
Abstract
Near neutral pH values are widely thought to be optimum for uptake of phosphate. This belief is based on an outdated view of soil phosphate chemistry. The literature on uptake by plants from solution and from soil, and especially on desorption by soil, are all consistent with a much lower pH optimum.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
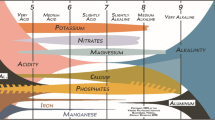
There is some confusion about the optimum soil pH for supply of phosphate to plants. Some textbooks advocate a near-neutral pH for “availability”. For example, “In most soils, phosphorus availability is at a maximum in the pH range 5.5 to 7” (Tisdale and Nelson 1966); “Liming acid soils to pH 6 – 7 should make (P) ions more available” (Leeper and Uren 1993); “Phosphorus is most available between pHW 6.0 to 7.0” (Price 2006); and Lambers et al. (2008) specify an optimum pH from 6.5 to 7.5 and again above about 8. The use of the word “available” implies integration of the supply side from the soil with the uptake side by the plant. Other textbooks are confined to the supply side indicating greater solubility in a similar pH range (Brady 1974; Tan 1992; Troch and Thompson 1993; Ashman and Puri 2003; White 2006). Where justification for these opinions is given, pH 6 to 7 is supposed to constitute a “valley” between “fixation” by iron and aluminium at lower pH and by calcium at higher pH. This is well illustrated by Price (2006) (Fig. 1). No data is given to substantiate this.

Redrawn Fig. 4.7 from Price (2006) purporting to show the effects of pH on the availability of phosphate (with permission from CSIRO Publishing)
The purpose of this short article is to review the available evidence and to show that the optimum pH is much lower.
The pH experienced by roots
It is common to quote a soil pH, but this is a convenient fiction: pH is a property of solutions, and soil is a solid. What is meant by this term is the pH of a solution in equilibrium with the soil. When the pH of soils was first measured, it seemed logical to use water as the solution. However, no matter what water:soil ratio is used, adding water results in a many-fold dilution of the soil solution. The pH so measured is appreciably higher than that of the soil solution at normal water contents, and consequently higher salt concentrations. The mnemonic is as follows: for negatively charged surfaces (as is the case for most soils) decreasing cation concentration means that more hydrogen ions are retained by the soil and the pH rises. When water is used, the value obtained is also vulnerable to small changes in the salt concentration in the soil solution. It is therefore now more common to measure pH using a solution of 0.01 M calcium chloride. This has the technical advantage of being much more stable, but it is also arguable that the value so obtained is closer to that experienced by plant roots. This is because the uptake of water by plants sweeps salts in the soil water towards the root surface. The amount of calcium so arriving is usually in excess of plant requirements and so calcium concentration increases close to the root surface. Even for a soil with a lower calcium concentration in the soil solution, a concentration of 0.01 M may be closer to the concentration at root surfaces. In order to differentiate such measurements from those using water pHW, the designation pH(CaCl2) is often used.
When calcium carbonate is present, pH(CaCl2) is about 7. This pH is set by the equilibrium between the calcium ion concentration, bicarbonate concentration in solution and the partial pressure of atmospheric carbon dioxide. Thus, the pH(CaCl2) commonly designated as near neutral is in fact characteristic of a calcareous soil and for most practical purposes, 7 is the maximum pH(CaCl2). At the other end of the pH scale, soils are fairly strongly buffered and pH(CaCl2) values lower than about 4 are uncommon in soils used for agriculture. Thus the practical range for the pH experienced by most roots of agriculturally important plants is from about 4 to about 7. Higher pH can occur in serpentine soils and in sodic soils, and lower values in forest soils subjected to acid rain. In this article, emphasis is therefore given to the pH(CaCl2) range between 4 and 7, but values outside this range are also considered, partly because they can occur but mainly because they help understand the mechanisms involved.
The effects of pH on uptake of phosphate by roots
“Availability” is a frequently used, but poorly defined term. Generally it indicates whether something can be accessed or used. I do not think it can be defined in terms of the supply side alone but must also consider the user. The effects of pH on uptake by roots are therefore relevant.
Between pH 4 and pH 7, phosphate adsorption by isolated barley or corn roots decreases markedly with increasing pH (Figs. 2 and 3). Similarly, the optimum pH for uptake by clover roots was found to be 4.3 (Dunlop and Dowling 1978). This occurs because roots take up phosphate as the \( {\mathrm{H}}_2{\mathrm{PO}}_4^{-} \) ion (Fig. 2). Fungi also take up the \( {\mathrm{H}}_2{\mathrm{PO}}_4^{-} \) ion (Smith et al. 2011) and the effects of pH on mycorrhizal plants would be expected to be similar.

Effect of pH on uptake of phosphate by barley roots expressed relative to the uptake at pH 4.1 which was taken as 100 % (Unbroken line). Seedlings of barley were grown in the dark at room temperature using 0.5 mM CaCl2. Roots were excised from plants 6–8 days old. Uptake experiments used about 1 g roots in 75 ml of 0.5 mM CaCl2 solution. pH was varied from 2.2 to 9.2 with Tris-HCl. The broken lines show the relative concentration of the indicated ions. Redrawn from Vange and Holmern (1974)

Effect of pH on the uptake of phosphate from solution by segments of corn roots. Roots were obtained from five day old corn seedlings grown in the dark at 25 °C on aerated 0.2 mM CaSO4. Three-cm segments of primary roots were taken 1 to 4 cm from the tip. Phosphate uptake was measured over five minutes using 32P labelled phosphate. Redrawn from Sentenac and Grignon (1985)
The effects of pH on phosphate sorption by soils
Terminology
The initial reaction between soil particles and phosphate is adsorption on variable charge surfaces. Evidence that variable charge surfaces are involved is obtained from the interaction between pH and salt concentration (Fig. 4a). At high pH, the reacting surfaces are negatively charged and increasing cation concentration facilitates increased adsorption of phosphate. At low pH, the reacting surfaces are positively charged and increasing anion concentration decreases adsorption of phosphate. Obviously, there is an intermediate pH at which there is no effect and this is assumed to be the point of zero charge for these conditions. The value of the point of zero charge decreases as the amount of adsorption increases (Barrow 1999) and it is also decreased by prior reaction with phosphate (Barrow and Debnath 2015). Values are usually in the range pH 3 to pH 5 and therefore for much of the pH range of interest, phosphate reacts with negatively charged surfaces rather than with positive surfaces as assumed by Hinsinger (2001).

Two aspects of the reaction between phosphate and Madan soil: the effects of salt concentration and the effects of time. a Interaction between salt concentration and pH in determining phosphate sorption. Drawn using data from Barrow and Debnath (2015). b Effect of time on sorption of phosphate and also showing desorption of phosphate after a further 48 h following sorption for 48 h. Drawn using data from Barrow and Debnath (2014)
The initial adsorption reaction is followed by a solid-state diffusion of the adsorbed phosphate ions down an electrochemical gradient into the adsorbing particles. Consequently, sorption increases with time and the sorption curves do not follow the same track as desorption curves (Fig. 4b). This category of phosphate is appropriately called “penetrated” and the sum of adsorbed and penetrated phosphate is appropriately called “sorbed phosphate”.
Effect of pH on the rate of adsorption
In most cases it is difficult to isolate the initial adsorption reaction from the subsequent penetration process. However, this can be done using “healed” goethite. This means that the faults in the crystal structure have been repaired by subjecting the goethite to high temperature for several hours. Penetration does not occur and the rates of the adsorption reaction can be seen in isolation.
Reaction of ions with a charged surface may comprise a number of steps, for example, the reaction of the ion with the surface, the displacement of a water molecule from the surface, gain or loss of protons, and/or the approach or departure of an electrolyte ion to balance the change in charge of the surface. We do not know from first principles, what the sequence of these reactions will be. However we assume that one of the reaction steps is appreciably slower than the others and that the rate of this reaction therefore determines the overall rate.
The equation to describe rate of reaction of ions with charged sites is:
where 0 t is the concentration of Occupied Sites at time t and is therefore a measure of the amount of adsorption, k 1 and k 2 are the rate constants, α is the fraction of the total phosphate dissociated to HPO4 2−, c t is the total concentration of phosphate in solution, m t is the concentration of vacant sites at time t, ψ is the electrical potential in the plane of adsorption at time t, F is the Faraday, R the gas constant and T (K) the temperature. The terms \( \frac{\leftarrow }{\alpha } \) and \( \frac{\to }{\alpha } \) are called transfer coefficients and are described by Bockris and Reddy (1970). The factors determining the magnitude of these coefficients include the number of electrons involved in the reaction and the position of the rate-determining step in the sequence of steps involved in the overall reaction (Barrow et al. 1981; Barrow 1987). The observed behaviour of decreasing rate of reaction with increasing pH is reproduced when \( \frac{\leftarrow }{\alpha } \) is 2 and \( \frac{\to }{\alpha } \) is zero. These values indicate that the rate-determining step precedes the electron transfer steps and does not itself involve an electron transfer.
Figure 5 shows that reaction of phosphate with healed goethite was far from instantaneous and that the rate of reaction decreased with increasing pH because \( \frac{\leftarrow }{\alpha } \) is 2. The corollary of this that because \( \frac{\to }{\alpha } \) is zero, rate of the desorption reaction (which is relevant when considering the supply of phosphate to plants) is not affected by pH.

Effect of pH on the rate of reaction of phosphate with a “healed” goethite (surface area 18 m2 g−1). For healed goethite there was little penetration reaction and the rate of the adsorption reaction can be observed. The fraction adsorbed is the amount adsorbed at a given time divided by the maximum adsorption at that pH. In the inset, the fraction adsorbed at the shortest period (15 min) is plotted against pH. (Drawn using data from Strauss et al. 1997)
Effect of pH on the amount of adsorption
The effects of pH on the magnitude of phosphate sorption is a complex subject with many sources of confusion, but can be understood using the proposal of Bowden et al. (1977) that sorption should be related not to concentration but rather to the surface activity function (S a ). This is given by:
where K i is the binding constant for the reacting ion (i), z i is its valency and the other symbols are as above. Equation (2) represents the equilibrium position of Equation (1).
Infra-red studies show that from pH 3.5 to 8 most of the links between phosphate and goethite are bidentate; at higher pH, and at high phosphate loading there is an increasing abundance of monodentate species (Hiemstra and Van Riemsdijk 1996). Nevertheless, the effects of pH and of concentration on phosphate sorption can be closely modelled assuming all the links are bidentate (Bowden et al. 1980; Hiemstra and Van Riemsdijk 1996; Strauss et al. 1997). When phosphate forms bidentate links to oxide surfaces, the reaction can be related to the concentration of divalent ions in solution (Barrow 1999). The value of z i is therefore −2. This is consistent with the development from Equation (1). The effects of increasing pH can be seen as a balance between increasing proportion of divalent ions and increasingly unfavourable electric potential. This is illustrated in Fig. 6. Up to pH values a little below 7, the concentration of divalent ions increases about 10 fold for unit increase in pH. Above this value the fraction present as divalent ions approaches unity, the decreasing potential then dominates, and sorption decreases rapidly with increasing pH.

Diagram to show how the potential term and the activity term of Equation (2) interact to determine the amount of sorption. The potential term is exp. (−z i F ψ /RT) and is dimensionless. The activity term is cα γ and has dimensions of μM. Their product predicts sorption as S a = K i cα γ exp (−ziFψ/RT). The meaning of the symbols is given in the text
When the effects of pH on phosphate sorption are studied with soils, the pH is usually raised using lime and dilute solutions of calcium chloride are used as a background electrolyte. Consequently, the pH range above about 7 is scarcely sampled (Fig. 7) and the large decrease in sorption expected to occur above pH 7 is not seen. There are only small effects of pH on phosphate sorption, with the shape of plots of sorption versus pH differing sightly between soils (Fig. 7). However, an important observation is that, for low-phosphate soils, sorption increased as the pH decreased from 5 to 3 (Fig. 7a–d) and there was often a minimum sorption near pH 5. The differences in the effects of pH on sorption were attributed to small differences in the effects of pH on the electric potential of the surface (Barrow 1984). One of the sources for differences in potential is prior reaction with phosphate. This decreases sorption and causes the pH at which least sorption occurs to move to lower pH (Figs. 7e-f, 8d).

Effect of pH and phosphate concentration in solution (mg L−1) as indicated to the right of each line, on phosphate sorption as measured in 0.01 M CaCl2 solutions. Soils 1 to 4 are unfertilized low-phosphate soils which differed widely in the amount of phosphate sorbed at a given concentration as indicated on the vertical axis. Soil 5 and 6 are from fertilized pasture. The pH was altered by incubating the soils with either lime or hydrochloric acid prior to measurement of sorption. The lines were fitted using a model in which the effects of concentration were described by assuming that adsorption occurred on sites with a normal distribution of electric potentials and the effects of pH were described using the same principles as in Fig. 6. From Barrow (1984)

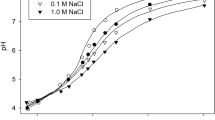
Effect of pH on phosphate desorption. Part a is drawn using data from Murrmann and Peech (1969). Data are for two soils, one limed the other not as shown. Both soils had previously received 215 kg P ha−1. The pH was altered using HCl or Ca(OH)2 and desorption was measured in 0.01 M CaCl2. Part b is drawn using data from Riley and Barber (1971). Soil was mixed with calcium chloride solutions at the indicated concentrations with different concentrations of hydrochloric acid. Part c and d are redrawn from Barrow (2002) (with permission from CSIRO Publishing). Soil 3 of Fig. 7 was incubated with the indicated concentrations of phosphate. Subsequently, samples of soil were further incubated with either lime or hydrochloric acid to change the pH. Sorption was then measured by mixing the soil samples within 0.01 M CaCl2 containing different concentrations of phosphate. At the same time, desorption was measured by mixing the soil with 0.01 M CaCl2 at a range of solution soil ratios. Sorption/desorption curves were closely described by the Freundlich equation: S = ac b – q. Where S is sorption and c is concentration. The index term (b) had a value of 0.4 in all cases. The parameter a is therefore a measure of the relative sorption. The parameter q estimates the amount of phosphate that could be desorbed at infinite dilution, that is, as the solution concentration approaches zero
Effects of pH on desorption
Desorption of phosphate is large at low pH, decreases to a minimum near pH 6–7 and increases again at pH above about 7 (Fig. 8a, b and c). This result is counterintuitive. One would expect the effects of pH on desorption to be the opposite of those on sorption. Sorption usually increases as the pH decreases below about 5 and that was the case for the soil represented in Fig. 8c (shown in.
Figure 8d). One would therefore expect desorption to decrease, but in fact it increased. How might that be explained?
There is no effect of pH on the rate of displacement of adsorbed phosphate (see earlier). The effects observed are therefore assumed to be on reverse diffusion of penetrated phosphate. The increase in desorption above about pH 7 can be assumed to occur because adsorbed phosphate is displaced; a concentration gradient from inside the particle to the outside is therefore established; and this induces movement of penetrated phosphate. However, as the pH is decreased from say 5 to say 3, adsorbed phosphate is likely to increase rather than decrease and a different explanation is required. Rather than inducing a chemical gradient, a potential gradient is induced. That is, the surface is less negative, or more positive, and it is this that induces movement of penetrated phosphate back towards the surface.
Effects of pH on uptake of phosphate from soil
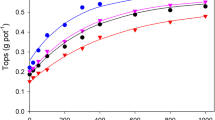
A linear increase in phosphate uptake by soybean was observed as the pH decreased (Fig. 9 Riley and Barber (1971).). This clear result was probably obtained because the pH of the soil closely adhering to the roots was measured. This pH differed from that of the bulk soil because of the differing effects of ammonium or nitrate uptake. The pH was measured in water. If CaCl2 solution had been used, values would have been lower and the range would have been from about pH 7 to about pH 4. For subterranean clover and for maize, there was often an increase in yield and phosphate uptake as the pH was increased above about 4. This was followed by a decrease as the pH was raised further (Fig. 10). For subterranean clover the effects of low pH were not due to soluble manganese (Barrow 1965) and may have been due to soluble aluminium. For maize the effects of low pH were probably due to soluble aluminium (Chen and Barber 1990). In contrast, yield of rye grass increased with increasing pH (Fig. 10b). This occurred because yield was limited by nitrogen supply rather than by phosphorus supply and increasing pH increased the rate of nitrogen mineralization (Barrow 1965).

Effect of pH on uptake of phosphate by soybean grown in a growth chamber in Chalmers silt loam from Indiana. The pH was altered by lime treatments and also by supplying nitrogen as ammonium or as nitrate. The pH was measured on roots, plus strongly adhering soil, after 5 min shaking with water at a water:material ratio of 2:1. Redrawn from Riley and Barber (1971)

Effect of pH on growth and uptake of phosphate in pot experiments. a The effects of pH and of phosphate supply on yield of Trifolium subterraneun cv Yarloop grown in Coolup sand from a fertilized pasture. The pH was altered by adding analytical grade calcium carbonate to the soil. pH was measured in 0.01 M CaCl2. (Drawn using data from Barrow (1965) with permission from CSIRO Publishing) b The effects of pH on yield of Trifolium subteraneum cvs Yarloop and Mount Barker and on Wimmera rye grass (Lolium rigidum). Conditions were similar to those of Fig. 10a except that 50 mg of phosphorus was added to the soil. (Drawn using data from Barrow (1965) with permission from CSIRO Publishing) c Phosphorus uptakes for the treatments represented in Fig. 10a. (Drawn using data from Barrow (1965) with permission from CSIRO Publishing) d Effect of pH on phosphorus uptake by maize. Plants were grown for 18.5 days in a growth chamber in Chalmers silt loam. The pH of the soil (originally 4.7) was altered by incubating the soil with either sulphuric acid or calcium carbonate. No fertiliser nitrogen was added. The method of measuring pH is not specified but judging from the values, it is assumed to be using water. (Data of Chen and Barber (1990)
Conclusions
It is difficult to understand why the model of soil phosphate chemistry illustrated in Fig. 1 has persisted. As is shown here, it fails the most fundamental test of science: it makes wrong predictions. In addition, there is very little evidence for the existence of the separate postulated sinks for phosphate. For example, Norrish and Rosser (1983) used an electron microprobe to show that in many Australian soils much of the phosphorus is associated with iron oxides and little with aluminium oxides. Further, when radioactive arsenate was reacted with soils, it was found that the reaction was largely with iron oxides (Fordham and Norrish 1979). Even in calcareous soils, hydrous oxides may provide the principle sites for reaction (Holford and Mattingly 1975). The model of Fig. 1 also has no facility for explaining other aspects of the behaviour of phosphate such as, for example, the effects of ionic strength let alone the differing effects of pH on sorption of other anions. In contrast, the observed effects of ionic strength provide direct evidence that phosphate ions react with variable charge surfaces. The effects of pH must therefore depend on the ions present in solution and on the potential of the surface. There is no need to postulate different mechanisms – such as “fixation” by iron and aluminium at low pH and by calcium at high pH (Price 2006): one simple mechanism is adequate for the pH range. Furthermore, the same principles can explain the different effects of pH on other anions such as fluoride, selenate, selenite, molybdate, and borate (Barrow 1999).
It is also difficult to understand how the idea that maximum phosphate availability occurs in the pH range 6 to 7 has persisted despite strong published evidence to the contrary, as indicated in Figs. 9 and 10. The essence of the problem is that there are three separate effects of pH. As the pH is decreased from say 6 to 4, the rate of uptake of phosphate by roots increases, the amount desorbed from soil increases, and the amount sorbed by soil often also increases. The first two effects increase availability; the third effect decreases it. To add to the complexity, the pH at the root surface may differ from that of the bulk soil and be significantly affected by the form of nitrogen supplied and by plant species. The relative contribution of the three effects might differ in different circumstances, but in the data available, the first two dominate and uptake increases as pH decreases. This is also consistent with the secretion of acids by cluster roots. These are structures that give improved access to phosphate in low-phosphate soils. Secretion of acid would be counter- productive if the optimum pH for availability of phosphate was between 6 and 7.
Soil pH affects the availability of many nutrients and of several toxic elements. The optimum pH for plant growth depends on which is the most limiting. This was shown in Fig. 10b in which the clovers were limited by phosphorus supply but the grass was limited by nitrogen supply. Consequently the optimum pH differed. This illustrates the kind of evaluation needed in many practical situations when deciding what pH to aim for. Raising the pH to say 6 to 7 might be justified for several reasons, but increasing the availability of phosphate is not one of them.
References
Ashman MR, Puri G (2003) Essential soil science, a clear and concise introduction to soil science. Blackwell Publishing, Carlton Victoria
Barrow NJ (1965) Further investigations of the use of lime on established pastures. Aust J Experimental Agric Anl Husb 5:442–449
Barrow NJ (1984) Modelling the effect of pH on phosphate sorption by soils. J Soil Sci 35:283–297
Barrow NJ (1987) Reactions with variable-charge soils. Martinus Nijhoff Publishers, Dordrecht
Barrow NJ (1999) The four laws of soil chemistry: the Leeper lecture 1998. Aust J Soil Res 37:787–829
Barrow NJ (2002) Influence of pH on a secondary effect of phosphate reaction: the decrease in sorption of newly-added phosphate. Aust J Soil Res 40:775–779
Barrow NJ, Debnath A (2014) Effect of phosphate status on the sorption and desorption properties of some soils of northern India. Plant Soil 378:383–395
Barrow NJ, Debnath A (2015) Effect of phosphate status and pH on sulphate sorption and desorption. Eur J Soil Sci 66:286–297
Barrow NJ, Madrid L, Posner AM (1981) A partial model for the rate of adsorption and desorption of phosphate by goethite. J Soil Sci 32:399–407
Bockris JO’M, Reddy AKN (1970) Modem electrochemistry: an introduction to an interdisciplinary area. Plenum Press, New York
Bowden JW, Posner AM, Quirk JP (1977) Ionic adsorptionon variable charge mineral surfaces. Theoretical-charge development and titration curves. Aust J Soil Res 15:121–126
Bowden JW, Nagarajah S, Barrow NJ, Posner AM, Quirk JP (1980) Describing the adsorption of phosphate, citrate and selenite on a variable charge mineral surface. Aust J Soil Res 18:49–60
Brady NS (1974) The nature and properties of soils, 10th edn. Macmillan, New York
Chen JH, Barber SA (1990) Soil pH and phosphorus and potassium uptake by maize evaluated with an uptake model. q 54:1032–1036
Dunlop J, Dowling JF (1978) Uptake of phosphate by white clover II. The effect of pH on the electrogenic phosphate pump J Exp Bot 2:1147–1153
Fordham AW, Norrish K (1979) Arsenate-73 uptake by components of several acid soils. Aust J Soil Res 17:307–316
Hiemstra T, Van Riemsdijk WH (1996) A surface structural approach to ion adsorption:the charge distribution (CD) model. J Colloid Interface Sci 179:488–508
Hinsinger P (2001) Bioavailability of soil inorganic P in the rhizosphere as affected by root-induced chemical changes: a review. Plant Soil 237:173–195
Holford ICR, Mattingly GEG (1975) The high- and low-energy phosphate adsorbing surfaces in calcareous soils. J Soil Sci 26:407–417
Lambers H, Chapin FS, Pons T (2008) Plant physiological ecology. Springer-Verlag, New York
Leeper GW, Uren NC (1993) Soil science: an introduction, 5th edn. Melbourne University Press, Carlton
Murrmann RP, Peech M (1969) Effect of pH on labile and soluble phosphate in soils. Soil Sci Soc Am Proc 33:205–210
Norrish K, Rosser H (1983) Mineral phosphate. In: CSIRO Div. of Soils (Editors), Soils, an Australian viewpoint. CSIRO, Melbourne
Price G. (2006) (Ed) Australian Soil Fertility Manual, 3rd Edition, Fertilizer Industry Federation of Australia and CSIRO. P 45
Riley D, Barber SA (1971) Effect of ammonium and nitrate fertilization on phosphorus uptake asrelated to root-induced pH changes at the root-soil interface. Soil Sci Soc Am Proc 35:301–306
Sentenac H, Grignon C (1985) Effect of pH on orthophosphate uptake by corn roots. Plant Physiol 77:136–141
Smith SE, Jakobsen I, Grønlund M, Smith FA (2011) Roles of arbuscular mycorrhizas in plant phosphorus nutrition: interactions between pathways of phosphorus uptake in arbuscular mycorrhizal roots have important implications for understanding and manipulating plant phosphorus acquisition. Plant Physiol 156:1050–1057
Strauss R, Brümmer GW, Barrow NJ (1997) Effects of crystallinity of goethite: II. Rates of sorption and desorption of phosphate. Eur J Soil Sci 48:101–114
Tan KH (1992) Principles of soil chemistry, 2nd edn. Marcel Dekker, New York
Tisdale SL, Nelson WL (1966) Soil fertility and fertilizers. The Macmillan Company, New York
Troch FR, Thompson LM (1993) Soils and soil fertility. Oxford University Press, Oxford
Vange MS, Holmern K, Nissen P (1974) Multiphasic uptake of sulfate by barley roots I. Effects of analogues, phosphate, and pH. Physiol Plant 31:292–301
White RE (2006) Principles and practice of soil science. Blackwell Publishing, Carlton Victoria
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: John Hammond.
Rights and permissions
About this article

Cite this article
Barrow, N.J. The effects of pH on phosphate uptake from the soil. Plant Soil 410, 401–410 (2017). https://doi.org/10.1007/s11104-016-3008-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-016-3008-9