape. The height of mature plants of the parental variety was evaluated.
Phenotypic evaluation of the pomegranate variety collection
The pomegranate collection was established in 1978, and since then, new accessions have been added to the orchard. Once each of the accessions reached the fruiting stage, the tree and fruit were characterized for at least 5 years. Characterization included, among other traits, date of maturity (when the fruit reached edible quality), fruit size (weight and diameter), peel and aril colors (visual evaluation), taste (organoleptic description), and TSS (by refractometer (ATC-1 Atago, Japan)). Total acid content was calculated as percentage of citric acid by titrating the pomegranate juice with a solution of NaOH 0.1 N. Five mature fruits were harvested from two different trees of each accession, from different parts of each tree. The characterizations of the different traits were averaged over 5 years (Online resource Table 1).
DNA extraction
The DNA extraction protocol was based on Porebski et al. (1997) with few modifications. Young leaves from the parental lines, the F1 parent of the F2 population, and the population plants were used for DNA preparation, 0.5 g resuspended in a 6-ml extraction buffer. The chloroform–octanol solution was replaced with chloroform–isoamyl alcohol. DNA was precipitated with sodium acetate instead of sodium chloride.
Statistical analysis
Means, standard deviations, trait distribution, pairwise correlation, ANOVA and Welch ANOVA, Shapiro–Wilk W test, and Wilcoxon/Kruskal–Wallis test (rank sums) analyses were conducted with the JMP program, v. 7.0 (SAS Institute Inc., Cary, NC).
Marker analysis and map construction
Array and probe design
SNP markers were discovered by whole-transcriptome sequencing of two mRNA samples that were extracted from the accessions Nana and Black (Ophir et al. 2014) and from the P.G.160-61 and P.G.100-1 accessions (unpublished data) on 454-GS FLX Titanium. Total RNA was extracted from a pool of tissues including leaves, roots, flowers (petals, anthers, ovaries), and fruit (stage 3 (Ben-Simhon et al. 2011)) of Nana and Black. The same pool of tissues, excluding the roots, was used for RNA extractions from the P.G.160-61 and P.G.100-1 accessions. De novo assembly was performed separately on pairs of accessions’ sequencing with MIRA (Chevreux et al. 2004), and polymorphic loci of low quality (Q < 30; PHRED scale; (Ewing et al. 1998)) were filtered out. The repertoire of SNPs for array design was composed of three SNP subsets: homozygous but polymorphic between Nana and Black (6001), homozygous but polymorphic between P.G.160-61 and P.G.101-2 (4311), and heterozygous at least in one of the parental accessions, i.e., Nana or Black (4800). Altogether the array was composed of 15,494 SNPs. Four probes were designed for each SNP to fill an Agilent 60K SurePrint array. Two replicate probes were designed for each allele, one to detect the forward DNA strand and the other to detect the reverse DNA strand. The probes were T m optimized, as described by Rubinstein et al. (2015).
Array hybridization and signal preprocessing
Labeling of DNA pools and hybridization were performed at the DNA array unit of the Weizmann Institute (Rehovot, Israel) following the Agilent CGH protocol for comparative genomic hybridization (http://www.agilent.com); hybridization temperature was modified to 55 °C. Emission intensities (signal values) were preprocessed in three steps: quantile normalization across arrays and dyes, removal of spatial slide defects, e.g., scratches, bubbles, etc. by the method described in Chai et al. (2010), and subtracting each probe by its median.
Genotype calling
An SNP genotype call was performed by k-means partition clustering. Differential signals (M = allele X–allele Y) were calculated for forward and reverse probes. All F2 hybridization signals per SNP were used as two vectors of forward-M and reverse-M, and a k-means clustering, with k = 3, was performed (see an example in Liu et al. (2003)). The genotype call of a cluster was called by projecting the medoid of the clusters on the diagonal of the forward-M and reverse-M axes. The cluster with the maximal medoid was typed as XX, the cluster with the median medoid was typed as XY, and the cluster with minimal medoid was typed as YY. Only SNPs with three distinct clusters, i.e., clusters with a minimal Bhattacharyya distance of 0.5 were selected for downstream analysis. In addition, SNPs were selected only if the parent F1 plant was heterozygous.
Map construction and QTL analysis
Mapping was performed using JoinMap® 3.0 software (Van Ooijen and Voorrips 2001). Markers were grouped at a minimum LOD score of 4.0 and a recombination frequency value of 0.4. JoinMap® 3.0 software uses Kosambi mapping functions to translate recombination frequency into map distance.
Using the MapQTL® 5 software (Van Ooijen 2004), QTLs and their significance were calculated using interval mapping (IM), multiple QTL model (MQM), and permutation analysis. A QTL was determined significant when its LOD score was higher than the calculated threshold (1000 permutation at p = 0.05).
Map association of traits within the germplasm collection
Association between traits and SNPs within the germplasm collection was analyzed using the TASSEL (version 3.0.164) program (Bradbury et al. 2007). The general linear model (GLM) function was used for association analysis. For each marker–trait combination, GLM finds the ordinary least squares solution. The p value was specified as less than 10−4. Associations within the germplasm collection were excluded if one of the allele compositions was presented in more than 80 % of the accessions within the collection. Significant associations were analyzed by the JMP program, v. 7.0 (SAS Institute Inc., Cary, NC) using an ANOVA test if the trait was normally distributed (fruit weight and TSS); otherwise, they were analyzed by Wilcoxon/Kruskal–Wallis tests (rank sums). In addition, mixed linear model (MLM) was analyzed using the TASSEL (version 3.0.164) program (Bradbury et al. 2007) that implement the kinship and structure analysis of the germplasm collection.
Results
Development and characterization of an F2 population
An F2 population, designated “Nana × Black,” was developed from a cross between Nana (P.G.232-243, P. granatum var. Nana) and Black (P.G.127-28) cultivars. The population was polymorphic in many characteristics (Fig. 1), including growth rate and fruit traits.

Polymorphism within the Nana × Black F2 population. The segregating population is polymorphic regarding plant height (a), fruit size (b), and aril color and size (c). Arrows indicate two low trees in a raw with high trees
The population was genotyped using the SNP CHIP. All the SNPs were newly developed markers from the pomegranate transcriptome (Ophir et al. 2014). Markers of 1835 were selected as described in genotype calling in the “Materials and methods” section. Two hundreds and fifteen of the SNPs were not polymorphic.
Although the size of the F2 population is not high, 3102 recombination events occurred in the 76 F2 plants. The number of recombination events per plant ranged from 25 to 73, with a mean of 45.6.
The heterozygosity in the population was 47 %, calculated as the average of the percent of heterozygous markers for each plant of the population. The percent of heterozygosity was lower than 30 % in only one progeny that had 28 % heterozygosity. Six plants had over 60 % heterozygosity; the highest percentage was 69 %. The alleles of the maternal parent were 1.3 times more abundant in the F2 population than the alleles of the paternal parent. Five progenies had more than twice the maternal alleles than the paternal alleles.
Development of an enriched linkage map
Total of 1625 SNP markers were analyzed by the JoinMap® 3.0 program. The segregation of 298 markers was not Mendelian segregation as was tested by chi-square test (χ 2 > 10.4, df = 2) (Van Ooijen and Voorrips 2001). In addition, 235 SNPs were removed since they were identically segregated. Overall, a total of 1092 SNP markers were used to construct the linkage map that was named Nana × Black (Fig. 2 and Online Resource Table 1).



Genetic map of pomegranate based on the Nana × Black F2 population including QTLs for fruit traits and plant height and data from map association of fruit quality traits in the ARO collection of varieties. LG numbers are marked above each group. Distances in centimorgans are written on the left side and marker names are on the right side of each linkage group. Markers that were genotyped within the variety collection are written in purple and end with _fl. Significant QTLs (by permutation test) analyzed by MQM analysis are represented by red lines. Rectangles and blue arrows indicate SNPs that are associated with fruit traits within the variety collection by MLM analysis
The Nana × Black F2 genetic map covers 960 centimorgans (cM) and is divided into seven major linkage groups (LGs), with an average of 1.17 cM per marker. The largest linkage group is LG1, which covers 155 cM (Online Resource Table 2). Interestingly, when 176 of the deviant distributed markers were included in the map calculations, four additional linkage groups were created; therefore, overall, the map covers 1141 cM. Two of the additional groups (LG8 and LG9) included 67 and 58 markers, respectively, and covered more than 60 cM each (Fig. 2). The additional deviant markers were located to LG2 (2), LG3 (5), LG6 (12), LG10 (27), and LG11 (5) (marked in Online Resource Table 3). The deviation from the Mendelian segregation was mainly due to a maternal homozygote genotype which was more frequent than the paternal genotype. In addition, there were 281 markers for which one of the parents was a heterozygote; they were spread all over the linkage groups.
Of the mapped SNPs, 233 were previously genotyped within the ARO collection (Ophir et al. 2014). The names of these markers are bolded and end with “_fl”. These markers were distributed all over the map (Fig. 2 and Online Resource Table 2 and 3).
Fruit traits
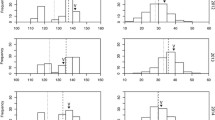
The F2 population was used to phenotypically characterize and map variation in fruit qualities such as size, taste, seed hardness, and color. The F2 plants as well as the F1 plant (parent of the mapping population) and its parental accessions were phenotyped during at least two growing seasons: the F1 in 2011, 2012, and 2014 and the F2 population in 2012 and 2013. The parent trees (Nana and Black) were characterized as part of the ARO collection for at least 5 years. In addition, they were characterized in 2011 and 2014. Mean and minimum and maximum values of the fruit traits for the parents and the F1 and the F2 population are presented in Table 1. Fruit perimeter, fruit weight, and aril weight were distributed as normal distribution in 2013 (Fig. 3) according to the Shapiro–Wilk goodness-of-fit test. The other three traits, aril color, seed hardness, and TSS, were not normally distributed (p ≤ 0.01) in 2013; however, in 2012, only seed hardness was not normally distributed (data not shown).

Distribution of nine traits within the F2 population. Seven fruit traits were evaluated in the segregating population in 2013: fruit perimeter (a), fruit weight (b), aril weight (c), aril color (d), seed hardness (e), and TSS (f). The results are averages of three fruits per plant. Plant height (g) and plant growth rate (h) were measured in the third year of growth. The distribution is accepted as normal when the prob >0.001 as was calculated by Shapiro–Wilk W test. Orange, purple, and green arrows point to the Nana, Black, and F1 parents values in 2014, respectively
The correlation between fruit perimeter and fruit weight was high, 0.86 and 0.94 for 2012 and 2013, respectively (Table 2). In addition, aril weight was also highly correlated with fruit weight, 0.53 and 0.68 for 2012 and 2013, respectively. The other fruit traits were correlated at lower levels (r ≤ 0.56).
The year effect was not significant according to Wilcoxon/Kruskal–Wallis Tests (p ≥ 0.01) for three traits: aril weight, color, and seed hardness. However, the year effect was significant (p ≤ 0.01) for three fruit traits: fruit perimeter, weight, and TSS, possibly since the trees were young. Young pomegranate trees tend to yield slightly different fruits than fully mature trees. Yield load can also affect these traits. The parental lines are mature trees; the year effect within the parents was not significant (p ≥ 0.01) for the various traits, except of fruit perimeter. Therefore, it may be assumed that fruit weight and TSS were affected by the age of the tree over the F2 population. Nevertheless, it may indicate a genotype × environment interaction.
Transgressive segregation was detected for five fruit traits (Fig. 3): fruit and aril weight, fruit perimeter, aril color, and TSS. Regarding the fruit weight trait, 16 plants had heavier fruits than the fruits of the Black variety, and only two plants with fruits lighter than Nana, were present in the population. With respect to aril weight, on the other hand, nine plants had fruits with smaller arils than Nana and only one had heavier arils than Black. Twenty-one plants had fruits with larger perimeters than Black and only one that was smaller than the fruits of Nana. The aril color of both parents was evaluated with high values, Nana arils are red and Black arils are dark pink. On the other hand, most of the plants in the population (61) were evaluated with lower scores for their aril color as compared to Nana. The TSS levels of Black were high (about 17.7) and the TSS levels of Nana were low (about 14.3). Ten of the plants had much lower TSS levels and three had higher TSS levels than Black.
Plant growth
The maternal parent of the segregating population was Nana (P.G.232-243, P. granatum var. Nana), which is a dwarf pomegranate. The mature plant height of this cultivar is 70 cm. The other parent, Black, has normal pomegranate plant height, about 3 m. The F1 plant was medium sized, about 2.7 m at 6 years after planting. The F2 population plant height was highly polymorphic (Fig. 1a), it was measured in the second and third year after planting. Plant height segregated from 0.6 to 3 m when measured 3 years from planting. During the third year, the growth rate distribution was from 2 to 187 cm/year. The distribution of the two traits was normal according to the Shapiro–Wilk goodness-of-fit test (p ≥ 0.01) (Fig. 3h, i). There was a high correlation (r = 0.75) between plant height in the fifth year of growth and in the third year of growth.
The correlations between plant height and fruit traits were low (r ≤ 0.42), as shown in Table 3.
Fruit traits within the pomegranate collection
The ARO pomegranate germplasm collection includes more than a hundred accessions from all over the world (Ophir et al. 2014). The fruits of each variety were evaluated during at least 3 years. Six fruit traits were analyzed within the collection (Table 4). Fruit weight, acid content, TSS to acid ratio, and seed hardness were highly polymorphic within the collection, as can be concluded from the traits’ high standard deviations (Table 4). However, TSS and aril weight were less variable within the collection. Fruit weight, TSS, and aril weight were normally distributed as was examined by Shapiro–Wilk W test (p ≥ 0.01).
Previous work divided the ARO pomegranate germplasm collection to two main groups by STRUCTURE analysis (Ophir et al. 2014); however, this division was not significant (p ≥ 0.05) for the fruit traits, except for seed hardness.
Identification of QTLs in the Nana × Black F2 population
QTLs were analyzed by MQM analysis, which narrows down the QTL around the most significant markers (Van Ooijen 2004). The QTL threshold base was calculated by the permutation analysis (1000 permutation at p = 0.05), ranging from 4.0 for aril color, aril weight, and fruit weight to 26.4 for seed hardness in the 2013 fruit season. Twenty-five QTLs were analyzed for seven different traits (Table 5 and Fig. 2), including plant growth rate, fruit weight and perimeter, aril weight and color, seed hardness, and TSS of the juice. LOD values varied from 39.45 to 3.97. The highest LOD value was 39.45 for seed hardness at LG1 (114–125 cM). The QTLs were analyzed at six linkage groups out of ten. Only two QTLs, one for seed hardness and one for fruit weight, were statistically significant for both 2012 and 2013 fruit seasons; all the other QTLs were significant for one fruit season only suggesting age effect on the traits within the population.
The highest significant QTL was for seed hardness (Seed hardness 1–1) at LG1 (114–125 cM) between the markers c22061_572 and c21761_335. It had a very high LOD value of 18.33 or 39.45 for the 2012 and 2013 fruit seasons, respectively, and can explain 65 or 94 % of the trait for the 2012 or 2013 fruit season, respectively. The hard seed trait was contributed by the Nana parent. Plants with Nana alleles at this locus are with low evaluation score, which means harder seeds.
Plant height was measured in the third and fifth year of growth. The same two QTLs were identified for both measurements. The more significant QTL (plant height 2-1) is at LG2 (51–80 cM), where the markers c6449_982, c15646_223, c12777_264, and c6269_275_fl are mapped. The LOD values of this QTL were 11.44 for the third year height, which explains 38.6 % of the trait, and 6.3, which explains 25 % of the trait for the fifth year height. The additional QTL (plant height 3-1) is at LG3 (37 cM) with a LOD value higher than 4.6, which explains about 15 % of the trait. Two QTLs for fruit size traits were identified in close proximity to the position of plant height 2-1 QTL. A QTL for fruit weight (fruit weight 2-1) was localized at LG2 (52–55 cM) with a LOD value of 5.97; the marker c12777_264 had the highest LOD value at this QTL. This QTL was analyzed for two successive fruit seasons. In addition, a QTL for fruit perimeter (fruit perimeter 2-1) was detected at LG2 (45–51 cM) with a LOD value of 6.49; the highest LOD value was by the marker c6449_982. At the same location, a QTL for aril weight (aril weight 2-2) was localized with a LOD value of 7.35. The juice TSS trait, as measured in the fruit season of 2013, also had a QTL (TSS 2-2) at LG2 (52 cM) with a LOD value of 6.76. It seems that the LG2 (50–55 cM) region contained sites for several fruit quality traits and for the plant height trait. However, all these fruit traits have more than one QTL.
There are two QTLs for fruit weight with almost the same LOD values, one (fruit weight 1-1) at LG1 (106 cM) with a LOD value of 7.64, the other one (fruit weight 3-1) at LG3 (28–30 cM) with a LOD value of 7.82. An additional smaller QTL (fruit weight 7-1) was located at LG7 (132 cM) with a LOD value of 3.97. Surprisingly, at this locus, the Nana alleles contributed to the existence of heavier fruits. The four QTLS for fruit weight were analyzed for the 2013 fruit season, each of the main three, at LG1, LG2, and LG3, which explains about 20 % of the trait. For the 2012 fruit season, only one QTL was analyzed at LG2 (52–55 cM), at the same location as in the 2013 season, which explains 32.1 % of the trait.
Fruit perimeter in the 2013 fruit season had two QTLs, one at LG2, fruit perimeter 2-1, as described, in the region (45–51 cM), and it explains 19.4 % of the trait. An additional QTL (fruit perimeter 2-3) with a lower LOD value was detected at another part of LG2 (121–124 cM). A single QTL was analyzed for the 2012 season (fruit perimeter 2-2) at LG2 (61 cM). This QTL was more significant than the QTLs identified for the 2013 fruit season, with a LOD value of 8.62, and it explains 42.3 % of the trait.
Six QTLs were identified for the aril weight trait. Only one of these was found for the 2013 season (aril weight 2-2), which explains the highest percent of the trait (36.5 %). The other five QTLs were analyzed in the 2012 fruit season, the most significant (LOD = 8.38) was aril weight 3-2 at LG3 (122 cM), close to SNP c15747_186. There is an additional QTL (aril weight 3-1) at LG3 with a lower LOD value (LOD = 3.79). Another highly significant QTL (LOD = 8.11) is positioned at LG10 (aril weight 10-1). Two separate QTLs (aril weight 2-1 and aril weight 2-3) were at LG2 (37 and 107 cM, respectively), both with a LOD value of about 5.5, these QTLs are not at the same position as aril weight 2-2.
Aril color was evaluated and scored from 1 to 5. The evaluation outcome was analyzed as a continuous trait. Five QTLs were analyzed for this trait. One of them (aril color 1-2) for the 2012 fruit season is at LG1 (67–69 cM). The other four aril color QTLs were analyzed for the 2013 fruit season, including an additional one at LG1 (aril color 1-1), but at a different position (22 cM), one at LG2 (aril color 2-1), one at LG3 (aril color 3-1), and one at LG5 (aril color 5-1). In QTL aril color 5-1, the Nana alleles reduced the evaluation score, while in all the other QTLs, the Nana alleles increased the evaluation scores, which meant increasing the aril red color.
TSS of the aril juice was assessed for the fruits of the population in the 2012 and 2013 fruit seasons. Two QTLs were analyzed at LG2, one for the 2012 fruit season (TSS 2-1) and TSS 2-2 in the 2013 fruit season. The two QTLs had a LOD value of about 6. Although the two QTLs are located on LG2, they are separated by 20 cM. The Black alleles contributed to higher TSS values.
Association study
The ARO germplasm collection was genotyped with 346 SNPs by Fluidigm-EP platform (Ophir et al. 2014). Association between fruit traits and polymorphic SNPs was analyzed by the TASSEL program using initially the GLM analysis (Table 6). SNPs found to be significantly associated by the GLM (p ≤ 10−4) were verified for normal allele distribution (p ≥ 0.01), and the significance of the association was confirmed by ANOVA or Wilcoxon/Kruskal–Wallis tests (Online Resource Table 4). The associations were among seed hardness, aril weight, fruit weight, and TSS. The associated traits were anchored to seven linkage groups on the genetic map by common markers (Fig. 2). A marker’s R 2 is the portion of total variation explained by that marker. The maximum R 2 value (0.3) was found between SNP c4861_219 at LG2 (22.141 cM) and seed hardness. Fruit weight trait was the only trait that overlapped between the markers that were associated by association mapping and linked by QTL mapping, at two different locations. The QTL fruit weight 1-1 at LG1 (106 cM) with a high LOD value of 7.64 is close to SNP c7552_803_fl positioned at LG1 (104 cM). The other common position was at LG2, where the QTL fruit weight 2-1 at LG2 (52–55 cM), which was significant for both the 2012 and the 2013 seasons, is close to the associated SNP c8107_922_fl at LG2 (43 cM). When TASSEL was used to take in account the kinship and Q value from structure analysis within the germplasm collection (MLM), the corrected results yielded the data presented in Table 7. It can be seen that only acid content and fruit weight were significantly associated (p ≤ 10−3).
Discussion
Based on novel SNP markers and an F2 population, we were able for the first time to assemble a genetic map for pomegranate. Although several types of genetic markers were used to genotype some of the world pomegranate collections (Currò et al. 2010; Hasnaoui et al. 2010, 2012; Pirseyedi et al. 2010; Basaki et al. 2011; Sarkhosh et al. 2011; Soriano et al. 2011; Jian et al. 2012; Noormohammadi et al. 2012; Zhang et al. 2012; Hajiahmadi et al. 2013; Singh et al. 2013; Ferrara et al. 2014; Orhan et al. 2014), these sets of markers were separately used for each collection and no general reference for relative position of the various markers was available. The Nana × Black genetic map provides for the first time an important reference tool of the pomegranate genome for positioning markers and traits. This genetic linkage map may be useful as a basic pomegranate map. Since this map includes more than 230 SNPs that are polymorphic within the variety collection, it can be of relevance for all pomegranate varieties. The current pomegranate genetic map is spread over 1141 cM. The markers are spread at an average of 1.17-cM intervals and the 1200 SNP markers are all positioned within transcribed genes. Because these genetic markers are within genes, they specify genes as anchors for genetic markers even in populations where the original SNPs themselves are not polymorphic.
The Nana × Black F2 population was chosen for the construction of the genetic map because the two parent accessions Nana and Black differ in many characteristics, and the resulting F2 population was expected to be genetically diverse. Nana and Black differ in important characteristics including fruit quality parameters, tree size and growth habit. Previous genetic classification divides the pomegranate germplasm collection into two statistically significant distinct genetic groups, G1 and G2; each of the parents of the population belongs to a different group. Nana belongs to the G1 group. The paternal parent, the Black accession, is part of the G2 group (Ophir et al. 2014).
The heterozygosity in the mapped F2 population was found to be 47 %, which is expected of an F2 population (Van Ooijen and Voorrips 2001). The maternal (Nana) alleles were 30 % more abundant within the F2 population. SNPs of 176 were abnormally distributed, mainly in four linkage groups. Deviating distortion is known in other mapped population of different plants. In maize, two F2 populations were compared before and after five generations of intermating; deviant segregation ratios were observed in both F2 populations; the overall allele frequencies were 51 % and 49 % for the different parents’ alleles (Lee et al. 2002). However, ratios of genotypic classes at 38 loci distributed among all chromosomes deviated from the expectations in F2. In a recombinant inbred line population of tomato, a significant deviation from the expected 1:1 ratio between the two homozygous classes was found in 73 % of the markers. In 98 % of the deviating markers, Lycopersicon esculentum alleles were present at a greater frequency than that of the Lycopersicon cheesmanii alleles; the skewed loci were scattered throughout the genome (Paran et al. 1995). Genotypic analysis of individual progenies within the pomegranate F2 population revealed that five plants contained more than twice the alleles of Nana. Phenotypic characterization of these individual progenies revealed that they were not dwarfed like Nana, nor did they resemble Nana in other phenotypic aspects. The possibility of obtaining such a high enrichment of parental alleles in F2 progenies is of high importance for breeding, as it provides a useful tool to hasten breeding towards a similarity of genotypes to one of the parents in advanced generations. Identification of shifts in allele frequencies within the genome can be important information for monitoring specific alleles or haplotypes and can be used to design appropriate breeding strategies (Collard and Mackill 2008).
QTL mapping allowed us to map several agriculturally important traits including TSS, fruit size, seed hardness, and plant size. While multiple genes were found to be responsible for some phenotypic traits in pomegranate (for example, fruit weight and aril weight), only one QTL was detected for fruit weight in peach (Dirlewanger et al. 1999) and two major QTLs were detected in tomato (Causse et al. 2002). For other traits such as TSS, seed hardness, and plant height, only two sites on the genetic map were found to be involved. Major genes were found for each of these three traits, suggesting the potential of the map to facilitate breeding for these traits. QTLS for TSS, seed hardness, and plant height were characterized in other plants as well. In peach, two major QTLs for TSS were also found (Dirlewanger et al. 1999). The seed hardness trait was mapped also in soybean (Zhang et al. 2008). Interestingly, seed hardness in soybean was also found to be mapped to two loci in the genetic map. QTL mapping of plant height was done in populus. In this plant, only one QTL was identified for plant height in the second year of growth (Bradshaw and Stettler 1995). In addition, in peach, growth habit was not mapped as one gene trait (Sajer et al. 2012).
A few QTLs were mapped in close proximity at LG2, including QTLs for plant height, fruit weight, fruit perimeter, aril weight, and TSS. One explanation for this phenomenon is that plant size has a pleiotropic effect on fruit traits. In rice, plant size was mapped to the same locus as grain yield, heading date (Zhang et al. 2006; Yan et al. 2011). Indeed, map-based cloning reveals that this locus encodes a putative HAP3 subunit of the CCAAT-box-binding transcription factor (Wei et al. 2010). In a complementary experiment, this gene increased plant height and the number of grains per panicle significantly. Another explanation is that the proximity of the QTLs is due to closely linked multiple QTLs. In tomato, it was shown that a cluster of QTLs of multiple characters such as fruit weight, fruit elasticity, color, sourness, aroma intensity, candy aroma, mealiness, dry matter weight, soluble solids, sugars, and eugenol content could be dissected by further genetic studies, such as fine mapping of this region (Causse et al. 2002).
Association studies for pomegranate were conducted within local collections, Basaki et al. (2011, 2013) who associated seven SSRs with 20 traits within the Iranian collection. Singh et al. (2015) analyzed the association of three traits, by GLM, with 44 microsatellites within the Indian collection. However, the map position of these SSRs was not determined. The present study used more than 200 mapped markers to analyze the association with fruit traits within a worldwide collection. When the structure of the collection was considered in the analysis, two traits (fruit weight and acid content) were found to be associated. The associated markers were mapped to two linkage groups.
The linkage map provides basic data about the order of markers and genes along the genome and detects QTLs for interesting trials. However, the QTLs might be significant only for a specific population due to exclusive genetic differences between the two parents. The association study within the variety collection took advantage of historic recombination events. In maize, for example, the nested association mapping population showed that there is a variation for recombination frequencies (McMullen et al. 2009); therefore, there could be different recombination events between two varieties that are not presented in the linkage map. Common mapped markers that were associated with traits within the collection of varieties can give information about the position of the associated markers even if no similar QTL was detected within the mapping population. Thus, population mapping and studies of associations within collections can yield a broader view of the number of loci involved in the control of complex traits. Moreover, QTLs that are co-localized with associated markers to the same trait, as was found for the two fruit weight QTLs, strongly suggest that these QTLs could be relevant in several populations and highly informative for breeding. Up till now, no draft of the pomegranate genome has been reported. Genome sequence is today a crucial tool for functional analysis of genes in many plant species and its absence severely inhibits the ability to associate gene function with important traits. A dense linkage map may be a platform to facilitate a new assembly of a genome (Mascher and Stein 2014). The pomegranate genetic map described in this study together with the de novo assembly of the pomegranate transcriptome published earlier could contribute significantly to the establishment of the pomegranate genome.
References
Basaki T, Choukan R, Nekouei SMK, Mardi M, Majidi E, Faraji S, Zeinolabedini M (2011) Association analysis for morphological traits in pomegranate (Punica geranatum L.) using microsatellite markers. Middle-East J Sci Res 9:410–417
Basaki T, Nejat MA, Nejad RJ, Faraji S, Keykhaei F (2013) Identification of simple sequences repeat (SSR) informative markers associated with important traits in pomegranate (Punica granatum L.). Int J Agron Plant Prod 4:575–583
Ben-Simhon Z, Judeinstein S, Nadler-Hassar T, Trainin T, Bar-Ya’akov I, Borochov-Neori H, Holland D (2011) A pomegranate (Punica granatum L.) WD40-repeat gene is a functional homologue of Arabidopsis TTG1 and is involved in the regulation of anthocyanin biosynthesis during pomegranate fruit development. Planta 234:865–881
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Bradshaw HD, Stettler RF (1995) Molecular genetics of growth and development in populus. IV. Mapping QTLs with large effects on growth, form, and phenology traits in a forest tree. Genetics 139:963–973
Causse M, Saliba-Colombani V, Lecomte L, Duffe P, Rousselle P, Buret M (2002) QTL analysis of fruit quality in fresh market tomato: a few chromosome regions control the variation of sensory and instrumental traits. J Exp Bot 53:2089–2098
Chai HS, Therneau TM, Bailey KR, Kocher JP (2010) Spatial normalization improves the quality of genotype calling for Affymetrix SNP 6.0 arrays. BMC Bioinforma 11:356
Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WE, Wetter T, Suhai S (2004) Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 14:1147–1159
Collard BCY, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc B Biol Sci 363:557–572
Currò S, Caruso M, Distefano G, Gentile A, La Malfa S (2010) New microsatellite loci for pomegranate, Punica granatum (Lythraceae). Am J Bot 97:e58–e60
Danin-Poleg Y, Tadmor Y, Tzuri G, Reis N, Hirschberg J, Katzir N (2002) Construction of a genetic map of melon with molecular markers and horticultural traits, and localization of genes associated with ZYMV resistance. Euphytica 125:373–384
Di Guardo M, Tadiello A, Farneti B, Lorenz G, Masuero D, Vrhovsek U, Costa G, Velasco R, Costa F (2013) A multidisciplinary approach providing new insight into fruit flesh browning physiology in apple (Malus x domestica Borkh.). PLoS One 8:e78004
Dirlewanger E, Moing A, Rothan C, Svanella L, Pronier V, Guye A, Plomion C, Monet R (1999) Mapping QTLs controlling fruit quality in peach (Prunus persica (L.) Batsch). Theor Appl Genet 98:18–31
Ewing B, Hillier L, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185
Fan S, Bielenberg DG, Zhebentyayeva TN, Reighard GL, Okie WR, Holland D, Abbott AG (2010) Mapping quantitative trait loci associated with chilling requirement, heat requirement and bloom date in peach (Prunus persica). New Phytol 185:917–930
Ferrara G, Giancaspro A, Mazzeo A, Giove SL, Matarrese AMS, Pacucci C, Punzi R, Trani A, Gambacorta G, Blanco A, Gadaleta A (2014) Characterization of pomegranate (Punica granatum L.) genotypes collected in Puglia region, Southeastern Italy. Sci Hortic 178:70–78
Graham IA, Besser K, Blumer S, Branigan CA, Czechowski T, Elias L, Guterman I, Harvey D, Isaac PG, Khan AM, Larson TR, Li Y, Pawson T, Penfield T, Rae AM, Rathbone DA, Reid S, Ross J, Smallwood MF, Segura V, Townsend T, Vyas D, Winzer T, Bowles D (2010) The genetic map of Artemisia annua L. identifies loci affecting yield of the antimalarial drug artemisinin. Science 327:328–331
Hajiahmadi Z, Talebi M, Sayed-Tabatabaei BE (2013) Studying genetic variability of pomegranate (Punica granatum L.) based on chloroplast DNA and barcode genes. Mol Biotechnol
Harel-Beja R, Tzuri G, Portnoy V, Lotan-Pompan M, Lev S, Cohen S, Dai N, Yeselson L, Meir A, Libhaber SE, Avisar E, Melame T, van Koert P, Verbakel H, Hofstede R, Volpin H, Oliver M, Fougedoire A, Stalh C, Fauve J, Copes B, Fei Z, Giovannoni J, Ori N, Lewinsohn E, Sherman A, Burger J, Tadmor Y, Schaffer AA, Katzir N (2010) A genetic map of melon highly enriched with fruit quality QTLs and EST markers, including sugar and carotenoid metabolism genes. Theor Appl Genet 121:511–533
Hasnaoui N, Buonamici A, Sebastiani F, Mars M, Trifi M, Vendramin G (2010) Development and characterization of SSR markers for pomegranate (Punica granatum L.) using an enriched library. Conserv Genet Resour 2:283–285
Hasnaoui N, Buonamici A, Sebastiani F, Mars M, Zhang D, Vendramin GG (2012) Molecular genetic diversity of Punica granatum L. (pomegranate) as revealed by microsatellite DNA markers (SSR). Gene 493:105–112
Holland D, Bar-Ya’akov I (2014) Pomegranate: aspects concerning dynamics of health beneficial phytochemicals and therapeutic properties with respect to the tree cultivar and the environment. In: Yaniv Z, Dudai N (eds) Medicinal and aromatic plants of the Middle-East. Springer, Netherlands, pp 225–239
Holland D, Hatib K, Bar-Ya’akov I (2009) Pomegranate: botany, horticulture, breeding. Hortic Rev 35:127–191
Jian Z-H, Liu X-S, Hu J-B, Chen Y-H, Feng J-C (2012) Mining microsatellite markers from public expressed sequence tag sequences for genetic diversity analysis in pomegranate. J Genet 91:353–358
Lee M, Sharopova N, Beavis W, Grant D, Katt M, Blair D, Hallauer A (2002) Expanding the genetic map of maize with the intermated B73 × Mo17 (IBM) population. Plant Mol Biol 48:453–461
Liu W, Di X, Yang G, Matsuzaki H, Huang J, Mei R, Ryder TB, Webster TA, Dong S, Liu G, Jones KW, Kennedy GC, Kulp D (2003) Algorithms for large-scale genotyping microarrays. Bioinformatics 19:2397–2403
Mascher M, Stein N (2014) Genetic anchoring of whole-genome shotgun assemblies. Front Genet 5
Masoud S, Khandan M, Nasre-Esfahani S (2005) Cytogenetical study of some Iranian pomegranate (Punica granatum L.) cultivars. Caryologia 58:132–139
McMullen MD, Kresovich S, Villeda HS, Bradbury P, Li H, Sun Q, Flint-Garcia S, Thornsberry J, Acharya C, Bottoms C, Brown P, Browne C, Eller M, Guill K, Harjes C, Kroon D, Lepak N, Mitchell SE, Peterson B, Pressoir G, Romero S, Rosas MO, Salvo S, Yates H, Hanson M, Jones E, Smith S, Glaubitz JC, Goodman M, Ware D, Holland JB, Buckler ES (2009) Genetic properties of the maize nested association mapping population. Science 325:737–740
Noormohammadi Z, Fasihee A, Homaee-Rashidpoor S, Sheidai M, Baraki SG, Mazooji A, Tabatabaee-Ardakani SZ (2012) Genetic variation among Iranian pomegranates (Punica granatum L.) using RAPD, ISSR and SSR markers. Aust J Crop Sci 6:268–275
Olukolu BA, Trainin T, Fan SH, Kole C, Bielenberg DG, Reighard GL, Abbott AG, Holland D (2009) Genetic linkage mapping for molecular dissection of chilling requirement and budbreak in apricot (Prunus armeniaca L.). Genome 52:819–828
Ophir R, Sherman A, Rubinstein M, Eshed R, Sharabi Schwager M, Harel-Beja R, Bar-Ya’akov I, Holland D (2014) Single-nucleotide polymorphism markers from de-novo assembly of the pomegranate transcriptome reveal germplasm genetic diversity. PLoS One 9:e88998
Orhan E, Ercisli S, Esitken A, Sengul M (2014) Molecular and morphological characterization of pomegranate (Punica granatum L.) genotypes sampled from Coruh Valley in Turkey. Genet Mol Res 13:6375–6382
Paran I, Goldman I, Tanksley SD, Zamir D (1995) Recombinant inbred lines for genetic mapping in tomato. Theor Appl Genet 90:542–548
Pirseyedi SM, Valizadehghan S, Mardi M, Ghaffari MR, Mahmoodi P, Zahravi M, Zeinalabedini M, Nekoui SMK (2010) Isolation and characterization of novel microsatellite markers in pomegranate (Punica granatum L.). Int J Mol Sci 11:2010–2016
Porebski S, Bailey LG, Baum BR (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Report 15:8–15
Rubinstein M, Katzenellenbogen M, Eshed R, Rozen A, Katzir N, Colle M, Yang L, Grumet R, Weng Y, Sherman A, Ophir R (2015) Ultra high-density linkage map for cultivated cucumber (Cucumis sativus L.) using a single nucleotide polymorphism genotyping array. PloS one In press
Sajer O, Scorza R, Dardick C, Zhebentyayeva T, Abbott AG, Horn R (2012) Development of sequence-tagged site markers linked to the pillar growth type in peach (Prunus persica). Plant Breed 131:186–192
Sarkhosh A, Zamani Z, Fatahi R, Hassani M, Wiedow C, Buck E, Gardiner S (2011) Genetic diversity of Iranian soft-seed pomegranate genotypes as revealed by fluorescent-AFLP markers. Physiol Mol Biol Plants 17:305–311
Seeram NP, Schulman RN, Heber D (2006) Pomegranates: ancient roots to modern medicine. CRC Press Taylor & Francis Group, Boca Raton
Singh SK, Meghwal PR, Pathak R, Gautam R, Kumar S (2013) Genetic diversity in Punica granatum revealed by nuclear rRNA, internal transcribed spacer and RAPD polymorphism. Natl Acad Sci Lett 36:115–124
Singh N, Abburi V, Ramajayam D, Kumar R, Chandra R, Sharma K, Sharma J, Babu KD, Pal R, Mundewadikar D, Saminathan T, Cantrell R, Nimmakayala P, Reddy U (2015) Genetic diversity and association mapping of bacterial blight and other horticulturally important traits with microsatellite markers in pomegranate from India. Mol Genet Genomics 290:1393–1402
Soriano J, Zuriaga E, Rubio P, Llácer G, Infante R, Badenes M (2011) Development and characterization of microsatellite markers in pomegranate (Punica granatum L.). Mol Breed 27:119–128
Van Ooijen JW (2004) MapQTL ® 5, Software for the mapping of quantitative trait loci in experimental populations. Kyazma B.V, Wageningen
Van Ooijen JW, Voorrips RE (2001) JoinMap ® 3.0, Software for the calculation of genetic linkage maps. Plant Research International, Wageningen
Wei XJ, Xu JF, Guo HN, Jiang L, Chen SH, Yu CY, Zhou ZL, Hu PS, Zhai HQ, Wan JM (2010) DTH8 suppresses flowering in rice, influencing plant height and yield potential simultaneously. Plant Physiol 153:1747–1758
Yan W-H, Wang P, Chen H-X, Zhou H-J, Li Q-P, Wang C-R, Ding Z-H, Zhang Y-S, Yu S-B, Xing Y-Z, Zhang Q-F (2011) A major QTL, Ghd8, plays pleiotropic roles in regulating grain productivity, plant height, and heading date in rice. Mol Plant 4:319–330
Zhang Y, Luo L, Xu C, Zhang Q, Xing Y (2006) Quantitative trait loci for panicle size, heading date and plant height co-segregating in trait-performance derived near-isogenic lines of rice (Oryza sativa). Theor Appl Genet 113:361–368
Zhang B, Chen PY, Chen CY, Wang DC, Shi AN, Hou AF, Ishibashi T (2008) Quantitative trait loci mapping of seed hardiness in soybean. Crop Sci 48:1341–1349
Zhang YP, Tan HH, Cao SY, Wang XC, Yang G, Fang JG (2012) A novel strategy for identification of 47 pomegranate (Punica granatum) cultivars using RAPD markers. Genet Mol Res 11:3032–3041
Acknowledgments
We thank Kamel Hatib for growing and maintaining the orchards. This research was supported by funding from the Chief Scientist Office, Ministry of Agriculture & Rural Development, http://www.science.moag.gov.il/, Agricultural Biotechnology 203-0753-09.
Authors’ contribution statement
R. H. B. conducted the phenotyping of the mapping population and the genetic and mapping analysis and wrote the paper; A. S. and R. E. conducted the sequencing, marker development, and the genotyping of the mapping population; M. R. and R. O. conducted the marker development, array and probe design, array hybridization and signal preprocessing, and genotype calling; I. B. Y conducted the phenotyping of the ARO collection and the mapping population; T. T. conducted the total RNA extraction and developed the method for DNA extraction from pomegranate; and D. H. wrote the paper and designed and supervised the study. All authors discussed the results and commented on the manuscript.
Data Archiving Statement
Data base on which this study was based was published before by Ophir et al. (2014) and deposited in GenBank (http://www.ncbi.nlm.nih.gov/sra/SRX395468[accn] and http://www.ncbi.nlm.nih.gov/sra/SRX395465[accn]).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Communicated by V. Decroocq
This article is part of the Topical Collection on Complex Traits
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource Table 1
(PDF 106 kb)
Online Resource Table 2
(PDF 8 kb)
Online Resource Table 3
(PDF 578 kb)
Online Resource Table 4
(PDF 19 kb)
Rights and permissions
About this article

Cite this article
Harel-Beja, R., Sherman, A., Rubinstein, M. et al. A novel genetic map of pomegranate based on transcript markers enriched with QTLs for fruit quality traits. Tree Genetics & Genomes 11, 109 (2015). https://doi.org/10.1007/s11295-015-0936-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-015-0936-0