
Abstract
Background and Aims
Selenium accumulation in vegetation has resulted in toxicity in livestock grazing on phosphate mine soils in Southeastern Idaho.
Methods
Plant and soil samples were collected from sites located near phosphate mines. Soil physicochemical properties, Se speciation, and Se distribution from a sequential extraction procedure (SEP) were examined in relation to bioavailability in the Se-hyperaccumulator, western aster (Symphyotrichum ascendens Lindl.). Selenium-hyperaccumulators are plants that can absorb over 1000 mg Se kg−1 DM (Dry Matter).
Results
Chemical analyses revealed that western aster contained Se exceeding 6000 mg kg−1 DM. Soil speciation results indicated that selenite (SeO3 2−) was dominant with lower levels of selenate (SeO4 2−) present. This was expanded using an SEP that accounted for six fractions. Regression analyses indicated a strong relationship for western aster Se and the water-soluble and phosphate-extractable SEP fractions combined (R 2 = 0.85). Once carbonate, amorphous Fe-oxide, organic, and residual Se fractions were factored into the analysis, the relationship decreased. A strong relationship between selenate and the water-soluble Se fraction was also observed (R2 = 0.83).
Conclusions
Soluble and phosphate-extractable Se were determined to be “bioavailable fractions” for western aster. Thus, simple water extractions can be used for quick assessment of Se bioavailability and provide a means to identify potentially hazardous areas locations.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Selenium (Se) is a trace element, with levels in natural soils ranging from 0.01 to 2.0 mg Se kg−1 (Shacklette and Boerngen 1984). Elevated levels of Se can result from a variety of anthropogenic processes, such as those associated with phosphate mining in southeast Idaho, where it is not uncommon to find soil Se levels much higher than normal background levels (McNeal and Balistrieri 1989). To date, there have been hundreds of reported grazing deaths due to livestock ingestion of plants growing on these contaminated soils (Buck and Jones 2002; Davis et al. 2012; Edmonson et al. 1993; Fessler et al. 2003). Western aster (Symphyotrichum ascendens Lindl.) is the plant responsible for the vast majority of these deaths in SE Idaho, with toxicity linked to its Se content (Pfister et al. 2013). Pfister et al. (2013) reported western aster tissues reaching levels as high as 4455 mg Se kg−1 DM, while western aster collected from mine wastes have tested as high as 13,000 mg Se kg−1 DM (Davis et al. 2011). Western aster is considered a Se-hyperaccumulator. Hyperaccumulator plants are defined as plants that uptake over 1000 mg Se kg−1 DM (Dry Matter) in above-ground biomass (White 2016). Deaths from grazing on this plant have occurred on previously reclaimed land, where Se-laden shale overburden was used as a replacement for topsoil prior to 1996 (Buck and Jones 2002; Davis et al. 2011). Mine reclamation is a process used to stabilize locations that have been disturbed and establish productive vegetation (Richards et al. 1998). During this time, environmental impact assessments did not identify Se levels as a concern in overburden materials. However, land reclaimed after 1996 used the original salvaged topsoil as the topsoil medium (Buck and Jones 2002). This change in practice was established in accordance with the Bureau of Land Management (BLM), a federal land holding agency, and the Clean Water Act (CWA), which regulates pollutant standards in waters of the US, in 1996 (Blanchard et al. 2002). In the Blackfoot River (ID, USA), Se levels have exceeded acute (0.02 mg L−1) and chronic (0.005 mg L−1) aquatic life criteria for the state of Idaho for a number of years due to snowmelt and base flow (IDEQ 2007; Meyers 2013). Mining throughout this watershed created pathways for contamination of waterways (Knudsen and Gunter 2004; Meyers 2013).
Selenium is considered an essential micronutrient to ruminants but becomes toxic at elevated levels (Davis et al. 2012; Davis et al. 2013; Edmonson et al. 1993). There is a narrow range between dietary and toxic dosages of Se in animals. Toxicity typically occurs when animals ingest between 3 to 8 mg Se kg−1 of body weight (NAS-NRC 1983). Because of this, Se toxicity occurs easily in Se-enriched soils and usually within a 24 to 72-h period (Davis et al. 2013; Shortridge et al. 1971).
In soil environments, Se can exist in four oxidation states (+6, +4, 0, and −2). At neutral to alkaline pH and in aerobic environments, the dominant species present are selenate (SeO4 2−) and selenite (SeO3 2−) (Spallholz 1994; Burau 1989). Soluble Se oxyanions are of particular concern because of their potential bioavailability or ability to be incorporated into biota (John and Leventhal 1995; Spallholz 1994). For example, western aster can take up greater than normal amounts of Se, primarily as selenate and, to a lesser extent, selenite (Spallholz 1994). This explains its ability to grow on seleniferous soils while nonaccumulating plants cannot (Davis et al. 2013). At high levels, Se phytotoxicity is typically related to Se-induced oxidative stress and distorted protein structures (Gupta and Gupta 2016).
It is understood that processes at the solid/water interface control the potential bioavailability of trace elements in the environment (Luoma 1983). Hence, sequential extraction procedures (SEP) are often employed to determine soil components associated with the target element of concern. For this study, we used a SEP described by Amacher (2010). Sequential extraction procedures involve a series of rinses and extractions with increasingly stronger reagents (Martens and Suarez 1997; Tessier et al. 1979; Zhao et al. 2005). They provide a more detailed understanding of the potential solubility and bioavailability of trace elements compared to the use of total concentrations. The fractions examined, using the Amacher (2010) procedure, are as follows: water-soluble Se, PO4 3−-extractable Se, carbonate associated Se, amorphous iron oxide associated Se, organic Se, and residual elemental forms of Se. These soil fractions presumably vary in their relation to Se bioavailability to plants. The water-soluble fraction accounts for soluble selenate and some selenite, while the PO4 3−-extractable fraction accounts for Se that is ligand exchangeable (Amacher 2010). Carbonate-associated Se is removed using ammonium acetate (NH4AOc), which has previously been used for exchangeable metals by Chapman (1965). Exchangeable metals are considered to be those that are bound to mineral and organic soil components but can be exchanged with one another (Chapman 1965). However, NH4AOc can also dissolve carbonate minerals, which causes metal solubilization (Tessier et al. 1979; Wagemann et al. 1977; Jackson 1958; Chapman 1965). Ammonium oxalate (C2H8N2O4), in the dark, was used to extract Se bound to amorphous iron oxides (Amacher 2010). This reagent was originally employed by Tamm (1922) to remove amorphous iron, aluminum, and silicon oxides from soils. At room temperature and in the dark, this reagent is specific to dissolving amorphous iron oxides (Gleyzes et al. 2002). Previous works indicate that organic Se comprises a large portion of Se in soil, either derived from parent material or partitioned within organic matter (Ryser et al. 2006; Ryser et al. 2005; Stillings and Amacher 2004; Yamada et al. 1998). In seleniferous soils, large quantities of Se are found in humified organic matter (Moreno Rodriguez et al. 2005). In the Amacher (2010) procedure, potassium persulfate (K2S2O8) was used to oxidize organic matter, releasing bound or occluded Se. Residual fractions are accounted for in the final SEP extraction, similar to Martens and Suarez (1997). This fraction is composed of trace elements bound or occluded in minerals that cannot be released for long periods of time (Tessier et al. 1979).
Understanding Se speciation and its various phases in soils is necessary to develop methods to reduce its solubility in contaminated soils. Accordingly, the objectives of this study were to:
-
(1)
Characterize the physicochemical properties of Se present in contaminated soils that affect its bioavailability
-
(2)
Determine Se speciation in soluble and exchangeable phases in soils because of its important influence on bioavailability, mobility, and toxicity.
-
(3)
Evaluate a soil sequential extraction procedure (SEP) that identifies six phases of Se in soils.
-
(4)
Relate Se levels from the SEP fractions and speciation to Se uptake in the Se-hyperaccumulator, western aster.
Materials and methods
Sample collection
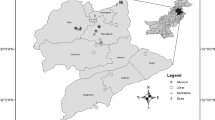
Five study sites associated with phosphate mining operations near Soda Springs, Idaho, USA were chosen. Study sites included three pre-1996 reclaimed locations, Sites A, B, and C, and two sites reclaimed post-1996, D and E (Fig. 1). Sites reclaimed prior to 1996 used the Se-laden shale overburden as the reclamation soil medium. Those reclaimed after 1996 used the original salvaged topsoil as the reclamation soil medium, as per regulatory changes that are in accordance with the BLM and CWA (Blanchard et al. 2002).

Map of five sites in southeastern Idaho. Note: Two sites are superimposed due to close proximity at this magnification
Sampling transects were established in a Y-shape for the five different locations (Fig. 2). In total, 78 samples were collected. Sixteen samples were collected from Sites A, B, C, and D locations and fourteen were collected from Site E.

Transect sampling diagrams for the five study locations. Black circles indicate soil sampling points, and dashed circles indicate vegetation sampling points. Note: Sites A-D contain 16 sampling points, and Site E contains 14. Diagrams are not to scale. a N = North; S = South; E = East; W = West; C = Center point
One center point was established at each study location where high Se levels were presumed to occur, determined by the presence of western aster. From the center point, transect lines were extended 30 m in three directions. Soil samples were collected every 6 m in each direction (0–20 cm depth). Soils were placed in plastic sampling bags that were stored in a plastic cooler for transport. Plant and soil samples were taken from the same transect points in order to establish a relationship between plant Se levels and associated soil fractions. Plant samples were collected from four points on each transect using a quadrat with dimensions of 0.5 by 1.0 m. Plants were identified by species by a field botanist, separated into paper bags by species, and washed. Fifty-seven plants were collected from Site A, 35 from Site B, 36 from the Site C, 47 from Site D, and 36 from Site E.
Following collection, soil samples were air-dried at ambient temperature and passed through a 2 mm sieve in order to remove coarse fragments. Prior to plant digestions, shoots were separated from roots and split into two groups. The first half of the shoots was washed with 0.50% sodium laurel sulfate followed by 0.10 M HCl. Shoots were then thoroughly rinsed with distilled water. The second half of the shoot was left unwashed. This was completed in order to confirm that Se was resultant of root absorption and not from soil deposition on the plant surface. The two separate halves (shoots) were placed in separate beakers, covered with aluminum foil, and dried for 48 h in a 70 °C oven. Once the two halves were dried, shoots were ground in a stainless steel coffee grinder (Capresso Coffee Grinder, Model 501).
Soil characterization
Soils were analyzed for the physicochemical properties particle size (sand, silt and clay), pH, total organic carbon (TOC), and total inorganic carbon (TIC) in duplicate. Prior to particle size analyses, soil samples were pretreated for removal of organic matter, carbonates, and secondary free iron oxides. Removal of these components can enhance separation of soil aggregates by decreasing particle coatings and cementing agents (Kunze and Dixon 1986). Pretreatments for organic matter were completed using hydrogen peroxide and magnesium chloride and for iron oxides using sodium-citrate dihydrate and sodium dithionite as described in procedures modified by Kunze and Dixon (1986). Carbonate pretreatments followed a procedure described by Saffari et al. (2009), using 0.5 M HCl. Following pretreatments, particle size was determined using the hydrometer method (Gee and Bauder 1986). Soils from Sites A, B, C, and D were silt loam and Site E soils were considered a loam.
Soil pH was determined in a 1:1 ratio with water using a pH 718 STAT Titrino (Metrohm, Herisau, Switzerland) equipped with a combination pH electrode (Thomas 1996). For carbon analyses, samples were ground to a fine powder and analyzed using a TOC analyzer that is capable of measuring TC, TIC, TOC, separately (Skalar PrimacSLC TOC Analyzer – Model CS22, De Breda, Netherlands).
Selenium sequential extraction procedure
Sequential extraction procedures generally follow a similar format to the Tessier et al. (1979) procedure. This was modified by Martens and Suarez (1997) for Se, which was further modified by Amacher (2010).
Six fractions of soil Se were separated through the following sequential extractions. For each sample in duplicate, 0.500 g of air-dried soil was weighed and placed into 50 mL polypropylene centrifuge tubes. The procedure is outlined as follows:
-
a.
Water-soluble Se. The first extraction was completed using 25.0 mL of deionized water, vortex mixed, and shaken for 2 h at 120 cycles/min using an Innova 4230 incubator shaker (New Brunswick Scientific, Edison, NJ). After extractions were complete, samples were centrifuged at 1500 rpm for ten minutes. Supernatants were collected following this (25 mL).
-
b.
Phosphate (PO 4 3−)-Extractable Se. The second extraction was completed on the residue from step a, using 25.0 mL of pH 7.2 0.01 M KH2PO4/K2HPO4 buffer, vortex mixed and shaken for 2 h at 120 cycles/min. After extractions were complete, samples were centrifuged at 1500 rpm for ten minutes. Supernatants were collected following this (25 mL).
-
c.
Carbonate-Associated Se. The third extraction was completed on residues from step b, using 25.0 mL of pH 5 1 M NH4OAc. This was left stay for 30 min to allow for initial reaction with carbonates to subside. Following this, the suspension was mixed using a vortex mixer and shaken for 24 h at 120 cycles/min. After extractions were complete, samples were centrifuged at 1500 rpm for ten minutes. Supernatants were collected following this (25 mL).
-
d.
Amorphous Iron oxide-Associated Se. The forth extraction was completed on residues from step c, using 25.0 mL of 0.2 M C2H8N2O4 + 0.2 M oxalic acid (C2H2O4) added to samples in the dark. The suspension was mixed using a vortex mixer and shaken for 2 h at 120 cycles/min with the shaker window covered to prevent light from interacting with soil solutions. After extractions were complete, samples were centrifuged at 1500 rpm for ten minutes, and 25 mL supernatants were collected. Following this, 5 mL of oxalate solution was added to centrifuge tubes, the residue was re-suspended using the vortex mixer, and centrifuged again for 5 min at 1500 g. The final supernatant (5 mL) was collected, added to the initial 25 mL supernatant, and diluted to a 50 mL volume.
-
e.
Organic Se Compounds. The fifth extraction was completed on residues from step d, using 25.0 mL of 0.1 M K2S2O8. The suspension was mixed using a vortex mixer. Samples were placed in an oven set to 90 °C for 2 h. After heating, samples were cooled to ambient temperature and centrifuged for 10 min at 1500 g. Following this, an additional 5 mL of 0.1 M K2S2O8 was added to the centrifuge tube, re-suspended using the vortex mixer, and centrifuged again for 5 min at 1500 g. The final supernatant (5 mL) was collected, added to the initial 25 mL supernatant, and diluted to a 50 mL volume.
-
f.
Residual Se. The final extraction was completed on residues from step e, using 2.5 mL of concentrated, trace metal grade nitric acid (HNO3). The suspension was mixed using a vortex mixer and placed in boiling water for 30 min. Following this, samples were cooled to ambient temperature and 20 mL of deionized water was added. Samples were vortex mixed and placed in a boiling water bath for 90 min. Samples were then cooled to ambient temperature again and centrifuged for 10 min at 1500 g. Supernatants were collected following this (25 mL). Deionized water (5 mL) was added to the centrifuge tubes, re-suspended via the vortex mixer, and centrifuged for 5 min at 1500 g. The final supernatant (5 mL) was collected, added to the initial 25 mL supernatant, and diluted to a 50 mL volume.
Following centrifugation, supernatants were filtered and saved for analysis. Residues were washed with 95% ethanol three times in between extractions and left to dry. Samples were analyzed using a Spectro Arcos ICP-AES (SPECTRO Analytical Instruments, Inc., Mahwah, NJ). Six method blanks were analyzed that corresponded with each fraction, with no detectable Se found.
Selenium speciation
Selenium speciation was determined for soils in duplicate from two points along each of the five transects. In order to remove soluble and exchangeable forms, Se was extracted from 1.0 g of soil using 25 mL of 0.05 M potassium phosphate dibasic (K2HPO4) in metal free polycarbonate centrifuge tubes and shaken for 24 h on an incubator shaker. The suspension was centrifuged at 9000 rpm, and the resulting supernatant was filtered through a 0.45 μm nitrocellulose membrane filter (Alam et al. 2000). The filtrate was analyzed for Se speciation using IC-ICP-MS. The IC system consisted of an Agilent 1200 series quaternary pump (Santa Clara, CA, USA) and a Hamilton PRP X-100 anion exchange column (Reno, NV, USA). The mobile phase used for transporting analytes through the IC column was pH 5 10 mM citric acid. The ICP-MS system consisted of an Agilent 7500cx. Instrument calibration was accomplished using prepared sodium selenate and selenite standards. The Se concentrations of the standards were determined against a NIST standard (Inorganic Ventures, Christiansburg, VA, USA) using ICP-MS with quantification using an m/z ratio of 77.
Prior to IC-ICP-MS analysis, samples were filtered a second time through 0.2 μm syringe filters. The sample was then analyzed by IC-ICP-MS for speciation. Quality controls for both Se speciation analysis and total Se analysis included blanks, random duplicates and spike recovery. Method detection limits for selenate and selenite were 0.8 and 0.9 μg L−1, respectively (Unrine et al. 2007).
Plant digestion
Dried plant shoots were digested in a 90 °C heat block using HNO3. Digested samples were collected and analyzed for Se using a Perkin Elmer NexION 300D ICP-MS (Shelton, CT, USA; Hall and Winger 2012). Quality assurance/quality control samples were obtained from a tomato leaf standard, and were digested in order to determine if Se was found in reagents or absorbed during filtration.
Statistics
Statistically significant differences between SEP fractions for all soils (78 total) from each site were compared using an analysis of variance (ANOVA) and least significant difference (LSD) to identify whether SEP Se fractions differed by site. The six SEP fractions (from 20 soils) were then related to above ground western aster (Symphyotrichum ascendens Lindl.) Se levels in soils from corresponding points along transects. Selenium speciation was related to the first two SEP fractions (water-soluble and PO4 3−-extractable). Analyses were completed using simple linear regression in the program, JMP® Pro 11.0.0 statistical software (SAS Institute Inc. 2013). Prior to analyses, the Shapiro-Wilk test confirmed approximate normality among specific data points used in the regression analysis. Following this, a regression equation was generated for western aster Se data from specific transect points and related to the sum of each fraction (water-soluble alone, water-soluble + PO4 3−-extractable Se, water-soluble + PO4 3−-extractable Se + carbonate associated Se, etc.). This was completed in order to compare relative bioavailability among fractions and identify any marked differences between successive phases.
Results
Physicochemical characteristics of reclaimed phosphate mine soil
Physicochemical properties of phosphate mine soils derived from overburden waste materials varied by site, which may be indicative of differences in reclamation practices (Table 1). Soils from Sites A, B, and C were reclaimed pre-1996 and Sites D and E were reclaimed after 1996. For example, Site D and E soils exhibited finer textures compared with the coarser textures of Sites A, B, and C. All soils were considered circumneutral to alkaline, with site average pH values ranging from 6.75 to 7.92. Site A, B and C soils were generally higher in pH than Site D and E soils, which was attributed to their much higher carbonate content or total inorganic carbon (TIC).
Soil selenium sequential extraction procedure
The distribution of Se phases in the six SEP fractions and total Se are summarized in Table 2, with mean comparisons determined from least significant difference (LSD) results. Total Se means, determined from the summation of SEP fractions, were highest for soils from Site B (217 mg Se kg−1 soil). The second highest mean was also observed in Site C soils (54.7 mg Se kg−1 soil). Selenium levels were lower for Sites A and E (35.6 and 40.2 mg Se kg−1 soil, respectively) and were especially low in soils sampled from Site D (21.1 mg Se kg−1 soil). Statistically, the Site B total Se mean differed from Sites A, C, D, and E. Sites D and E soils were reclaimed post-1996 with the original salvaged topsoil that contained low levels of Se compared to sites A, B, and C that were reclaimed pre-1996 using Se rich overburden material (Blanchard et al. 2002).
For the individual SEP fractions, we define Se levels up to 2 mg Se kg−1 soil as low and between 3 and 15 mg Se kg−1 as moderate. Selenium levels above 16 mg Se kg−1 soil are considered high. In general, lower levels of Se were observed in water-soluble fractions or were below the limit of detection of the ICP-AES (Table 2). Water-soluble Se was not detected (ND) in any of the sixteen soils from both Sites D and E. Nine, eleven, and three of the sixteen soils from Site C, A, and B, respectively, showed no detectable water-soluble Se. The maximum water-soluble Se content was observed at Site B (10.7 mg Se kg−1 soil). The Site B water-soluble Se mean was also considered statistically different from those observed in Sites A, C, D, and E.
Like the water-soluble Se fraction, PO4 3−-extractable Se was lower than the other examined fractions or was not detected in most soils. Similar to its water-soluble fraction, soils from Site B exhibited the maximum levels of PO4 3−-extractable Se (3.33 mg Se kg−1 soil). The Site B mean was also considered statistically different from means observed in the other sites. Thirteen soils from Sites A and C, and three from Site B contained no detectable Se in the PO4 3−-extractable form. One soil from Site E and two soils from Site D contained no detectable Se in this fraction.
Selenium levels within the carbonate fraction were higher and more appreciable than the water-soluble and PO4 3−-extractable fractions for many of the soils. Again, moderate Se values were noted in soils from Site B, especially at the center point (15.7 mg Se kg−1 soil), whereas carbonate Se was only detected in three Site D soils and one Site E soil. Site B mean soil Se was considered statistically different from Se found in the other four sites for this particular fraction.
Levels in the amorphous iron oxide fraction were comparable to carbonate fractions. However, Se associated with amorphous iron oxides was not detected in many of the soils examined. This phase was not observed in any of the soils sampled from Site D. Only one soil sampled from Site A contained Se within this fraction and three within Site E. Interestingly, the maximum level of Se was indicated in soils from Site E for this fraction (10.3 mg Se kg−1 soil). Moderate levels of Se were also observed in Site B soils with a maximum of 9.74 mg Se kg−1 soil. Selenium in this fraction was not detected in six and five of the soils sampled from Sites C and B, respectively. For soils with detectable levels of Se in this fraction, levels were between 3.87 and 10.3 mg Se kg−1 soil. Unlike previous fractions, the Site B mean Se was not considered different from Sites C and E. Sites C and E mean Se amorphous iron oxide values were also considered statistically the same as those found in Sites A and D.
Most of the soils exhibited moderate to high abundances of organic Se, with the exception of six Site C soils and one soil from Site A, where they were not detected. Comparisons between all site means indicated that Sites A, C, D, and E were not considered statistically different. Sites B, C, and E were also not statistically different. The maximum organic Se level was observed in Site B (44.2 mg Se kg−1 soil). Site C soils were also high in organic Se along some segments of the transect line as well, with a maximum content of 35.2 mg Se kg−1 soil.
Residual Se was the dominant fraction for most of the soils sampled, having the highest abundance of Se. No detectable Se for this fraction was observed in two of the Site C soils, which were also below detection throughout the entire SEP. Excluding those below detection, seven of the Site C soils were below 10 mg Se kg−1. Eight out of the sixteen soils sampled from Site B were well above 200 mg Se kg−1 soil for residual Se, with a maximum of 385 mg Se kg−1 soil observed. Soils from Site B were considered to be statistically different from the other 4 sites. One of the Site C soils from the first northwest point along the transect exceeded 100 mg Se kg−1 soil.
Soil selenium speciation
Selenium speciation was determined for soils extracted with K2HPO4. This extraction includes both water-soluble and exchangeable Se species (Zhao et al. 2005). Overall, selenite predominated with lower levels of selenate present (Fig. 3). Highest levels of selenite were observed in soils from Site B point NE2 (6.21 mg Se kg-1 soil), and highest selenate levels were observed in the center point of Site B (1.16 mg Se kg−1 soil). Intermediate levels of selenite were observed in Sites A and C soils. Lowest selenite levels were found in Sites D (0.06 mg Se kg−1 soil) and E (0.07 mg Se kg−1 soil), which are sites that have been reclaimed post-1996. No selenate was detected in these soils.

Speciation for selenium in soils from two points from each transect. Insert shows maximized view for Sites D and E. *A = Site A; B = Site B; C = Site C; D = Site D; E = Site E; CO = transect center points. Refer to Fig. 2 for sampling plan diagram for each point
Selenium speciation results were related to the water-soluble and PO4 3−-extractable SEP fractions using simple linear regression. A statistically significant relationship was observed for selenate and water-soluble Se (R2 = 0.83; P = 0.0002; Fig. 4a), with no significant relationship observed for selenite (R2 = 0.30; P = 0.1007). Weak relationships were observed for selenate and the PO4 3−-extractable Se fraction (R2 = 0.49; P = 0.0244) and for selenite and the PO4 3−-extractable Se fraction (R2 = 0.57; P = 0.0117; Fig. 4b).

a and b: (a) Simple linear regression analysis demonstrating the relationship of soil selenate from speciation analyses and the water-soluble selenium fraction from the soil sequential extraction procedure (SEP) and (b) the relationship of soil selenite and the phosphate (PO4 3−) extractable fraction from the soil SEP
Plant shoot selenium
Selenium levels were determined for plant shoots found at the five sites. The only plant species sampled that was considered to be a hyperaccumulator was western aster, which is a native perennial forb. Selenium levels in western aster and other predominating plants found are summarized in Table 3. Many of these species are considered Se secondary accumulators and can contain hundreds of mg Se kg−1 in shoots (Ellis and Salt 2003).
Overall, Se levels were consistently higher in plants throughout the Site B transect. Of the plants sampled, the western aster species exhibited the highest Se levels. The highest level was observed at the center point of Site B (6823 mg Se kg−1 DM). In secondary accumulators, the highest Se level was observed in crested wheatgrass (Agropyron cristatum L.; 644 mg Se kg−1 DM). High alfalfa (Medicago sativa L.) Se levels were also observed along all three lines of this transect, especially at point NW3 (364 mg Se kg−1 DM). The highest Se level in brome grass (Bromus) was observed at the second southern point along this transect (267 mg kg−1 DM).
The other two pre-1996 reclaimed sites, A and C, also contained elevated Se levels in plant samples. Western asters found at the center point of Site A were elevated in Se levels (2635 mg Se kg−1 DM), while those sampled from Site C were highest at the center point (1972 mg Se kg−1 DM). Of the more predominant secondary accumulators, the highest Se levels were observed in bulbous bluegrass (Poa bulbosa L.; 353 mg kg−1 DM) at the Site A center point. Elevated alfalfa levels were also indicated at this same center point as well (307 mg kg−1 DM).
Lower plant Se levels were observed in the post-1996 reclaimed sites, D and E. Only two western aster plants were sampled along the Site D transect and one along the Site E transect. The maximum levels were 35.9 mg Se kg−1 DM in Site E and 17.5 mg Se kg−1 DM in Site D western asters, which also corresponds with lower Se soil levels measured in these sites.
Western aster Se and soil Se fractionation relationship
The relationship between the hyperaccumulator, western aster, shoot Se levels, and Se levels in the six SEP fractions was evaluated using simple linear regression. Multiple linear regression was not used for this analysis due to high multicollinearity between fractions. For non-detectable Se where no values could be determined by ICP-AES, a proxy value of the limit of detection (LOD) divided by the square root of two was used in the analysis. This is a standard replacement technique for values below the quantification level, when they are considered small but not known (Hopke et al. 2001). This technique has been shown to reduce the overall error caused by replacement, compared to methods such as replacement with LOD/2 or with zero (Croghan and Egeghy 2003).
A bivariate regression analysis was completed that involved a progressive summation of the six SEP fractions together (Table 4). This was employed in order to determine fractions that promote an increase in Se bioavailability. For water-soluble Se alone, a highly significant relationship was observed (Table 4 and Fig. 5). This suggests that 81% of Se found in western aster can be explained by the water-soluble fraction of Se in soils. For the summation of water-soluble and PO4 3−-extractable Se, a highly significant positive relationship with a slight increase was also observed (85%).

Simple linear regression analysis demonstrating the relationship of western aster shoot Se levels with the water-soluble fraction from a soil sequential extraction procedure (SEP)
Once carbonate associated Se was summed with the previous two fractions, the relationship decreased (79%). Further large decreases in the relationship were observed once amorphous iron oxide-associated Se, organic, and residual fractions were summed with the previous fractions. Because the residual fraction acts as a total Se value, this may explain the slight increase in the observed coefficient of determination.
Discussion
Selenium sequential extraction procedure
Differences in soil topsoil materials used and soil properties were speculated to have an affect on Se behavior and bioavailability. However, differences in SEP fractions between site were only statistically significant for Site B soils. Higher levels provide an explanation for the correspondingly higher Se levels found in plant shoots, particularly for the water-soluble and PO4 3−-extractable forms of Se. Likewise, non-detectable Se in most of the Sites D and E soils in these fractions also explains lower plant levels as well. According to Zhao et al. (2005), KH2PO4 extractions provide an estimate of Se that is available for uptake. Competition from oxyanions, such as phosphate or dissolved organic carbon (DOC) acids, could cause Se to partition into more soluble phases (Dhillon and Dhillon 2000). Above normal levels of plant-available phosphorus have also been indicate by Bond (1999) in phosphate mines in the western US. Phosphate has been noted to compete with Se on soil mineral surfaces (Balistrieri and Chao 1990; Goldberg and Traina 1987; Neal et al. 1987).
An appreciable abundance of Se within the carbonate SEP fraction indicates that carbonate minerals are possible means of Se sequestration via adsorption or co-precipitation. Selenium is particularly high within this fraction for many soils reclaimed pre-1996, which we speculate may pertain to either high pH or a higher abundance of Se overall. As indicated by Goldberg and Glaubig (1988), selenite sorption increases from pH 6 to 8 and plateaus from pH 8 to 9 on calcite surfaces. The pH in all soils examined were within this range and were especially high in soils from Site B. According to Zhao et al. (2005), calcium carbonate was the best determinant for decreasing Se bioavailability in soils through Van der Waals surface attraction.
Data also indicate that amorphous iron oxides sequester Se in soils reclaimed pre-1996, particularly at Sites B and C. In general, both selenite and selenate sorb to oxyhydroxides surfaces (Chapman et al. 2010). Soils generally do not sorb selenate above pH 7 on iron oxides (Cornell and Schwertmann 2003). Work from Balistrieri and Chao (1990) indicates that selenite is largely bound by amorphous iron oxides at pH relevant to our soils (pH 6–8) with subsequent decreases after pH 8. This occurs as strong inner-sphere complexes according to Goldberg (2013). Because of this, we speculate that iron oxide sorbed Se in the soils examined occurs as strong complexes that are not likely bioavailable.
An abundance of Se in the organic fraction was noted, possibly as selenide and/or selenite that are occluded within organic matter (Martens and Suarez 1997). Residual elemental Se was in the greatest abundance. The abundance of the two aforementioned fractions indicates the presence of Se that has not yet weathered from primary minerals (Ryser et al. 2006). They are not as soluble, have low oxidation potential, and hence, are generally not considered bioavailable (Jacobs 1989; Neal 1995). Because of this, we believe that the proportion of both phases is particularly important for soils reclaimed after 1996, suggesting that Se is largely not bioavailable.
Selenium speciation
Selenite was the dominant species identified in K2HPO4 extracts, with appreciable quantities of selenate also indicated in Sites A, B, and C. Selenate was not detected in soils reclaimed after 1996. This corroborates with high plant Se levels in Sites A, B, and C and lower levels in those from Sites D and E. Higher levels of selenite compared with selenate is also supported by work from Ryser et al. (2006). Using μ-XANES and fluorescence microprobe mapping, their results indicated that organic Se, elemental Se, and selenite were the dominant species observed in soils from the Conda Mine in Caribou National Forest, ID. Selenate was only dominant in one of the sixteen points of interest for the three soils examined. Ryser et al. (2006) indicate that reduced Se species oxidize to selenate over time. It was speculated that selenate was quickly lost in soils via leaching and/or plant uptake (Ryser et al. 2006).
Strong relationships were identified using regression between water-soluble Se from the SEP and selenate from speciation analyses, indicated in Fig. 4a. The relationship with selenite was considered insignificant. This corresponds with strong associations of selenite with solid phases at pH <7, while selenate is typically more soluble (Fordyce 2013). Our findings also suggest that selenate comprises a large portion of the water-soluble SEP fraction.
Plant selenium
Previous work from Zhao et al. (2005) has indicated that Se bioavailability to plants can be verified by examining plant uptake. Hyperaccumulators sampled from Sites A, B, and C were highly elevated in Se. These species can tolerate high quantities of Se and can accumulate thousands of mg Se per kg−1 DM (White 2016). This is thought to be because of the conversion of Se in plants to methylselenocysteine (Freeman et al. 2006). Methylselenocysteine is not as toxic as selenate, which is also taken up by plants (Freeman et al. 2009; de Souza et al. 1998; Neuhierl and Bock 1996). Secondary accumulating plant species typically do not exceed several hundred mg Se kg−1 DM, and therefore, are expected to have lower Se levels than hyperaccumulators (NAS-NRC 1983).
Highest plant Se levels in Site B soils were expected as soils from this site also contained the highest Se levels out of the five sites. The presence of more hyperaccumulating than nonaccumulating/accumulating plants species in highly seleniferous soils was expected. Nonaccumulators generally do not inhabit seleniferous soils because they can only tolerate levels between 10 to 100 μg Se g −1 DM, while secondary accumulators can only tolerate levels of around 1 mg Se g −1 DM (Dhillon and Dhillon 2009; Fordyce 2013; Moreno Rodriguez et al. 2005; Rosenfeld and Beath 1964; White et al. 2004; White 2016). Hyperaccumulator tolerance of Se may be related to the Se species stored and area of storage within the plant (de Souza et al. 1998; Freeman et al. 2006; Quinn et al. 2011; Van Hoewyk et al. 2005). These species can store Se as organic methylselenocysteine, while non-accumulators and accumulators store Se as selenate (de Souza et al. 1998; Freeman et al. 2006; Van Hoewyk et al. 2005). Hyperaccumulators also store Se in leaf epidermis and in pollen, ovules, and seeds, while plants that do not hyperaccumulate store Se in leaf vascular tissues (Freeman et al. 2006; Quinn et al. 2011). According to El Mehdawi et al. (2012), hyperaccumulators exhibit improved growth with increasing supply of Se from soils. This may suggest that these species can benefit from Se uptake via increased anti-oxidant activity (El Mehdawi et al. 2012; Cartes et al. 2005; Hartikainen 2005).
Western Aster Se and a soil sequential extraction procedure
The summation of Se in the first two SEP fractions (water-soluble and PO4 3−-extractable Se) exhibited the strongest relationship. Slight decreases in this relationship were observed when carbonate-associated Se was summed with the previous fractions. Work from Zhao et al. (2005) showed a highly correlated relationship for PO4 3−-extractable Se with plant uptake, with an R2 value of 0.91 for both field and greenhouse studies. Further decreases in relationships were observed following the inclusion of the amorphous Fe oxide-associated and organic Se. Their weak relationship suggests a possible decrease in their contribution to Se bioavailability to western asters and that this fraction is more recalcitrant.
The regression analysis suggests that Se bioavailability increases when Se is water-soluble and PO4 3−-extractable, and begins to decrease with carbonate associations. Carbonate fractions of Se are considered more recalcitrant and require acidification for release. Therefore, the first two fractions were identified as “bioavailable fractions.” Our Se speciation results also suggest that selenate is the form of Se present in the water-soluble fraction, and hence, the form taken up by western aster. Several studies have indicated that soil selenate is immediately accumulated by plants in comparison with selenite (Broadley et al. 2006; Fordyce 2013; Pilbeam et al. 2015). Selenate additions in soils have also resulted in ten-fold more uptake in plant than selenite (Jacobs 1989; Neal 1995). Previous work from Sharmasarkar and Vance (1995) have noted strong relationships between grass species and selenate, suggesting that selenate is the main solution species absorbed. Our regression analyses suggest that other fractions of Se in the SEP (after carbonate bound Se extractions) do not greatly contribute to Se bioavailability. The water-soluble extract, in conjunction with Se levels in shoots of western aster, can be used to identify potentially problematic soil and predicting Se levels in western aster. Simple water extractions can be employed to assess areas where Se levels are particularly toxic in plants to grazing livestock. This can be a useful, quick tool during remediation efforts. For example, a soil water-soluble extract value of 3 mg kg−1 would correspond to a predicted Se level in western aster of approximately 5000 mg kg−1 (Fig. 5). This level definitely sets a warning threshold that indicates that these areas should be avoided by grazing animals and targeted for remediation.
Conclusions
This study examined Se contamination in soils resulting from previous phosphate mining reclamation practices. Sequential extractions indicated that the dominant phase of Se observed was in the residual fraction, followed by the organic Se fraction. Notable amounts of carbonate- and amorphous iron oxide-associated Se were also observed in most soils, demonstrating that a portion of Se was sequestered via adsorption or co-precipitation reactions. Lower levels of water-soluble and PO4 3−-extractable Se were indicated in the SEP. Findings associated with higher abundances of bound forms of Se corroborate with our speciation result because a higher level of selenite was present. Selenite is typically bound or exchangeable. Lower levels of the more soluble Se species, selenate, were also present in speciation results. This corresponds with low abundances of selenate in the water–soluble SEP fraction. A highly significant relationship between selenate and the water-soluble Se fraction was confirmed with a lower, but still significant, relationship observed for selenite and the PO4 3−-extractable Se fraction.
Plant shoot Se levels were reflective of soil Se levels. High levels of Se were found in Se accumulating and hyperaccumulating (western aster) vegetation sampled from Sites A, B, and C, which were reclaimed before 1996, with lower levels noted for plants sampled from Sites D and E reclaimed post-1996. This was especially true for western aster. Regression data suggests that a strong relationship exists for water-soluble and PO4 3−-extractable Se with increasing plant uptake. This relationship decreases once other SEP fractions are factored into the model and decrease even further with the inclusion of organic Se fractions. Simple water extractions could be beneficial tools, as indicated by the relationship between the water-soluble SEP fraction and selenate speciation and the water-soluble fraction and western aster shoot Se. This work identifies a method for quick assessment of Se bioavailability in soils and for relating this to Se accumulation in western aster found in problematic locations.
References
Alam MGM, Tokunaga S, Maekawa T (2000) Extraction of selenium from a contaminated forest soil using phosphate. Environ Technol 21:1371–1378
Amacher M (2010) Sequential extraction of selenium. USDA-Forest Service Open File Report. pp. 155–159
Balistrieri LS, Chao TT (1990) Adsorption of selenium by amorphous iron oxyhydroxide and manganese dioxide. Geochim Cosmochim Acta 54:739–751
Blanchard T, Baun C, Stone L, Gudgell D, Hayes J (2002) Hardrock and phosphate mining in Idaho. A report by the Idaho Conservation League and Boulder-White Clouds Council. www.wildwhiteclouds.org/news_mining_IdahoMiningRpt.pdf. Accessed July 2016
Bond MM (1999) Characterization and control of selenium releases from mining in the Idaho phosphate region. MS Thesis. University of Idaho, Moscow
Broadley MR, White PJ, Bryson RJ, Meacham MC, Bowen HC, Johnson SE, Hawkesford MJ, McGrath SP, Zhao F, Breward N, Harriman M, Tucker M (2006) Biofortification of UK food crops with selenium. Nutrition Society 65:169–181
Buck BW, Jones JL (2002) Interagency/industry coordination to respond to selenium contamination at phosphate mines in southeastern Idaho. http://citeseerx.ist.psu.edu/viewdoc/download?. Accessed July 2016
Burau RG (1989) Selenium in arid and semiarid soils. J Irrig and Drain E-ASCE 115:42–47
Cartes P, Gianfreda L, Mora ML (2005) Uptake of selenium and its antioxidant activity in ryegrass when applied as Selenate and selenite forms. Plant Soil 276:359–367
Chapman HD (1965) Total exchangeable bases. Soil science Society of America. Inc. and American Society of Agronomy, Inc., Madison
Chapman PM, Adams WJ, Brooks ML, Delos CG, Luoma SN, Maher WA, Ohlendorf HM, Presser TS, Shaw DP (2010) Ecological assessment of selenium in the aquatic environment. CRC Press, New York
Cornell RM, Schwertmann U (2003) The iron oxides: structure, properties, reactions, occurrences and uses. Wiley-VCH, Weinheim
Croghan CW, Egeghy PP (2003) Methods of dealing with values below the limit of detection using SAS. Southeastern SAS User Group, St. Petersburg
de Souza MP, Pilon-Smits EAH, Lytle CM, Hwang S, Tai J, Honma TSU, Yeh L, Terry N (1998) Rate-limiting steps in selenium assimilation and volatilization by Indian mustard. Plant Physiol 117:1487–1494
Davis TZ, Stegelmeier BL, Green BT, Welch KD, Panter KE, Hall JO (2011) Acute toxicity of selenium compounds commonly found in selenium accumulator plants. CAB International, Cambridge
Davis TZ, Stegelmeier BL, Panter KE, Cook D, Gardner DR, Hall JO (2012) Toxicokinetics and pathology of plant-associated acute selenium toxicosis in steers. J vet Diagn Investig 24:319–327
Davis TZ, Stegelmeier BL, Welch K, Pfister JA, Panter KE, Hall JO (2013) Comparative oral dose toxicokinetics of selenium compounds commonly found in selenium accumulator plants. J Anim Sci 91:4501–4509
Dhillon SK, Dhillon KS (2000) Selenium adsorption in soils as influenced by different anions. J Plant Nutr 163:577–582
Dhillon KS, Dhillon SK (2009) Accumulation and distribution of selenium in some vegetable crops grown in selenate-se treated clay loam soil. Front Agric China 3:366–373
Edmonson AJ, Norman BB, Suther D (1993) Survey of state veterinarians and state veterinary diagnostic laboratories for selenium deficiency and toxicosis in animals. J am vet med Assoc 202:865–872
El Mehdawi AF, Cappa JJ, Fakra SC, Self J, Pilon-Smits EAH (2012) Interactions of selenium hyperaccumulators and nonaccumulators during cocultivation on seleniferous or nonseleniferous soil – the importance of having good neighbors. New Phytol 194:264–277
Ellis DR, Salt DE (2003) Plants, selenium and human health. Curr Opin Plant Biol 6:273–279
Fessler AJ, Moller G, Talcott PA, Exon JH (2003) Selenium toxicity in sheep grazing reclaimed phosphate mining sites. Vet hum Toxicol 45:294–298
Fordyce FM (2013) Selenium deficiency and toxicity in the environment. In: Selinus O (ed) Essentials of medical geology, Revised edn. Springer, Netherlands
Freeman JL, Quinn CF, Marcus MA, Fakra S, Pilon-Smits EAH (2006) Selenium-tolerant diamondback moth disarms Hyperaccumulator plant defense. Curr Biol 16:2181–2192
Freeman JL, Quinn CF, Lindblom SD, Klamper EM, Pilon-Smits EAH (2009) Selenium protects the hyperaccumulator Stanleya pinnata against black-tailed prairie dog herbivory in native seleniferous habitats. Am J bot 96:1075–1085
Gee GW, Bauder JW (1986) Particle-size analysis. In: Klute A (ed) Methods of soil analysis: part 1-physical and mineralogical methods. 5 edn. Soil Science Society of America, Inc. and American Society of Agronomy, Inc., Madison
Gleyzes C, Tellier S, Astruc M (2002) Fractionation studies of trace elements in contaminated soils and sediments: a review of sequential extraction procedures. TrAC Trends Anal Chem 21:451–467
Goldberg S (2013) Modeling selenite adsorption envelopes on oxides, clay minerals, and soils using the triple layer model. Soil Sci Soc am J 77:64–71
Goldberg S, Glaubig RA (1988) Anion sorption on a calcareous, Montmorillonitic soil-selenium. Soil Sci Soc am J 52:954–958
Goldberg S, Traina SJ (1987) Chemical modeling of anion competition on oxides using constant capacitance model-mixed ligand approach. Soil Sci Soc am J 51:929–932
Gupta M, Gupta S (2016) An overview of selenium uptake, metabolism, and toxicity in plants. Front Plant Sci 7:2074
Hall JO, Winger H (2012) Toxicology ICP/MS sample digestion and preparation procedure for Forage & Feed. Utah Veterinary Diagnostic Laboratory, Logan
Hartikainen H (2005) Biogeochemistry of selenium and its impact on food chain quality and human health. J Trace Elem med Biol 18:309–318
Hopke PK, Liu C, Rubin DB (2001) Multiple imputation for multivariate data with missing and below-threshold measurements: time-series concentrations of pollutants in the Arctic. Biometrics 57:22–33
IDEQ (2007) Selenium project, Southeast Idaho phosphate mining resource area. Water Quality Sampling for Metals - Blackfoot River and Tributaries (HUC 17040207), Selected Bear River Tributaries (HUC 16010201), and Selected Salt River Tributaries (HUC 1704105). Idaho Department of Environmental Quality, Pocatello Regional Office, Pocatello. https://www.deq.idaho.gov/media/468229-_water_data_reports_surface_water_water_bodies_selenium_southeast_idaho_2007.pdf. Accessed July 2016
Jackson ML (1958) Soil chemical analysis. Prentice-Hall, Englewood Cliffs
Jacobs LW (1989) Selenium in agriculture and the environment. Soil Science Society of America Special Publication 23. SSSA, Madison
John DA, Leventhal JS (1995) Ch 2 bioavailability of metals. In: du Bray EA (ed) USGS Open File Report 95-831: Preliminary compilation of descriptive geoenvironmental mineral deposit models. U.S. Department of the Interior - U.S. Geological Survey, Denver. https://pubs.usgs.gov/of/1995/0831/report.pdf. Accessed July 2016
Knudsen AC, Gunter ME (2004) Chapter 7: the effects of weathering on the mineralogy of the phosphoria formation, Southeast Idaho. In: James RH (ed) Handbook of exploration and environmental geochemistry. Elsevier Science B.V, Amsterdam
Kunze GW, Dixon JB (1986) Pretreatment for mineralogical analysis. In: Klute A (ed) Methods of soil analysis: part 1-physical and mineralogical methods. 5 edn. Soil science Society of America, Inc. and American Society of Agronomy, Inc., Madison
Luoma SN (1983) Bioavailability of trace metals to aquatic organisms - a review. Sci Total Environ 28:1–22
Martens DA, Suarez DL (1997) Selenium speciation of soil/sediment determined with sequential extractions and hydride generation atomic absorption spectrophotometry. Environ Sci Technol 31:133–139
McNeal JM, Balistrieri LS (1989) Geochemistry and occurrence of selenium: an overview. In: Jacobs LW (ed) Selenium in agriculture and the environment. Soil Science Society of America and American Society of Agronomy, Madison
Meyers T (2013) Remediation scenarios for selenium contamination, Blackfoot watershed, southeast Idaho, USA. Hydrogeol J 21:655–671
Moreno Rodriguez MJ, Cala Rivero V, Jiménez Ballesta R (2005) Selenium distribution in Topsoils and plants of a semi-arid Mediterranean environment. Environ Geochem Health 27:513–519
NAS-NRC (1983) Selenium in nutrition, Revised edn. National Acadamies Press, Washington DC
Neal RH (1995) Selenium. Blackie Academic & Professional, London
Neal RH, Sposito G, Holtzclaw KM, Traina SJ (1987) Selenite adsorption on alluvial soils: II. Solution composition effects. Soil Sci Soc Am J 51:1165–1169
Neuhierl B, Bock A (1996) On the mechanism of selenium tolerance in selenium-accumulating plants. Purification and characterization of a specific selenocysteine methyltransferase from cultured cells of Astrugulus bisculatus. Eur J Biochem 239:235–238
Pfister JA, Davis TZ, Hall JO (2013) Effect of selenium concentration on feed preferences by cattle and sheep. J Anim Sci 91:5970–5980
Pilbeam DJ, Greathead HMR, Drihem K (2015) Selenium. CRC Press, Boca Raton
Quinn CF, Prins CN, Freeman JL, Gross AM, Hantzis LJ, Reynolds RJB, Yang S, Covey PA, Banuelos GS, Pickering IJ, Pilon-Smits EAH (2011) Selenium accumulation in flowers and its effects on pollination. New Phytol 192:727–737
Richards RT, Chambers JC, Ross C (1998) Use of native plants on Federal Lands: policy and practice. J Range Manag 51:625–632
Rosenfeld I, Beath OA (1964) Selenium: Geobotany, biochemistry, toxicity and nutrition. Academic Press, New York
Ryser AL, Strawn DG, Marcus MA, Johnson-Maynard JL, Gunter ME, Möller G (2005) Micro-spectroscopic investigation of selenium-bearing minerals from the western US phosphate resource area. Geochem Trans 6:1–11
Ryser AL, Strawn DG, Marcus MA, Fakra S, Johnson-Maynard JL, Möller G (2006) Microscopically focused synchrotron X-ray investigation of selenium speciation in soils developing on reclaimed mine lands. Environ Sci Technol 40:462–467
Saffari M, Yasrebi J, Karimian N, Shan XQ (2009) Effect of calcium carbonate removal on the chemical forms of zinc in calcareous soils by three sequential extraction methods. Research Journal of Biological Sciences 4:858–865
SAS Institute Inc. (2013) JMP 11.0.0. SAS Institute Inc., Cary
Shacklette HT, Boerngen JG (1984) Element concentrations in soils and other surficial materials of the conterminous United States. US geological survey professional paper 1270. United States Government Print Office, Washington
Sharmasarkar S, Vance GF (1995) Characterization and correlation of soil and plant selenium in some range and coal mine environments of Wyoming. Commun Soil Sci Plant Anal 26:2577–2591
Shortridge EH, O'Hara PJ, Marshall PM (1971) Acute selenium poisoning in cattle. New Zeal vet J 19:47–50
Spallholz JE (1994) On the nature of selenium toxicity and carcinostatic activity. Free Radic Biol med 17:45–64
Stillings L, Amacher M (2004) Selenium attenuation in a wetland formed from mine drainage in the Phosphoria formation, southeast Idaho. Life cycle of the Phosphoria formation: from deposition to the post-mining environment. Elsevier B.V., Amsterdam
Tamm O (1922) Eine method zur bestimmung der anorganischen komponenten des gelkomplex in boden. Medd Statens Skogforsokanst 19:385–404
Tessier A, Campbell PGC, Bisson M (1979) Sequential extraction procedure for the speciation of particulate trace metals. Anal Chem 51:844–851
Thomas GW (1996) Soil pH and acidity. American Society of Agronomy, Madison
Unrine JM, Jackson BP, Hopkins WA (2007) Selenomethionine biotransformation and incorporation into proteins along a simulated terrestrial food chain. Environ Sci Technol 41:3601–3606
Van Hoewyk D, Garifullina GF, Ackley AR, Abdel-Ghany SE, Marcus MA, Fakra S, Ishiyama K, Inoue E, Pilon M, Takahashi H, Pilon-Smits EAH (2005) Overexpression of AtCpNifS enhances selenium tolerance and accumulation in Arabidopsis. Plant Physiol 139:1518–1528
Wagemann R, Brunskill GJ, Graham BW (1977) Composition and reactivity of some river sediments from Mackenzie Valley, NWT Canada. Environ Geol 1:349–358
White PJ (2016) Selenium accumulation by plants. Ann bot 117:217–235
White P, Bowen H, Parmaguru P, Fritz M, Spracklen WP, Spiby RE, Meacham MC, Mead A, Harriman M, Trueman LJ, Smith BM, Thomas B, Broadley MR (2004) Interactions between selenium and sulphur nutrition in Arabidopsis thaliana. J Exp Biol 55:1927–1937
Yamada H, Yumei K, Aso T, Hiroshi U, Fujimura T, Yonebayashi K (1998) Chemical forms and stability of selenium in soil. Soil Sci Plant Nutr 44:385–391
Zhao CY, Ren JH, Xue CZ, Lin E (2005) Study on the relationship between soil selenium and plant selenium uptake. Plant Soil 277:197–206
Acknowledgements
Funding for this research was provided by the United States Department of Agriculture-Agricultural Research Service Grant No. A26232.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Juan Barcelo.
Electronic supplementary material
ESM 1
(DOCX 134 kb)
Rights and permissions
About this article

Cite this article
Favorito, J.E., Eick, M.J., Grossl, P.R. et al. Selenium geochemistry in reclaimed phosphate mine soils and its relationship with plant bioavailability. Plant Soil 418, 541–555 (2017). https://doi.org/10.1007/s11104-017-3299-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-017-3299-5