
Abstract
The heat shock protein 70 (Hsp70) family members are known as molecular chaperones. They play a crucial role in protecting plant cells and tissues from thermal or abiotic stress through protein folding and in assembly, stabilization, activation, and degradation processes. Although many studies have been performed to identify molecular functions of individual family members, there is a limited study on genome-wide identification and characterizations of Hsps in the Populus model tree genus. We have identified 34 poplar Hsp70 genes, which were phylogenetically clustered into three major groups. Gene structure and motif composition are relatively conserved in each group. Mainly tandem and infrequently segmental duplications have a significant role in poplar Hsp70 gene expansion. The in silico microRNA (miRNA) and target transcript analyses identified that a total of 19 PtHsp70 genes were targeted by 27 plant miRNAs. PtHSP70-14 and PtHSP70-33 are the most targeted by miR390 and miR414 family members, respectively. For determination of drought response to Hsp70 genes, publicly available RNA-seq data were analyzed. Poplar Hsp70s are differentially expressed upon exposure to different drought stress conditions. Expression analysis of PtHsp70 genes was also examined under drought stress in drought-sensitive and drought-resistant Populus clones with quantitative real-time PCR (qRT-PCR). PtHsp70-16 and PtHsp70-26 genes might provide adaptation to drought stress for both clones. Because of high expression responses to drought in only resistant Populus clone, PtHsp70-25 and PtHsp70-33 genes might be used for determination of drought-tolerant clones for molecular breeding studies. This research provides a fundamental clue for contribution of PtHsp70s to drought tolerance in poplar.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Populus (poplar) is a vital genus of trees. Because of their broad dispersion and usage as a source for paper production and as a bioenergy source supported by rapid growth and high genetic diversity, poplar plays a crucial role in economy and ecosystem (Jansson and Douglas 2007). The species of Populus presents splendid opportunities to examine stress responses toward drought which affect not only survival but also biomass accumulation (Marron et al. 2002; Monclus et al. 2006). In recent years, there has been an increasing interest in genotype, transcriptome, and drought response relations in Populus trees (Caruso et al. 2008; Song et al. 2012). Black cottonwood (Populus trichocarpa Torr. & Gray) is a woody deciduous plant, which lives for long years and cultivated basically in western North America. It is the first tree species whose genome was sequenced and published (Tuskan et al. 2006). When economic importance of wood and wood products is considered, availability of the poplar genome plays an important role for molecular processes of growth, advances, and responses toward environmental changes seen in trees. The poplar genome also provides a valuable information for evolutionary comparisons between herbaceous and woody plants.
A number of researchers (Schlesinger 1990; Schöffl et al. 1998; Kotak et al. 2007; Lund 2001; Kampinga and Craig 2010) have reported that heat shock proteins (Hsps) are classified into five major families based on functions and molecular mass, viz., small Hsps, Hsp60, Hsp70, Hsp90, and Hsp100. They are highly conserved and well characterized in a few model plants such as the Arabidopsis, rice, and poplar (Hu et al. 2009; Krishna and Gloor 2001; Hill and Hemmingsen 2001; Scharf et al. 2001; Lee et al. 2007; Zhang et al. 2013). Hsps play a crucial role in maintaining protein homeostasis, behaving as molecular chaperones and supporting protein refolding when stress conditions are available (Vierling 1991; Hendrick and Hartl 1993; Boston et al. 1996; Hartl 1996; Waters et al. 1996; Török et al. 2001). In recent years, Zhang et al. (2014) showed that many Hsp70 genes may play important roles in fiber development processes including fiber initiation and elongation in cotton. This makes Hsps a valuable resource for researchers studying their response of different stress conditions, their functions for protecting plants against abiotic stresses, and for development mechanisms.
Recently, researchers have shown an increased interest in functional analysis of Hsp70 family members in different organisms such as Arabidopsis and rice (Lin et al. 2001; Sung et al. 2001; Wang et al. 2014; Sarkar et al. 2013; Jung et al. 2013). Although Hsp90 gene family members in poplar were identified (Zhang et al. 2013), other Hsps including Hsp70s still have not been fully characterized in poplar and little is known about their interactors (Jung et al. 2013). Furthermore, existing research studies recognize the critical role of Hsp70 genes which enhanced the plant’s tolerance to environmental stresses. Knockout mutations in Arabidopsis stromal 70-kD heat shock proteins (cpHSC70-1 and cpHSC70-2) caused formation of defective phenotypes and decrease in thermo-tolerance of germinating seeds (Su and Li 2008). A recent study by Jungkunz et al. (2011) involved in generation of AtHSP70-15 gene deficient Arabidopsis plants. This resulted in drastic increase in mortality after heat treatment. So, it can be concluded that AtHSP70-15 plays an essential role for heat response. In the same study, overexpression of AtHSP70-1 leads to increase in stress tolerance in Arabidopsis (Jungkunz et al. 2011). A similar series of experiments to show that alternation in expression of the Arabidopsis thaliana cytosolic/nuclear HSC70-1 molecular chaperone directly influenced development and abiotic stress tolerance was performed by Noel et al. (2007). In another major study, BIP gene (encoding Hsp70) from A. thaliana was responsible for development of female gametophyte (Maruyama et al. 2010) and the same gene in Nicotiana tabacum protected plant against water stress (Alvim et al. 2001). Researchers conducted a series of studies related with BIP1/OsBIP3 gene functions in rice. This gene regulated XA21-mediated immunity (Park et al. 2010), seed development (Wakasa et al. 2011), and programmed cell death (Qi et al. 2011). However, the biological functions of many Hsp70 family members have not yet been identified in many organisms including poplar.
Omics technologies are very helpful for detection of new genes and determination of their function (Feist and Palsson 2008). Although the recent developments for gene discovery studies have significantly increased, there is little known about the genome-wide survey and expression patterns of Hsp70 gene family in poplar. The genome-wide analysis and identification studies from Arabidopsis (Lin et al. 2001; Sung et al. 2001), rice (Sarkar et al. 2013; Jung et al. 2013), cotton (Zhang et al. 2014), and poplar (Zhang et al. 2015) are a few examples for Hsp genes. Due to various factors, including a relatively small genome size, fast-growing industrial materials and the release of the latest Populus trichocarpa genome sequence data, v3.0, gave us an opportunity to identify and further analyze the poplar Hsp70 gene family. Although the poplar Hsf and Hsp gene families were previously identified on a genome-wide level (Zhang et al. 2013; Zhang et al. 2015), we made a detailed study for identification, comparison, functionally characterization, and expression analysis of Hsp70 genes in poplar. In addition, there is a limited study on genome-wide identification and characterizations of Hsps in the poplar as well as other plants’ genome. Therefore, this study makes a major contribution to research on function of Hsp family members. Here, a comprehensive set of Hsp70 genes (34) was identified based on the complete genome sequence of poplar. Subsequently, chromosomal localization, motif analysis, exon–intron organization, homology, and phylogenetic analysis were also investigated. Finally, we examined the expression patterns of Hsp70 family members from the publicly available transcriptome data and experimental data. This research serves as a base for future studies and provides a fundamental clue for exploration into the functions of this significant gene family. In addition, identified genes presented here can be used for cloning studies in agricultural applications
Materials and Methods
Analysis and Identification of Poplar Hsp70 Genes
Different Hsp70 protein members (about 259 amino acid sequences) from diverse organisms (A. thaliana, Cucumis sativus, Glycine max, Hordeum vulgare, Medicago truncatula, N. tabacum, Oryza sativa, Physcomitrella patens, Ricinus cummunis, Solanum lycopersicum, Sorghum bicolor, Triticum aestivum, Vigna radiate, Vitis vinifera, and Zea mays) at Heat Shock Protein Database Information Resource (http://pdslab.biochem.iisc.ernet.in/hspir/index.php) (Kumar et al. 2012) were downloaded to identify potential members of Poplar Hsp70 proteins. BLASTP at PHYTOZOME v10.3 database (www. phytozome.net) and The Hidden Markov Model (HMM) search at Pfam database (http://pfam.sanger.ac.uk) were performed against the poplar genome with default parameters (Goodstein et al. 2012). Identified poplar HSP70 proteins were also used as query in NCBI BLASTP for characterization of hypothetical or uncharacterized proteins in poplar. Redundant sequences were removed using the decrease redundancy tool (web.expasy.org/decrease_redundancy). Each non-redundant sequence was again analyzed to check the presence of Hsp70 domains by SMART (http://smart.emblheidelberg.de) (Letunic et al. 2012) and Pfam (http://pfam.sanger.ac.uk) searches. Theoretical isoelectric points (pI), molecular weights, and instability index were calculated using ProtParam Tool (http://web.expasy.org/protparam).
Physical Location, Gene Structure Classifications, and Analysis of Poplar Hsp70 Proteins
Specific chromosomal locations, intron numbers, and sizes (bp) were determined by Phytozome database. The PtHsp70 genes were plotted on all poplar chromosomes from the short-arm telomere to the long-arm telomere and finally visualized with MapChart (Voorrips 2002). Segmental and tandem duplications were determined by Plant Genome Duplication Database (PGDD; http://chibba.pgml.uga.edu/duplication/index/blast) (Tang et al. 2008). In detail, BLASTP search was performed against all predicted Hsp70 proteins of Populus trichocarpa, and the first five matches with ≤1e-05 was considered as potential anchors. Collinear blocks were evaluated by MCScan, and alignments with ≤1e-10 were selected as important matches (Tang et al. 2008; Du et al. 2013). Tandem duplications were also characterized as adjacent genes of same subfamily located within 10 predicted genes apart or within 30 kbp of each other (Du et al. 2013; Shiu and Bleecker 2003). The exon–intron analysis of the PtHsp70 proteins was generated by Gene Structure Display Server (GSDS) software (gsds.cbi.pku.edu.cn) (Guo et al. 2007). The coding sequences and genome sequences were used for prediction of gene structure of the poplar Hsp70 genes.
Phylogenetic Analysis and Identification of the Conserved Domains
Phylogenetic analysis was conducted using the neighbor-joining method with bootstrap analysis for 1000 iterations. Multiple sequence alignments corresponding to conserved motif regions, characteristic of the Hsp70 protein members, were determined by ClustalW with a gap open and gap extension penalties of 10 and 0.1, respectively (Thompson et al. 1997). The alignment file was firstly imported into MEGA5 (Tamura et al. 2011) and used to construct an unrooted phylogenetic tree.
The Multiple EM for motif elicitation (MEME) (http://meme.nbcr.net/meme3/meme.html) (Bailey et al. 2006) was used to identify motifs in candidate sequences. The parameters for the analysis were as follows: number of repetitions, any; maximum number of motifs, 20; and optimum width of motif, ≥2 and ≤300. Discovered MEME motifs with ≤1e-30 were searched in the InterPro database with InterProScan (Quevillon et al. 2005).
GO Annotation
The functional annotation of Hsp70 protein sequences and the analysis of annotation data were performed by using Blast2GO (http://www.blast2go.com) (Conesa and Götz 2008). First, all identified PtHsp70 amino acid sequences were introduced into Blast2GO program. Then, functional annotation was achieved in three steps: (i) BLASTp to find homologous sequences, (ii) MAPPING to retrieve GO terms related with the BLAST results, and (iii) ANNOTATION of Gene Ontology (GO) terms for selection reliable functions to given amino acid sequences. The program provides the output defining three categories of GO classification, namely, biological processes, cellular components, and molecular functions.
Comparative Physical Mapping of Hsp70 Protein Member Between Poplar and Other Species
For identification of orthologous relationship between poplar PtHsp70 amino acid sequences and Hsp70s from four species including Arabidopsis, rice, maize, and grape, BLASTP search was conducted in Phytozome database (www.phytozome.net). Hits with ≤1e-5 and at least 80 % identify were considered significant. Orthologous Hsp70 genes among poplar, Arabidopsis, rice, maize, and grape were placed on corresponding species chromosomes which were finally visualized with MapChart.
Estimating the Rates of Synonymous and Non-synonymous Substitution
For estimation of the synonymous (Ks) and non-synonymous (Ka) substitution rates, the amino acid sequences belonging to duplicated protein-encoding PtHsp70 protein members and orthologous gene pairs between poplar and Arabidopsis, rice, maize, and grape were firstly aligned with CLUSTALW based on multiple sequence alignment tool. Then, PAL2NAL program (http://www.bork.embl.de/pal2nal) (Suyama et al. 2006) was used for alignment of the amino acid sequences and their respective original complementary DNA (cDNA) sequences of PtHsp70 genes. This program converts a multiple sequence alignment of proteins and the corresponding DNA sequences into a codon alignment and finally estimated the synonymous (Ks) and non-synonymous (Ka) substitution rates. Time (million years ago, Mya) of duplication and divergence of each Hsp70 genes were also calculated with a formula as T = Ks/2λ (λ = 6.5 × 10 e-9) (Lynch and Conery 2000; Yang et al. 2008).
In Silico Analysis of miRNA Targets in PtHsp70 Genes
MicroRNA (miRNA) target analysis helps to understand miRNA regulatory mechanisms. Previously known plant pre-miRNA sequences obtained from miRBase v20.0 (http://www.mirbase.org) and plant miRNA database (http://bioinformatics.cau.edu.cn/PMRD) were utilized for identification of miRNAs targeting the PtHsp70 genes. Poplar Hsp70 gene targets and plant miRNAs were characterized by aligning them using the web-based psRNA Target Server (http://plantgrn.noble.org/psRNATarget) with default parameters. Alignment between all known plant miRNAs and its PtHsp70 gene target(s) were evaluated by the parameters described by Zhang (2005). Further analysis of the computationally identified miRNA targets were performed by BLASTX searches with ≤1e-10 against poplar EST sequences at NCBI database for identification and confirmation of putative gene homologous.
Homology Modeling of Hsp70 Proteins
All poplar Hsp70 protein sequences were scanned at Protein Data Bank (PDB) (Berman et al. 2000) by using BLASTP to determine the similar sequence and known best sample which have three-dimensional structure. Phyre2 database (Protein Homology/Analogy Recognition Engine; http://www.sbg.bio.ic.ac.uk/phyre2) was used for prediction of 3D protein structure of Hsp70 proteins (Kelley and Sternberg 2009). Predicted protein structures of poplar Hsp70s were evaluated in terms of confidence level (>90 %) and percentage residue level (80 to 100).
Expression Analysis of the PtHsp70 Genes in Transcriptome Data
All Illimuna HiSeq readings and Roche454 RNA-Seq data were obtained from Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) database at the NCBI under the following accession numbers: (i) SRP005997 (experiment accession nos. SRX047542 for the control and SRX047543 for the treatment), (ii) SRP033028 (experiment accession nos. SRX377987 for the control and SRX472726 for the treatment), and (iii) SRP024267 (experiment accession nos. SRX297950, SRX297104, SRX29795, and SRX297952 for the control; SRX297957, SRX297955, SRX297954, and SRX297953 for the moderately dehydrated leaves; and SRX297964, SRX297963, SRX297962, and SRX297961 for severely dehydrated leaves) (Tang et al. 2013; Tang et al. 2014; Cossu et al. 2014). All readings were downloaded in raw sequencing data as “.sra” format and converted to “fastq” format for Illimuna and “sff” format for Roche 454 by the NCBI SRA Toolkit’s fastq-dump command. After discarding low-quality readings (Phred quality (Q) score <20) and trimming adapters by using FASTX toolkit, all clean readings were subjected to FastQC analysis for checking reading qualities in terms of per-base sequence qualities, per-sequence quality scores, per-base nucleotide content, and sequence duplication levels. The raw count data were transformed and normalized using CLC Genomic Workbench version 7.5. Then, gene expression measurement and hierarchical clustering heat map were constructed based on log2 RPKM values by PermutMatrix software (Caraux and Pinloche 2005).
Plant Materials, Growth Conditions, and Treatments
Poplar clones were kindly obtained from Behiçbey Forest Nursery (Ankara Regional Directorate of Forestry, Ankara, Turkey). Physiological, morphological, and biochemical responses of Populus nigra clones to drought stress were previously evaluated. Clone R and Clone S poplar clones were determined as drought resistant and drought sensitive, respectively (Yildirim 2013). Plant materials were collected from nursery clonally propagated 1-year-old Populus nigra clones, Clone R (drought resistant) and Clone S (drought sensitive), grown under a natural photoperiod, humidity, and temperature in the field. These Populus clones were used as a single maternal plant. Rooted cuttings from these maternal Populus Clone R and Clone S were cultivated in 20 × 20 cm2 pots containing packaged potting soil, peat moss, and vermiculite (2:2:1, v/v) in the greenhouse, under controlled environmental conditions (25 °C day/20 °C night, 16-h light/8-h dark photoperiod) with relative humidity from 55 to 80 %. The trees were well watered until the drought treatments began. In other words, they were watered to reach field capacity every day until uniformly developed trees (with 80–100 cm in height) were obtained for the water stress treatments. For control, trees were normally watered by one to three waterings per day to compensate field capacity. For drought stress application, soil relative extractable water (REW) was controlled by water supply four times a day. Soil REW was maintained for 10 days as a drought stress. Fully expanded leaves at approximately 6–10 internodes from apex from drought-resistant and drought-sensitive clones were harvested at 10 days of control and drought-stress-treated trees (Supplementary Fig. S1). The experimental design was as follows: 3 clones (biological replicates) × 2 treatments (control and drought stress) × 2 clones (drought resistant and drought sensitive).
RNA Isolation and Quantitative Real-Time PCR Analysis
About 150–200 mg leaf samples from control and stress-treated trees was homogenized with liquid nitrogen. Three milliliters of pre-heated extraction buffer (2 % [w/v] CTAB, 2 % [w/v] PVP, 100 mM Tris/HCl pH 8.0, 25 mM EDTA, 2 M NaCl, 0.5 g/L spermidine, 2.7 % [v/v] 2-ME) was mixed with the frozen leaf powder and incubated at 65 °C for 10 min. Two separate extraction steps with 3 mL ice-cold chloroform/isoamylalcohol (24:1, v/v) were performed. A total of 0.25 volumes of ice-cold 10 M LiCl was added to precipitate RNA and incubated at 4 °C for 18 h. After centrifugation at 16,000×g, 4 °C, 60 min, the pellets were incubated with 4 mL ice-cold 75 % ethanol at −80 °C for 60 min, followed by centrifugation at 16,000×g, 4 °C, 20 min. Ethanol was removed from tubes. Pellets were dried and dissolved in RNase-free water. RNA concentrations and integrity were determined using a Multiskan™ GO Microplate Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and agarose gel electrophoresis, respectively. DNA contamination in samples was removed with DNase I (Fermentas, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
To examine expression profiles of 13 members of Hsp70 genes in leaf tissues of control and drought-stress-treated poplar clones, quantitative real-time PCR (qRT-PCR) was conducted as previously reported (Turktas et al. 2013) using SYBR Green I Master Kit (Roche, Germany) on LightCycler 480 Instrument II (Roche, Germany). Primers for the PtHsp70 genes were designed considering the conserved regions of Hsp70 members. A list of the primers used in qRT-PCR is presented in Supplementary Table S9. The qRT-PCR was carried out in 96-well optical plates. PCR reactions were performed in a total volume of 20 μL containing 0.1 μL reverse and forward primers (100 pmol), 2 μL of cDNA, and 10 μL FastStart SYBR Green I Master Mix, and nuclease-free water was added up to 20 μL. The 18S rRNA gene was used as the internal control (Wang et al. 2010; Budak et al. 2013). The qRT-PCR conditions were set up as follows: preheating at 95 °C for 5 min, followed by 55 cycles of 95 °C for 10 s, 53 or 55 °C for 20 s, and 72 °C for 10 s. The melting curves were adjusted to 95 °C for 5 s and 55 °C for 1 min and then cooled to 40 °C for 30 s. All reactions were repeated three times with triple biological replicates. The expression levels were calculated as the mean signal intensity across the three replicates. Relative gene expression was calculated using ΔΔCT values obtained from the formulas ΔCT = CT target − CT reference and ΔΔCT = ΔCT treated sample − ΔCT untreated sample (0-h treatment). For all chart preparations, selected RNA relative amount was evaluated for gene expression level using the 2− ΔΔCT (Livak and Schmittgen, 2001; Baloglu et al. 2014a). In addition, the standard errors of mean among replicates were calculated. Student’s t test was used to obtain the statistical significance of the difference between treated samples and untreated samples (0-h treatment under abiotic stress). If P values <0.01, we considered the PtHsp70 genes as differentially expressed genes.
Results and Discussion
Characterization of Hsp70 Protein Coding Sequences in Poplar
Hsp70 protein sequences belonging to 15 plant genomes—A. thaliana, C. sativus, G. max, H. vulgare, M. truncatula, N. tabacum, O. sativa, Physcomitrella patens, R. cummunis, Solanum lycopersicum, Sorghum bicolor, T. aestivum, Vigna radiate, Vitis vinifera, and Z. mays—were used as queries for identification of putative poplar Hsp70 genes. We performed BLAST, hidden Markov model (HMM), and keyword querying searches in relevant databases. Hsp70-related domains were searched in Pfam and SMART databases for validation of presence of them. After removing redundant sequences, we identified 34 putative PtHsp70 genes in the genome of Populus trichocarpa (Table 1). For convenience, the Hsp70 genes were named from PtHsp70-01 to PtHsp70-34 based on scientific name of poplar (Populus trichocarpa) and ordered on the chromosomes from 1 to 19. The particularization of poplar Hsp70 proteins is listed in Table 1 which includes number of amino acids (length), molecular weight, isoelectric point (PI), and NCBI annotation. According to the detailed information, the lengths of PtHsp70 protein sequence ranged from 99 residues (PtHsp70-30) to 972 residues (PtHsp70-07), while the isoelectric point (pI) ranged from 4.77 (PtHsp70-22) to 9.94 (PtHsp70-05).
The protein sequences in the representative genomes of 15 plant species was searched for comparative genomic analyses. A total of 259 genes encoding Hsp70 proteins were identified in these selected plant species. The density of PtHsp70 is about 0.0804 which is higher than in most of the analyzed plants. A. thaliana (0.2889) and O. sativa (0.0941) are plant species that showed the highest density when compared to ratio of the number of PtHsp70 to genome size (Supplementary Table S1). Although individual Hsp70 genes have been identified in different plant species such as maize (Rochester et al. 1986), barley (Chen et al. 1994), and pea (Dhankher et al. 1997), identification of this family genes on genomic level has been firstly performed in Arabidopsis which contains 18 Hsp70 genes (Lin et al. 2001; Sung et al. 2001). In a recent study, characterization of 32 Hsp70 genes from rice was announced (Rouard et al. 2011; Sarkar et al. 2013; Jung et al. 2013). We also found similar gene numbers in poplar genome with 34 Hsp70 genes which is in contrast to Zhang et al.’s (2015) findings. Greenphyl phylogenomics database (GreenPhyl v4) (Rouard et al. 2011) also indicates the same Hsp70 gene numbers for poplar. This database contains a catalogue of gene families based on gene predictions of plant genomes. So, it can be concluded that we found an exact number of Hsp70 genes based on Pfam, SMART domain searches, and GreenPhyl v4 and Phytozome v10.3 databases.
Chromosomal Distribution, and Tandem and Segmental Duplications
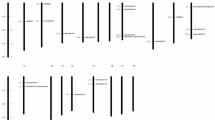
The position of all 34 Hsp70 genes was mapped on chromosomes of poplar (Fig. 1 and Supplementary Fig. S2). The distribution of the Hsp genes on chromosomes was not uniform. Some chromosomes and chromosomal regions have high density of the Hsp70 genes than other regions. Chromosome 1 (20.5 %) contained the highest number of Hsp70 gene among all chromosomes. Conversely, ten chromosomes (chromosome 2, 4, 6, 9, 11, 12, 14, 15, 16, and 19) possessed only one Hsp70 gene (2.94 %) and showed the lowest density. The exact position (in bp) of each PtHsp70 on poplar chromosome is indicated in Table 1. Gene distribution pattern on chromosomes revealed that PtHsp70 genes located on chromosomes 10 and chromosomes 8 and 13 appear to be congregate at the lower end and upper end of the arms, respectively (Fig. 1).

Physical locations of the poplar Hsp70 genes. The chromosome numbers (numbered 1–19) are shown at the top of each chromosome (Chr; represented as bars). Tandem duplicated genes on a particular chromosome are indicated in the box. Chromosomal distances are given in Mbp
It is known that segmental and tandem duplication has played a role in the evolution and expansion of gene families in plants (Cannon et al. 2004). Tandem and segmental duplication of PtHsp70 gene members was also determined (Supplementary Tables S2 and S3). The highest numbers of tandem duplication were observed in chromosome 10. Several direct tandem repeats were found on chromosome 1 (PtHsp70-01, PtHsp70-02, PtHsp70-03), chromosome 3 (PtHsp70-12, PtHsp70-13), and chromosome 8 (PtHsp70-16, PtHsp70-17) (Fig. 1).
Events of gene duplication occur frequently and cause evolution of related genes in organisms (Mehan et al. 2004). Earlier studies show that two genome-wide duplication events called as eurosid and salicoid have occurred in poplar genome. This resulted in a series of chromosomal reorganizations that involve reciprocal tandem/terminal fusions and translocations (Tuskan et al. 2006). Overall, there are 20 segmental duplicated poplar Hsp70 genes detected, which equals to approximately 59 % (20/34) of total PtHsp70 genes (Supplementary Table S3). The most surprising aspect of the data is high segmental duplicated ratio which is firstly shown for Hsp70 genes in different organisms including poplar. In rice, two segmental duplicates of Hsp70 genes (cHsp70-1 with cHsp70-6, and cHsp70-7 with uHsp70-2) were observed (Sarkar et al., 2013). This demonstrated that tandem and segmental duplications have a significant role in poplar HSP70 gene expansion. This interprets that evolution of those genes might have proceeded quickly thorough specific gene duplications or through integration into genomic region following a reverse transcription (Lecharny et al. 2003).
Phylogenetic Classification of PtHsp70 and Identification of Domain Conservation, and Gene Structure
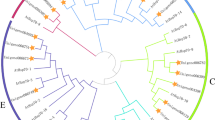
An elaborative phylogenic analysis was conducted to understand the evolutionary distinction of domain structure in Hsp70 proteins. The phylogenetic tree was constructed using 34 PtHsp70 proteins through neighbor-joining (NJ) method. The phylogenetic analysis classified all PtHsp70 into three main clusters (clusters I to III) comprising of 6, 5, and 23 proteins, respectively (Fig. 2). Cluster III was then further split into three subgroups (subgroups IIIa, IIIb, and IIIc). Because a good count of the internal branches were found to have high bootstrap values, it was clear by bootstrap analysis of 1000 replicates. A great number of internal branches also had high bootstrap values, demonstrating statistically reliable pairs of potential homologous derivation. Construction of a phylogenetic tree has been also performed for functional prediction of Hsp70 proteins in other species like Arabidopsis and rice. Members of the Hsp70 proteins have been separated into two large groups with seven subgroups in Arabidopsis. In Arabidopsis, members of the Hsp70 and the Hsp110, which is a subfamily of Hsp70 superfamily and structurally very similar to Hsp70, constituted of two large groups (Lin et al. 2001). Phylogenetic tree analysis of rice Hsp70 proteins indicated that four well-supported clades, called as A, B, C, and D, were separated with each other (Sarkar et al. 2013). These results agree with the findings of other studies, in which different domains including endoplasmic reticulum (ER) retention signal (HDEL sequence), and classical cytoplasmic Hsp70 characteristic C-terminal signal (EEVD sequence) were shown in rice and Arabidopsis. In our study, we also characterized ER Hsp70 protein members in clusters IIIa and IIIb. In addition, cytoplasmic poplar Hsp70 proteins (PtHsp70-01, PtHsp70-02, PtHsp70-13, PtHsp70-16, PtHsp70-17, PtHsp70-23, PtHsp70-24, PtHsp70-25, and PtHsp70-26) mainly cluster on cluster IIIc. So, it can be concluded that certain members of groups were separated from their clusters.

Phylogenetic tree of poplar Hsp70 proteins. The sequences were aligned by CLUSTALW at MEGA5 and the unrooted phylogenetic tree was deduced by neighbor-joining method. The proteins were classified into three distinct clusters. Each family was assigned a different color according to well-known members in other species
To check reliability of the phylogeny, motif compositions were also examined. MEME software was used to determine motifs through complete amino acid sequences of HSP70 proteins. Based on domain compositions of Hsp70, a total of 15 distinct motifs were identified. Conserved amino acid compositions of identified motifs are shown in Supplementary Table S4. The majority of the closely correlated items have common motif composition, providing potential functional similarity among the Hsp70 proteins (Supplementary Fig. S3). For instance, all Hsp70 genes that are tandem duplicated (PtHsp70-01, PtHsp70-02, PtHsp70-03, PtHsp70-12, PtHsp70-13, PtHsp70-16, PtHsp70-17, PtHsp70-19, PtHsp70-23, PtHsp70-24, PtHsp70-25, PtHsp70-26, PtHsp70-28) were found in the cluster IIIc. Those genes demonstrate that there were 15 motifs conserved among these sequences. In addition, other Hsp70 proteins in the phylogenetic tree also have similar motif structure. However, this type of motif sequence conservation or variation between the proteins specifies a functional equivalence or diversification in respect to the different biological functions (Puranik et al. 2012). On the other hand, certain motifs were also defined and clustered into different clades. They might be species specific for poplar. Other than the Hsp70 domain region, Hsp70 proteins also contain some additional conserved motifs that may demonstrate possible function sites or take part in activation of the Hsp70 protein functions. The results obtained from this study also match those observed in earlier studies. In the rice, Sarkar et al. (2013) found that C-terminus and ATP binding domain has high motif similarity. They also indicated similar motif composition in closely related Hsp70 proteins in the phylogenetic tree (Sung et al. 2001). In the beginning of the N-terminal, a highly conserved ATP-binding motif (GID) was also indicated in Arabidopsis (Sung et al. 2001). This motif was the same as the ATP-binding motif of Arabidopsis Hsp70 proteins and defined as motif 2 in our study.
We also analyzed the exon–intron organization of 34 poplar Hsp70 genes to gather some insight information for gene structure (Supplementary Fig. S4). We found a total of six Hsp70 genes without intron, which equals to 17.64 % of overall PtHsp70 genes. The maximum intron numbers was observed in PtHSP70-07 gene with 23 introns. Examination of the intron–exon organization indicated that family members of Hsp70 within the same cluster shared similar gene structures in respect to intron number or exon length. Especially, PtHsp70 genes found in cluster 3b and 3c showed similar exon–intron patterns. The present findings seem to be consistent with those of Sung and colleagues (2001) who found that the intron–exon structure of the Arabidopsis Hsp70 genes differed from proteins which were targeted to different subcellular locations. This also accords with earlier observations, which showed that Hsp70 genes in rice showed similar intron–exon arrangement in their respective phylogenetic clades (Sarkar et al. 2013).
Gene Ontology Annotation
For determination of the functional annotation of identified Hsp70 genes, blast2GO Gene Ontology package was used (Conesa et al. 2005). The GO slim analysis demonstrated the putative involvement of 34 Hsp70 proteins in diverse biological processes, molecular function, and cellular localization (Fig. 3 and Supplementary Table S5). A total of 11 and 4 categories were determined for biological process and molecular function, respectively. The highest represented categories in biological processes were biological regulation, response to stimulus/abiotic stimulus, and cellular/developmental processes. Although they are fewer in number, conditions related to secondary response to metal ion, response to reactive oxygen species, and response to biotic stimulus and protein folding were also observed. Mostly represented categories in molecular functions were binding activity, transcription factor activity, and enzyme regulator activities. Cellular localization prediction indicated that 17 Hsp70 proteins were localized in the cell and its sections including cytoplasm, membrane, cell wall, cytosol, and nucleus. Remaining poplar Hsp70 proteins were found in organelle such as chloroplast, apoplast, Golgi apparatus, and endoplasmic reticulum (Fig. 3 and Supplementary Table S5).

Gene Ontology (GO) distributions for the Hsp70 proteins. The Blast2Go program provides the gene ontology terms under three categories including biological processes, molecular functions, and cellular component
Orthologous Relationships of Hsp70 Genes Between Poplar and Other Species
Physically mapped PtHsp70 genes were compared with those in chromosomes of Arabidopsis, rice, maize, and grapevine for comparative mapping to obtain orthologous relationships of Hsp70s (Supplementary Fig. S5). When compared to these organism’s genomes, specific orthologous relationships could be derived on an average for 45 % proteins for the identified 34 PtHsp70 protein-encoding genes in poplar. Maximum orthology of PtHsp70 genes annotated on the poplar chromosomes was obtained with maize (53 %), followed by rice (47 %), Arabidopsis (45 %), and grapevine (35 %). These findings further support the idea of chromosomal rearrangements which are mainly responsible for shaping the distribution and organization of PtHsp70 genes in poplar, Arabidopsis, rice, maize, and grapevine genomes. According to these data, we can infer that comparative mapping can provide a useful information for understanding the evolutionary process of Hsp70 genes among poplar and other plant species. The present results are also significant for isolation and cloning of similar Hsp70 genes from poplar, using the map-based genomic information of other related plant species for genetic enhancement.
Duplication and Divergence Rate of the PtHsp70 Genes
Some gene families which are composed of multiple copies of genes could possibly evolve primarily through tandem duplication and infrequently large-scale segmental duplications. Gene duplications including segmental or tandem have been reported in many plant TF gene families such as bZIP, NAC, MBF, and bHLH as well as HSPs (Nijhawan et al. 2008; Baloglu et al. 2014b; Wang et al. 2011; Kavas et al. 2015; Puranik et al. 2012; Cannon et al. 2004; Jain et al. 2007). Therefore, we investigated relation of Darwin’s positive selection in divergence and duplication of Hsp70 genes to understand family expansion of the important family members. To elaborate this, non-synonymous (Ka) versus synonymous (Ks) substitution rate ratios (Ka/Ks) were predicted for 13 tandem and 20 segmentally duplicated gene pairs, as well as between orthologous gene pairs of PtHsp70 with those of grapevine (seven pairs), Arabidopsis, rice, and maize (for each five pairs) (Fig. 4). Ka/Ks ratios for tandem duplication differed from 0.02 to 3.01, with an average of 1.07 (Supplementary Table S2), while Ka/Ks for segmentally duplicated gene pairs varied from 0.0096 to 0.1682 with an average of 0.07 (Supplementary Table S3). These results provide further support for the hypothesis that duplicated PtHsp70 genes are under strong purifying selection pressure because their Ka/Ks ratios were below 1 (i.e., <1). Furthermore, duplication action of these tandemly and segmentally duplicated genes can be estimated to have arisen from 0–4 and 2–60 Mya, respectively (Fig. 4). It can be seen from the data in Supplementary Table S6 that the maximum and minimum Ka/Ks values among the orthologous gene pairs of poplar Hsp70 with grapevine (0.08) and poplar Hsp70 with rice–maize (for each 0.02) were obtained, respectively. Even though synonymous substitution rates between rice–poplar and maize–poplar Hsp70 genes were the same, previous divergence was estimated around 24–77 Mya from rice–poplar, when compared to maize–poplar Hsp70 genes (23–134 Mya). Estimated tandem and segmental duplication period (average of 1.25 and 16.03 Mya, respectively) for poplar Hsp70 genes can be used for evolutionary studies with –Arabidopsis (22.5 Mya), –rice (45 Mya), –grapevine (9.8 Mya), and –maize (67.7 Mya) orthologous Hsp70 gene pairs. There were significant differences between tandem (Ka/Ks = 1.07) and segmental (Ka/Ks = 0.07) duplication events of HSP70 gene pairs. Tandemly duplicated genes showed more recent duplication events (average 1.25 Mya), whereas those estimations for segmentally duplicated gene pairs were average of 16.03 Mya. This study therefore suggested that tandem duplication events in poplar Hsp70 genes have played a more predominant role in evolution than segmental duplication events. It can be concluded that combination of information about tandem and segmental duplications in poplar and different organisms help us to understand evolution and maintenance of members of the Hsp70 gene family.

Time of duplication and divergence (MYA) of Hsp70 genes. This is based on synonymous substitution rate (Ks), which estimated using duplicated Hsp70 gene pairs of poplar and orthologous Hsp70 gene pairs between poplar and Arabidopsis or rice or maize or grapevine
Identification of miRNAs Targeting HSP70T transcripts
We implemented the grading schema based on miRU to score the complementarity between miRNA and their target transcript (Zhang 2005). The maximum expectation which is the threshold of the score and an UPE defined as maximum energy to unpair the target site are two important parameters for determination of targets. The maximum expectation threshold value was adjusted to 3.0. A miRNA/target site pair has been discarded if its score is greater than the threshold. The accessibility of messenger RNA (mRNA) target site to miRNA was determined to be one of the important factors involved in target recognition. The psRNATarget server uses RNA for calculation of target accessibility, which is represented by the energy needed to disassociate secondary structure around target. The lesser energy means the higher possibility of small RNA binding and cleavage to target mRNA. There were 19 PtHsp70 genes (PtHsp70-03, PtHsp70-04, PtHsp70-07, PtHsp70-08, PtHsp70-11, PtHsp70-12, PtHsp70-14, PtHsp70-15, PtHsp70-16, PtHsp70-18, PtHsp70-19, PtHsp70-21, PtHsp70-23, PtHsp70-24, PtHsp70-26, PtHsp70-27, PtHsp70-28, PtHsp70-32, PtHsp70-33) targeted by 27 plant miRNAs were found in poplar genome through psRNATarget: A Plant Small RNA Target Analysis Server. On the other hand, certain plant miRNAs did not demonstrate any gene target. PtHsp70-14 and PtHsp70-33 are the most abundant transcripts among the target genes, which were targeted by all 27 plant miRNAs (Supplementary Table S7). The miR390, one of most abundant identified miRNA in different species, regulated several auxin-responsive factors through TAS3-derived tasiARFs (Axtell et al. 2006) and targeted to PtHsp70-14 in our study. It is therefore likely that connections exist between functions of PtHsp70-14 gene (protein folding and oxidation–reduction process) and miR390. Most of the targets identified in our study were responsible for plant growth, development, metabolism, and defense responses to environmental changes. For example, PtHsp70-33 plays important roles in oxidation–reduction process, protein folding, and response to heat/hydrogen peroxide. The miR414, whose target was PtHsp70-33, has been also a widely found miRNA in our study. The miR414 primarily targets transcriptional regulators and transcription factors such as bZIP, WRKY, MYB, B3 family transcription factors, scarecrow, heat shock proteins, and TCP (Guleria and Yadav 2011; Eulgem et al. 2000; Gurley 2000; Jakoby et al. 2002; Suo et al. 2003; Romanel et al. 2009). It can thus be suggested that identification of miRNAs and their targets play a crucial roles for understanding of Hsp70 gene family functions.
Homology Modeling of HSP70 Proteins
BLASTP search was conducted against PDB to build the homology pattern. A total of 28 Hsp70 proteins (PtHsp70-01-02-03-05-06-08-09-10-12-13-14-16-17-18-19-20-21-22-23-24-25-26-27-28-29-30-32-34) with a higher homology were selected. Detection rate was used for estimation of homology modeling in Phyre2, which employs the alignment of hidden Markov models through HMM-HMM search (Söding 2005) in order to remarkably improve accuracy of the alignment. The intensive mode of Phyre 2 utilizes the multi-template modeling to achieve a higher accuracy. In addition, it integrates a new ab initio folding simulation termed as Poing (Jefferys et al. 2010) to model areas of proteins without any significant homology for known structures. All 28 PtHsp70 proteins were modeled at >90 % reliability, and the residue percentage varied from 80 to 100 (Fig. 5 and Supplementary Table S8). The secondary structures were predominantly constituted of α helices and have rare incurrence of β sheets. Thus, all suggested protein structures are assessed to be highly reliable which offers a preliminary basis for understanding the molecular function of PtHsp70 proteins.

Predicated 3D structures of Hsp70 proteins. The structure of 28 Hsp70 proteins with >90 % confidence level is shown
Differential Expression Patterns of Hsp70 Genes in Populus
One of the fundamental objectives of a gene expression profiling on a genomic scale is to identify the genes that are differentially expressed within the organism being examined. This can provide useful clues for the functions of these genes. To acquire information about the drought response of PtHsp70 genes in poplar, a RNA-Seq approach was implemented to data sets obtained from SRA database. Following normalization and transformation analysis, PtHsp70 genes were scored from the highest to the lowest based on their differential expression under control and drought stress conditions. Cossu et al. (2014) used Illumina sequencing technology to obtain a global view of the molecular responses of poplar hybrid to drought. In that study, hybrids between Populus deltoides (L155-079, female) and Populus nigra (71077-2-308, male) were utilized with three treatments (control, C; moderate, D1; and severe drought, D2) for transcriptome analysis. According to high-throughput tag sequencing analysis, we found some PtHsp70 genes (PtHsp70-01-02-03-05-06-20 and 21) whose expression was increased in at least one of the D1 and D2 drought stress treatments (Fig. 6a). Expression level of remaining poplar Hsp70 genes decreased after moderate or severe drought stress application. We also examined RNA-seq data from Tang et al. (2014) who investigated leaf transcriptome derived from Populus trichocarpa seedlings grown in normal condition (control; well watered) and drought stress (D1; water-limited). Based on their transcriptome data, PtHsp70-03-04-07-10-16-20-21-32 and 33 genes were induced by drought stress (Fig. 6b). In addition, we realized that some PtHsp70 genes including PtHsp70-03-20 and 21 had similar expression patterns for both studies. In other words, these genes were significantly upregulated in different poplar species such as poplar hybrids (between Populus deltoides and Populus nigra) and Populus trichocarpa. Lastly, Tang et al. (2013) investigated the responses of the Populus euphratica to soil water deficit using pyrosequencing approach. According to high-throughput sequencing data, only three PtHsp70 genes (PtHsp70-12-23 and 26) gave the response to drought stress with increase in their expression levels after moderate drought stress application (Fig. 6c). It can be concluded that PtHsp70s genes are differentially expressed upon exposure to different drought stress conditions and different Populus species such as hybrid (between Populus deltoides and Populus nigra), Populus trichocarpa, and Populus euphratica.

Heat maps of the differentially expressed Hsp70 genes under control and different drought stress conditions. Three different images constructed based on studies. a Cossu et al. 2014 and C: control (plant 85_4), D1: moderate drought stress (plant 85_12), and D2: severe drought stress (plant 85_42); b Tang et al. 2014 C: control (well watered) and D1: drought stress (water-limited); and c Tang et al. 2013 C: control (well-watered), and D: moderate drought stress summarize expression pattern of 34 PtHsp70. Note that expression values mapped to a color gradient from low (plain green) to high expression (dark red)
Drought Stress Responses of Hsp70 Genes in Populus
In order to reveal the responses of poplar Hsp70 genes to drought stress, we analyzed the expression profiles of PtHsp70s in leaf tissues of two Populus nigra L. clones, Clone S (drought sensitive) and Clone R (drought resistant), with qRT-PCR. A total of 13 PtHsp70 genes including PtHsp70-03-04-09-10-12-16-17-20-23-24-25-26-33 were selected for expression analysis. Based on the literature search, highly expressed Hsp70 genes under the drought stress were determined for quantitative real-time PCR (Neill et al. 1999; Cho and Choi 2009; Song et al. 2009; Cohen et al. 2010). Expression of PtHsp70-03, PtHsp70-04, PtHsp70-09, PtHsp70-10, PtHsp70-12, PtHsp70-17, PtHsp70-20, PtHsp70-23, and PtHsp70-24 was found to be repressed. All downregulated PtHsp70 genes shared similar expression patterns in both controls of Clone S and Clone R. However, a substantial difference between the susceptible and resistant controls was observed. The transcript concentrations of all downregulated PtHsp70 genes in control samples of Clone R are higher than Clone S ones. This result may be explained by the fact that all downregulated PtHsp70 genes in Clone R (PtHsp70-03, PtHsp70-04, PtHsp70-09, PtHsp70-10, PtHsp70-12, PtHsp70-17, PtHsp70-20, PtHsp70-23, PtHsp70-24) might contribute to different biological processes as molecular chaperones under normal condition. In addition, PtHsp70-16 and PtHsp70-26 were viewed to be upregulated in both leaf tissues of poplar, i.e., compared to control samples of Clone S and Clone R, an increased response at the transcription level of PtHsp70-16 and PtHsp70-26 genes was observed after drought stress treatment. So, it is possible to hypothesize that PtHsp70-16 and PtHsp70-26 genes might likely play a role in drought stress response for both sensitive and resistant Populus nigra L. clones.
Although PtHsp70-25 and PtHsp70-33 were downregulated in Clone S, they were induced in Clone R (Fig. 7). PtHsp70-25 and PtHsp70-33 genes might be used for determination of drought-tolerant clones for molecular breeding studies because high expression responses to drought stress for these genes were only observed in Clone R, resistant poplar clone. So, it can be also suggested that PtHsp70-25 and PtHsp70-33 genes might provide adaptation to drought stress for resistant poplar clone. The expression pattern of PtHsp70-16, PtHsp70-26, and PtHsp70-33 genes detected by qRT-PCR is generally consistent with the RNA-seq results. For example, PtHsp70-16 and PtHsp70-33 genes were induced after drought treatment for both Populus nigra, in our study, and Populus trichocarpa in leaf transcriptome study (Tang et al. 2014) (Fig. 6b). In addition, increase in gene expression level of PtHsp70-26 gene was observed in Populus nigra and Populus euphratica for our study and study performed by Tang et al. (2013) (Fig. 6c), respectively. Genotypic variation in the expression response of these genes to drought stress is an interesting observation in this study. The different expression patterns of PtHsp70s imply that PtHsp70 members in different Populus species may be involved in response to drought stress.

Expression profiles of 13 PtHsp70 genes in leaf tissues of two Populus nigra L. clones, Clone S (drought sensitive) and Clone R (drought resistant) with qRT-PCR. Three biological replicates each with three technique replicates were performed and bars represent standard error of the mean for the replicates. SC: clone S-control; ST: clone S-drought stress treatment; RC: clone R-control; RT: clone R-drought stress treatment
Understanding the plant responses at molecular level is crucial to improve the stress tolerance and productivity. In this study, we measured the expression of 13 PtHsp70 genes to analyze their possible drought-responsive roles. Differential expression profiles of the Hsp70 genes under drought stress suggest that some other genes functioning in water deficiency might also be regulated by this family. In other biotic and abiotic stress studies, similar results were reported (Neill et al. 1999; Cho and Choi 2009; Song et al. 2009; Cohen et al. 2010).
In a recent study, expression patterns of rice OsctHsp70 genes including Os05g38530, Os01g62290, Os03g16920, and Os03g16860 were significantly upregulated by salt and drought treatments, indicating that they might have roles in various abiotic stress responses (Jung et al. 2013). These results match those observed in our study, in which PtHsp70-16 and PtHsp70-26 genes were induced with the same pattern under the drought stress conditions for both poplar clones. The present findings seem to be consistent with recent research which found an increase in expression level of PtHsp90 group I, PtHsp90-1a, PtHsp90-1b, and PtHsp90-3 in both the Soligo and Carpacio poplar genotypes under drought stress conditions (Zhang et al. 2013). The results of these studies indicate that some other genes might also be regulated by water deficiency. These differentially expressed gene families including PtHsp70 gene members deserve further investigation into their potential role in different abiotic stresses.
References
Alvim FC, Carolino SM, Cascardo JC, Nunes CC, Martinez CA, Otoni WC, Fontes EP (2001) Enhanced accumulation of BiP in transgenic plants confers tolerance to water stress. Plant Physiol 126:1042–1054
Axtell MJ, Jan C, Rajagopalan R, Bartel DP (2006) A two-hit trigger for siRNA biogenesis in plants. Cell 127:565–577
Bailey TL, Williams N, Misleh C, Li WW (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34:W369–373
Baloglu MC, Inal B, Kavas M, Unver T (2014a) Diverse expression pattern of wheat transcription factors against abiotic stresses in wheat species. Gene 550(1):117–122
Baloglu MC, Eldem V, Hajyzadeh M, Unver T (2014b) Genome-Wide analysis of the bZIP transcription factors in Cucumber. PLoS One 9(4):e96014. doi:10.1371/journal.pone.0096014
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
Boston RS et al (1996) Molecular chaperones and protein folding in plants. Plant Mol Biol 32:191–222. doi:10.1007/BF00039383
Budak H, Akpinar BA, Unver T, Turktas M (2013) Proteome changes in wild and modern wheat leaves upon drought stress by two-dimensional electrophoresis and nanoLC-ESI–MS/MS. Plant Mol Biol 1–15
Cannon SB, Mitra A, Baumgarten A, Young ND, May G (2004) The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol 4:10
Caraux G, Pinloche S (2005) PermutMatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics Applications Note 21(7):1280–1281. doi:10.1093/bioinformatics/bti141
Caruso A, Chefdor F, Carpin S, Depierreux C, Delmotte FM, Kahlem G, Morabito D (2008) Physiological characterization and identification of genes differentially expressed in response to drought induced by PEG 6000 in Populus canadensis leaves. J Plant Physiol 165:932–941. doi:10.1016/j.jplph.2007.04.006
Cho EK, Choi YJ (2009) A nuclear-localized HSP70 confers thermoprotective activity and drought-stress tolerance on plants. Biotechnol Lett 31:597–606
Cohen D, Bogeat-Triboulot MB, Tisserant E, Balzergue S, Martin-Magniette ML, Lelandais G, Ningre N, Renou JP, Tamby JP, Le Thiec D et al (2010) Comparative transcriptomics of drought responses in Populus: a metaanalysis of genome-wide expression profiling in mature leaves and root apices across two genotypes. BMC Genomics 11(1):630
Conesa A, Götz S (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 619832. doi: 10.1155/2008/619832
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21(18):3674–3676
Cossu RM, Giordani T, Cavallini A, Natali L (2014) High-throughput analysis of transcriptome variation during water deficit in a poplar hybrid: a general overview. Tree Genetics & Genomes 10:53–66. doi:10.1007/s11295-013-0661-5
Dhankher OP, Drew JE, Gatehouse JA (1997) Characterisation of a pea Hsp70 gene which is both developmentally and stress-regulated. Plant Mol Biol 34(2):345–52
Du D, Zhang Q, Cheng T, Pan H, Yang W et al (2013) Genome-wide identification and analysis of late embryogenesis abundant (LEA) genes in Prunus mume. Mol Biol Rep 40(2):1937–46. doi:10.1007/s11033-012-2250-3
Eulgem T et al (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci 5:199–206
Chen F, Hayes PM, Mulrooney DM, Pan A (1994) Nucleotide sequence of a cDNA encoding a heat-shock protein (HSP70) from barley (Hordeum vulgare L.). Plant Physiol 106(2):815
Feist AM, Palsson BO (2008) The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotechnol 26:659–667. doi:10.1038/nbt1401
Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD et al (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40:D1178–D1186. doi:10.1093/nar/gkr944
Guleria P, Yadav SK (2011) Identification of miR414 and expression analysis of conserved miRNAs from Stevia rebaudiana. Genomics Proteomics Bioinformatics 9(6):211–217. doi:10.1016/S1672-0229(11)60024-7
Guo AY, Zhu QH, Chen X, Luo JC (2007) GSDS: a gene structure display server. Yi Chuan 29:1023–1026. doi:10.1360/yc-007-1023
Gurley WB (2000) HSP101: a key component for the acquisition of thermotolerance in plants. Plant Cell 12:457–460
Hartl FU (1996) Molecular chaperones in cellular protein folding. Nature 381:571–580. doi:10.1038/381571a0
Hendrick JP, Hartl F (1993) Molecular chaperone functions of heat-shock proteins. Annu Rev Biochem 62:349–384. doi:10.1146/annurev.bi.62.070193.002025
Hill JE, Hemmingsen SM (2001) Arabidopsis thaliana type I and II chaperonins. Cell Stress Chaperon 6(3):190–200
Hu W, Hu G, Han B (2009) Genome-wide survey and expression profiling of heat shock proteins and heat shock factors revealed overlapped and stress specific response under abiotic stresses in rice. Plant Sci 176(4):583–590
Jain M, Nijhawan A, Arora R, Agarwal P, Ray S et al (2007) F-Box Proteins in Rice. genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol 143:14671483. doi:10.1104/pp. 106.091900
Jakoby M, Weisshaar B, Droge-Laser W, Vicente-Carbajosa J, Tiedemann J et al (2002) bZIP transcription factors in Arabidopsis. Trends Plant Sci 7(3):106–111. doi:10.1016/s1360-1385(01)02223-3
Jansson S, Douglas CJ (2007) Populus: a model system for plant biology. Annu Rev Plant Biol 58:435–458. doi:10.1146/annurev.arplant.58.032806.103956
Jefferys BR, Kelley LA, Sternberg MJE (2010) Protein folding requires crowd control in a simulated cell. J Mol Biol 397:1329–1338. doi:10.1016/j.jmb.2010.01.074
Jung KH, Gho HJ, Nguyen MX, Kim SR, An G (2013) Genome-wide expression analysis of HSP70 family genes in rice and identification of a cytosolic HSP70 gene highly induced under heat stress. Funct Integr Genomics 13(3):391–402. doi:10.1007/s10142-013-0331-6
Jungkunz I, Link K, Vogel F, Voll LM, Sonnewald S, Sonnewald U (2011) AtHsp70-15-deficient Arabidopsis plants are characterized by reduced growth, a constitutive cytosolic protein response and enhanced resistance to TuMV. Plant J 66:983–995
Kampinga HH, Craig EA (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11:579–592. doi:10.1038/nrm2941
Kavas M, Baloğlu MC, Atabay ES, Tanman Ziplar U, Yıldız Daşgan H, Ünver T (2015) Genome-wide characterization and expression analysis of common bean bHLH transcription factors in response to excess salt concentration. Mol Genet Genomics. doi:10.1007/s00438-015-1095-6
Kelley LA, Sternberg MJE (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371
Kotak S, Larkindale J, Lee U, von Koskull-Döring P, Vierling E, Scharf KD (2007) Complexity of the heat stress response in plants. Curr Opin Plant Biol 10:310–316. doi:10.1016/j.pbi.2007.04.011
Krishna P, Gloor G (2001) The Hsp90 family of proteins in Arabidopsis thaliana. Cell Stress Chaperone 6(3):238–246
Kumar R, Nagarajan NS, Arunraj SP et al (2012) HSPIR: a manually annotated heat shock protein information resource. Bioinformatics Applications Note 28(21):2853–2855. doi:10.1093/bioinformatics/bts520
Lecharny A, Boudet N, Gy I, Aubourg S, Kreis M (2003) Introns in, introns out in plant gene families: a genomic approach of the dynamic of gene structure. J Struct Funct Genomics 3:111–116
Lee U, Rioflorido I, Hong SW, Larkindale J, Waters ER, Vierling E (2007) The Arabidopsis ClpB/Hsp100 family of proteins: chaperones for stress and chloroplast development. Plant J 49(1):115–127
Letunic I, Doerks T, Bork P (2012) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. doi:10.1093/nar/gkr931
Lin BL, Wang JS, Liu HC, Chen RW, Meyer Y, Barakat A, Delseny M (2001) Genomic analysis of the Hsp70 superfamily in Arabidopsis thaliana. Cell Stress Chaperones 6(3):201–208
Livak JK, Schmittgen TD (2001) Analysis of relative gene expression data using realtime quantitative PCR and the 2−ΔΔCt method. Methods 25:402–408
Lund PA (2001) Molecular chaperones in the cell. Oxford University Press, Oxford
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155. doi:10.1126/science.290.5494.1151
Marron N, Delay D, Petit JM, Dreyer E, Kahlem G, Delmotte FM, Brignolas F (2002) Physiological traits of two Populus×euramericana clones, Luisa Avanzo and Dorskamp, during a water stress and re-watering cycle. Tree Physiol 22:849–858. doi:10.1093/treephys/22.12.849
Maruyama D, Endo T, Nishikawa S (2010) BiP-mediated polar nuclei fusion is essential for the regulation of endosperm nuclei proliferation in Arabidopsis thaliana. Proc Natl Acad Sci USA 107:1684–1689
Mehan MR, Freimer NB, Ophoff RA (2004) A genome-wide survey of segmental duplications that mediate common human genetic variation of chromosomal architecture. Hum Genomics 1(5):335–344
Monclus R, Dreyer E, Villar M, Delmotte FM, Delay D, Petit JM, Barbaroux C, Thiec D, Brechet C, Brignolas F (2006) Impact of drought on productivity and water use efficiency in 29 genotypes of Populus deltoides×Populus nigra. New Phytol 169:765–777. doi:10.1111/j.1469-8137.2005.01630.x
Neill SJ, Burnett EC (1999) Regulation of gene expression during water deficit stress. Plant Growth Regul 29:23–33. doi:10.1023/a:1006251631570
Nijhawan A, Jain M, Tyagi AK, Khurana JP (2008) Genomic survey and gene expression analysis of the basic leucine zipper transcription factor family in rice. Plant Physiol 146:333–350. doi:10.1104/pp. 107.112821
Noel LD, Cagna G, Stuttmann J, Wirthmuller L, Betsuyaku S, Witte CP, Bhat R, Pochon N, Colby T, Parker JE (2007) Interaction between SGT1 and cytosolic/nuclear HSC70 chaperones regulates Arabidopsis immune responses. Plant Cell 19:4061–4076
Park CJ, Bart R, Chern M, Canlas PE, Bai W, Ronald PC (2010) Overexpression of the endoplasmic reticulum chaperone BiP3 regulates XA21-mediated innate immunity in rice. PLoS One 5:e9262. doi:10.1371/journal.pone.0009262
Puranik S, Sahu PP, Srivastava PS, Prasad M (2012) NAC proteins: regulation and role in stress tolerance. Trends Plant Sci 17:1360–1385. doi:10.1016/j.tplants.2012.02.004
Qi Y, Wang H, Zou Y, Liu C, Liu Y, Wang Y, Zhang W (2011) Overexpression of mitochondrial heat shock protein 70 suppresses programmed cell death in rice. FEBS Lett 585:231–239
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N et al (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33:W116–W120. doi:10.1093/nar/gki442
Rochester DE, Winer JA, Shah DM (1986) The structure and expression of maize genes encoding the major heat-shock protein, Hsp70. EMBO J 5:451–458
Romanel EA et al (2009) Evolution of the B3 DNA binding superfamily: new insights into REM family gene diversification. PLoS One 4:e5791
Rouard M, Guignon V, Aluome C, Laporte MA, Droc G, Walde C, Zmasek CM, Perin C, Conte MG (2011) GreenPhylDB v2.0: comparative and functional genomics in plants. Nucleic Acids Res 39:D1095–D1102
Sarkar NK, Kundnani P, Grover A (2013) Functional analysis of Hsp70 superfamily proteins of rice (Oryza sativa). Cell Stress Chaperones 18(4):427–437
Scharf KD, Siddique M, Vierling E (2001) The expanding family of Arabidopsis thaliana small heat stress proteins and a new family of proteins containing alpha-crystallin domains (Acd proteins). Cell Stress Chaperones 6(3):225–237
Schlesinger MJ (1990) Heat shock proteins. J Biol Chem 265(21):12111–12114
Schöffl F, Prandl R, Reindl A (1998) Regulation of the heat shock response. Plant Physiol 117:1135–1141. doi:10.1104/pp.117.4.1135
Shiu SH, Bleecker AB (2003) Expansion of the receptor-like kinase/pelle gene family and receptor-like proteins in Arabidopsis. Plant Physiol 132:530–543. doi:10.1104/pp. 103.021964
Söding J (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951–960. doi:10.1093/bioinformatics/bti125
Song H, Zhao R, Fan P, Wang X, Chen X, Li Y (2009) Overexpression of AtHsp90.2, AtHsp90.5 and AtHsp90.7 in Arabidopsis thaliana enhances plant sensitivity to salt and drought stresses. Planta 229(4):955–964
Song YP, Wang ZL, Bo WH, Ren YY, Zhang ZY, Zhang DQ (2012) Transcriptional profiling by cDNA-AFLP analysis showed differential transcript abundance in response to water stress in Populus hopeiensis. BMC Genomics 13:286. doi:10.1186/1471-2164-13-286
Su PH, Li HM (2008) Arabidopsis stromal 70-kD heat shock proteins are essential for plant development and important for thermotolerance of germinating seeds. Plant Physiol 146:1231–1241
Sung DY, Vierling E, Guy CL (2001) Comprehensive expression profile analysis of the Arabidopsis Hsp70 gene family. Plant Physiol 126(2):789–800
Suo J et al (2003) Identification of GhMYB109 encoding a R2R3MYB transcription factor that expressed specifically in fiber initials and elongating fibers of cotton (Gossypium hirsutum L.). Biochim Biophys Acta 1630:25–34
Suyama M, Torrents D, Bork P (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 34:W609–W612. doi:10.1093/nar/gkl315
Tamura K, Peterson D, Peterson N, Stecher G, Nei M et al (2011) MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Tang H, Bowers JE, Wang X, Ming R, Alam M et al (2008) Synteny and collinearity in plant genomes. Science 320:486–488. doi:10.1126/science.1153917
Tang S, Dong Y, Liang D et al (2014) Analysis of the drought stress-responsive transcriptome of Black Cottonwood (Populus trichocarpa) using deep rna sequencing. Plant Mol Biol Rep. doi:10.1007/s11105-014-0759-4
Tang S, Liang H, Donghui Y et al (2013) Populus euphratica: the transcriptomic response to drought stress. Plant Mol Biol 83:539–557. doi:10.1007/s11103-013-0107-3
Thompson JD, Gibson TJ, Plewniak F et al (1997) The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882. doi:10.1093/nar/25.24.4876
Török Z, Goloubinoff P, Horwáth I et al (2001) Synechocystis HSP17 is an amphitropic protein that stabilizes heat-stressed membranes and binds denatured proteins for subsequent chaperone-mediated refolding. Proc Natl Acad Sci USA 98:3098–3103. doi:10.1073/pnas.051619498
Turktas M, Inal B, Okay S, Erkilic EG et al (2013) Nutrition metabolism plays an important role in the alternate bearing of the olive tree (Olea europaea L.). PLoS ONE 8(3):e59876. doi:10.1371/journal.pone.0059876
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A et al (2006) The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313(5793):1596–1604. doi:10.1126/science.1128691
Vierling E (1991) The role of heat shock proteins in plants. Annu Rev Plant Phys 42:579–620. doi:10.1146/annurev.pp. 42.060191.003051
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78. doi:10.1093/jhered/93.1.77
Wakasa Y, Yasuda H, Oono Y, Kawakatsu T, Hirose S, Takahashi H, Hayashi S, Yang L, Takaiwa F (2011) Expression of ER quality control-related genes in response to changes in BiP1 levels in developing rice endosperm. Plant J 65:675–689
Wang Y, Lin S, Song Q, Li K, Tao H et al (2014) Genome-wide identification of heat shock proteins (Hsps) and Hsp interactors in rice: Hsp70s as a case study. BMC Genomics 15:344. doi:10.1186/1471-2164-15-344
Wang J, Zhou J, Zhang B, Vanitha J, Ramachandran S et al (2011) Genome-wide expansion and expression divergence of the basic leucine zipper transcription factors in higher plants with an emphasis on sorghum. J Integr Plant Biol 53(3):212–231. doi:10.1111/j.1744-7909.2010.01017.x
Wang X, Liu W, Chen X, Tang C et al (2010) Differential gene expression in incompatible interaction between wheat and stripe rust fungus revealed by cDNA -AFLP and comparison to compatible interaction. BMC Plant Biol 10:9. doi:10.1186/1471-2229-10-9
Waters ER, Lee GJ, Vierling E (1996) Evolution, structure and function of the small heat shock proteins in plants. J Exp Bot 47:325–338. doi:10.1093/jxb/47.3.325
Yang Z, Gu S, Wang X, Li W, Tang Z et al (2008) Molecular evolution of the cpp-like gene family in plants: insights from comparative genomics of Arabidopsis and rice. J Mol Evol 67:266–277. doi:10.1007/s00239-008-9143-z
Yildirim K (2013) Transcriptional and physiological responses to drought stress in Populus nigra L. Dissertation, Middle East Technical University
Zhang Y (2005) miRU: an automated plant miRNA target prediction server. Nucleic Acids Res 33 (Web Server issue) W701–4
Zhang J, Li J, Liu B, Zhang L, Chen J, Lu M (2013) Genome-wide analysis of the Populus Hsp90 gene family reveals differential expression patterns, localization, and heat stress responses. BMC Genomics 14:532. doi:10.1186/1471-2164-14-532
Zhang Y, Wang M, Chen J, Rong J, Ding M (2014) Genome-wide analysis of HSP70 superfamily in Gossypium raimondii and the expression of orthologs in Gossypium hirsutum. Yi Chuan 36(9):921–33. doi:10.3724/SP.J.1005.2014.0921. (Article in Chinese)
Zhang J, Liu B, Li J, Zhang L, Wang Y, Zheng H, Lu M, Chen J (2015) Hsf and Hsp gene families in Populus: genome-wide identification, organization and correlated expression during development and in stress responses. BMC Genomics 16:181. doi:10.1186/s12864-015-1398-3
Acknowledgments
This work was financially supported by the Kastamonu University Scientific Research Project Management Coordination Unit under Grant No: KÜBAP-01/2014-09. We would like to thank Dr. Tuncay PORSUK from Director of Inner Anatolia Forestry Research Institute for his help in providing the Populus nigra L. clones.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
Fig. S1 Examples of P. nigra drought resistant and sensitive clones used in the experiments. SC = Sensitive clone-control, ST = Sensitive clone-drought treated, RC = Resistant clone-control, RT = Resistant clone-drought treated. Asterisks indicate leaves (healthy, fully expanded, at approximately 6–10 nodes from the apex) collected for RNA isolation. (JPEG 377 kb)
ESM 2
Fig. S2 Percentage of Hsp70 genes on each poplar chromosome to show their distribution abundance. (JPEG 36 kb)
ESM 3
Fig. S3 Schematic representation of conserved motifs (obtained using MEME) in Hsp70 proteins. Different motifs are represented as differently-colored boxes at the N-terminal and C-terminal region for the transcription regulatory region. (JPEG 197 kb)
ESM 4
Fig. S4 Exon/intron structures of the PtHsp70 genes. The Hsp70 family members were classified according to Fig. 2. The values in parentheses show the number of corresponding classes of Hsp70 genes. Green boxes represent exons and black lines represent introns. (JPEG 274 kb)
ESM 5
Fig. S5 Comparative physical mapping of Hsp70 genes in different organisms. Comparative physical mapping revealed high degree of orthologous relationships of Hsp70 genes located on chromosomes of poplar with (A) Arabidopsis, (B) rice, (C) maize and (D) grapevine. (JPEG 223 kb)
ESM 6
Table S1. Summary of Hsp70 gene family factors of plants (XLSX 11 kb)
ESM 7
Table S2. The Ka/Ks ratios and estimated divergence-time for tandemly-duplicated Hsp70 proteins. (DOCX 18 kb)
ESM 8
Table S3. The Ka/Ks ratios and estimated divergence time for segmentally-duplicated Hsp70 proteins. (DOCX 29 kb)
ESM 9
Table S4. Amino acid composition of the poplar Hsp70 motifs. Single letters of the motif match that letter; and the letter groups in square brackets match any letters in the group. (DOCX 15 kb)
ESM 10
Table S5. Blast2Go annotation details of Hsp70 protein sequences. (XLSX 15 kb)
ESM 11
Table S6. The Ka/Ks ratios and estimated divergence time for orthologous Hsp70 proteins between poplar, Arabidopsis, rice, maize and grapevine. (DOCX 16 kb)
ESM 12
Table S7. miRNA targets identified by psRNA Target. (XLSX 16 kb)
ESM 13
Table S8. Reliability and the residue percentage of 28 candidate HSP 70 proteins chosen for homology modeling. (DOC 36 kb)
ESM 14
Table S9. List of primers used in quantitative real-time-PCR expression analysis of Hsp70 genes. (DOC 35 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Yer, E.N., Baloglu, M.C., Ziplar, U.T. et al. Drought-Responsive Hsp70 Gene Analysis in Populus at Genome-Wide Level. Plant Mol Biol Rep 34, 483–500 (2016). https://doi.org/10.1007/s11105-015-0933-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-015-0933-3