
Abstract
Plants utilize a plethora of peptide signals to regulate their immune response. Peptide ligands and their cognate receptors involved in immune signaling share common motifs among many species of vascular plants. However, the origin and evolution of immune peptides is still poorly understood. Here, we searched for genes encoding small secreted peptides in the genomes of three bryophyte lineages—mosses, liverworts and hornworts—that occupy a critical position in the study of land plant evolution. We found that bryophytes shared common predicted small secreted peptides (SSPs) with vascular plants. The number of SSPs is higher in the genomes of mosses than in both the liverwort Marchantia polymorpha and the hornwort Anthoceros sp. The synthetic peptide elicitors—AtPEP and StPEP—specific for vascular plants, triggered ROS production in the protonema of the moss Physcomitrella patens, suggesting the possibility of recognizing peptide ligands from angiosperms by moss receptors. Mass spectrometry analysis of the moss Physcomitrella patens, both the wild type and the Δcerk mutant secretomes, revealed peptides that specifically responded to chitosan treatment, suggesting their role in immune signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plants cope with constantly changing environmental conditions and numerous threats, such as phytopathogens, throughout their life. Because of their ability to sense different molecules, pattern-recognition receptors (PRRs) are considered to be a part of the innate immune system (Couto and Zipfel 2016; Boutrot and Zipfel 2017). PRRs consist of receptor-like kinases (RLKs) with an intracellular kinase domain and receptor-like proteins (RLPs) that comprise only extracellular and transmembrane domains. To date, different ectodomains binding the ligands have been described, such as leucine-rich repeats (LRRs), lysine motifs, lectin-like motifs as well as epidermal growth factor (EGF)-like domains (Couto and Zipfel 2016; Boutrot and Zipfel 2017). LRR-RLKs are the most abundant RLK family. Over two hundred of LRR-RLKs that regulate various processes from growth and development to immune response were found in Arabidopsis thaliana (Smakowska-Luzan et al. 2018). It was shown that the canonical RLK architecture consisting of kinase domain, transmembrane domain and extracellular domain was present after the divergence of land plants (Han 2019). It was proposed that RLK receptors participating in plant-microorganism symbiotic interactions have been already present in the most recent common ancestor of extant land plants and green algae (Delaux et al. 2015).
Genomes of bryophytes also contain LRR-RLK genes; however, their average number is lower compared with that of angiosperms (Ponce de León and Montesano 2013; Bressendorff et al. 2016). As such, 134 and 81 LRR-RLKs genes have been identified in moss and spikemoss, respectively (Dufayard et al. 2017). In particular, Physcomitrella patens encodes LysM-RLKs CERK1 homolog involved in the perception of fungal chitin (Bressendorff et al. 2016; Galotto et al. 2020). It is assumed that CERK1 was probably involved in plant immune signaling in the last common ancestor of all land plants (Fürst-Jansen et al. 2020). However, FLS and EFR receptors, responsible for the detection of proteaceous ligands, such as bacterial flagellin and elongation factor Tu, have not been found in P.patens (Bressendorff et al. 2016).
Another class of receptors that are able to detect pathogens inside plant cells are named nucleotide-binding and leucine-rich repeat domain (NBS-LRR) proteins. They are fairly studied in angiosperms, but for bryophytes the number of corresponding studies is much lower (Han 2019; Gao et al. 2018). The NBS-LRR proteins have been found in both some algae and bryophytes, suggesting the probable presence of such immune system components already in the earliest land plants (Gao et al. 2018; de Vries et al. 2018).
Pathogen-associated molecular patterns (PAMPs) that are sensed by plant PRRs trigger defense response cascades and the release of the plant’s own damage-associated immune signals (DAMPs). The known PAMPs include peptides flg22 from flagellin (Felix et al. 1999), elf18 from EF-Tu (Kunze et al. 2004), specific for rice RaxX (Luu et al. 2019), percepted by the first predicted pattern recognition receptor XA21 (Song et al. 1995), as well as liposaccharides, lipoproteins, nucleic acids and chitin, percepted by a LysM RLK chitin-elicitor receptor kinase 1 (CERK1) (Miya et al. 2007; Wan et al. 2008; Bigeard et al. 2015).
To date, several dozens of DAMP peptides that are involved in the immune signaling have been identified. For example, the CAP-derived Peptides (CAPE) are derived from functional precursor protein PR-1b and are involved in pathogen defense pathways (Chen et al. 2020). One of the most well-studied and diverse groups of DAMP peptides is the plant elicitor peptides (PEP) family. PEPs have been identified in various families of angiosperms and are generated from inactive protein precursors. For example, in Arabidopsis thaliana, a 23-aa AtPEP1 is released from a 92-aa inactive protein precursor and is recognized by PEPR1 and PEPR2, which trigger immune signaling cascades (Huffaker et al. 2006; Yamaguchi et al. 2010). An increase in reactive oxygen species (ROS) levels in response to PEP treatment has been shown in several studies on Arabidopsis (Huffaker et al. 2006; Ma et al. 2013). There are 8 PEPs identified in Arabidopsis thaliana, and PEPs within plant families are highly conserved. Although a comparison of PEPs from dicots and monocots (AtPEP1 and ZmPEP1, respectively) revealed a sequence identity of 20.8 %, which is the percentage of the PEPs sequence conservation among different plant families (Lori et al. 2015). PEPs have not been identified in bryophytes and whether this DAMP signaling pathway is conserved among different plant lineage is unknown (Lori et al. 2015).
Antimicrobial peptides provide the first barrier against pathogenic bacteria and fungi, but a number of regulatory peptides are also involved in the immune responses in plants. Antimicrobial and signaling immune peptides in both plants and animals activate cascades of biochemical reactions in cells and induce expression of the corresponding defense genes (Czyzewicz et al. 2013; Oh et al. 2018). Such peptides have been found in many plant species, their precursor sequences contain conserved regions and are predicted from a number of genomes (Lease and Walker 2006; Butenko et al. 2009). These peptides belong to a group of small secreted peptides (SSPs) that usually represent sequences of preproteins of approximately 100 to 250 amino acids. SSPs are usually processed into bioactive peptides consisting of up to 50 amino acids (Lease and Walker 2006; Breiden and Simon 2016; Bang et al. 2017) and often work at nanomolar physiological concentrations (Murphy et al. 2012). Based on their origin and structure, SSPs are classified as Cys-rich, non-Cys-rich, post-translationally modified (PTM), non-PTM, functional precursor and encoded by short open reading frames (sORFs). The characterization and annotation of the SSPs in plant genomes is still ongoing. Several approaches have been developed to identify SSPs gene candidates in plant genomes in recent years (Ohyama et al. 2008; Pan et al. 2013; Zhou et al. 2013; Li et al. 2014; Ghorbani et al. 2015; Bang et al. 2017). Because there are similarities between the structure and sequences of functional peptides, the pipelines based on hidden Markov models (HMMs) of known families have been used to scan genome assemblies for SSP genes (Pan et al. 2013; Zhou et al. 2013). Recent evidence suggests that of all the tools used for prediction of potential SSP SPADA is currently the most efficient computational pipeline, which combines multiple approaches to identify new small peptides in plants (Zhou et al. 2013, 2020; Bang et al. 2017). Databases of known SSPs for various plant species are being created; the most recent one is MtSSPdb (https://mtsspdb.noble.org/) (Bang et al. 2017; Boschiero et al. 2020). Another database, PlantSSP, contains more than 39,000 small proteins derived from 32 plant species (Ghorbani et al. 2015). Machine learning has also been shown to be an efficient method for predicting genes of signaling peptides with conserved domains such as Clavata/Embryo Surrounding Region (CLE) (Zhang et al. 2020b). Recent research also shows that analysis of both the transcriptome and proteome followed by bioinformatic screening improves the identification accuracy (Wang et al. 2020) on per with a direct peptidomic approach because mature signaling peptides are often post-modified, and in silico analysis alone is not enough (Luo et al. 2019). The role of predicted new SSPs in plant defense responses has also been verified using a microarray assay (Zhou et al. 2020).
The knowledge gap surrounding immune system components of bryophytes such as P.patens or Marchantia polymorpha began to fill in recent studies (Bressendorff et al. 2017; Carella et al. 2019; Peñuelas et al. 2019; Galotto et al. 2020). Recently, it was reported that M.polymorpha has the components of the whole jasmonic acid (JA)-signaling cascade (Peñuelas et al. 2019; Monte et al. 2019). It was also reported that A.thaliana and M.polymorpha both have a functionally conservative jasmonoyl-isoleucine (JA-Ile) receptor COI1, although they recognize different ligands (Monte et al. 2018). In addition, a comparison between a liverwort M.polymorpha and an angiosperm Nicotiana benthamiana has shown a number of shared defense genes activating a phenylpropanoid metabolic pathway upon oomycetes infection (Carella et al. 2019). The key participants in this pathway have also been found in streptophyte algae (de Vries et al. 2017). There is also a clear antagonism of JA and salicylic acid (SA) pathways in M.polymorpha (Matsui et al. 2019). These findings indicate the functional conservation of key components of plant immunity across a wide range of plant lineages. However, the evolution of peptide immune signals as well as their signaling cascades components is still poorly studied. It is unclear if there are conserved immune peptide signaling pathways among non-vascular and vascular plants. The Physcomitrella patens secretome comprises hundreds of endogenous peptides and stress hormones treatment results in changes in peptide pools (Fesenko et al. 2019b; Filippova et al. 2019). It is possible that some of these peptides may be involved in immune signaling and are recognized by LRR-RLK receptors. Through many experiments, it was shown that the moss P.patens respond to attack from a wide range of phytopathogens such as oomycetes, bacteria and fungi (Overdijk et al. 2016; Ponce de León et al. 2007, 2012; Oliver et al. 2009; Ponce de León and Montesano 2013; Reboledo et al. 2015; Bressendorff et al. 2016, 2017). Bacterial pathogens trigger cell death, cytoplasmic shrinkage, chloroplast browning, induction of defense genes such as PR-1, PAL, CHS and to a lesser extent LOX in moss P.patens gametophores, similar to vascular plants (Ponce de León et al. 2007; Ponce de León and Montesano 2013). Cell wall reinforcement is accompanied by an accumulation of different ROS types, fungal infection, in particular, induces production of H2O2 and hydroxyl radicals in moss cells, as well as promotes defense genes expression (Ponce de León and Montesano 2013; Lehtonen et al. 2009, 2012). In angiosperms ROS accumulation is associated with stress signaling. Respiratory Burst Oxidase Proteins (RBOHs) catalyze the production of superoxide anion free radical (O2−) from oxygen and considered the main source of ROS in the apoplast under biotic stress. Then, superoxide dismutase catalyzes the dismutation of O2− radical into the most long-lived ROS - hydrogen peroxide (H2O2) (Qi et al. 2017; Shapiguzov et al. 2012). Apoplastic peroxidases can also lead to the production of Н2О2 under biotic stress. It was shown that Н2О2 from the apoplast might enter the cell through aquaporins (Qi et al. 2017; Shapiguzov et al. 2012). The cellular organelles - chloroplasts, mitochondria, and peroxisomes can also produce ROS that regulate the immune response (Vellosillo et al. 2010; Shapiguzov et al. 2012).
Aside from fundamental studies of immune response mechanisms, a vast majority of research is focused on practical application of that knowledge for growing sustainable crops. Targets of such studies are usually pattern-recognition receptors; gene transfer of pattern-recognition receptors from donor plants, including model plants such as Arabidopsis, to recipients that lack such receptors leads to resistance of plants to pathogens to which they were initially susceptible (Lacombe et al. 2010; Schoonbeek et al. 2015; Schwessinger et al. 2015; Hao et al. 2016; Saur et al. 2016). In addition, genes of existing receptor-like kinases that recognize pathogen effectors within plant cells have been modified at the binding site to improve ligand recognition (De la Concepcion et al. 2019). Thus, these studies reveal a common principle of the immune response among a wide range of phylogenetically distant plant species. The gradual complication of the pathways and the addition of new specific components of the immune system in the process of evolution leads to differences in the ways different plants resist the pathogen attacks (Han 2019). However, the aforementioned examples indicate that the study of the conservative components of plant’s immune system on non-vascular plant models is of significant importance for understanding and regulating defense responses in vascular plants.
The genomes of the bryophyte model organisms are fully annotated, as three high-quality genomes of Anthoceros hornworts have also been recently analyzed, thus broadening the availability of suitable model systems in the bryophyte lineage (Zhang et al. 2020a). In this study, we searched for possible homologs of receptor-like kinases and plant SSPs in the genomes of several bryophytes using bioinformatic tools such as SPADA (https://github.com/ZhaoBioinformaticsLab/PlantSSPProtocols) and HMMER (http://hmmer.org). We also conducted a comparative analysis of secreted endogenous peptide pools from a wild type model plant moss Physcomitrella patens and mutants with knocked-out cerk gene upon chitosan treatment. The data obtained were compared with our previous data from experiments with salicylic acid and methyl jasmonate (Fesenko et al. 2019b; Filippova et al. 2019). We also found the increase of ROS release upon the treatment of wild-type moss and ∆cerk mutants with chitosan, as well as the well-known plant peptide elicitors and our signal peptide candidates.
Materials and methods
Plant cultivation and elicitor treatment
Protonemata of the moss Physcomitrella patens subsp. patens Gransden 2004 (Freiburg, Germany) wild type and mutant lines were grown in 200 mL liquid Knop medium containing 500 mg/L ammonium tartrate under white light with a photon flux of 61 µmol/m2 s under a 16-hour photoperiod at 24°C. Moss ∆cerk mutant lines were provided by Dr Bressendorff (Bressendorff et al. 2016). For mass spectrometry analysis, 5-day-old protonemata were treated with 0.1 mg/mL chitosan (Mw = 50,000–190,000 Da, 75–85 % deacetylated; Sigma-Aldrich, USA) and incubated for 3 h. Chitosan was dissolved in water with 0.001 % glacial acetic acid and pre-filtered through a 0.50 µm membrane filter (Millipore). To analyze ROS accumulation, 5-day old protonemata were treated with 1 mg/mL chitosan for 1 min or synthetic 5 µM peptides for 15 min. For qRT-PCR analysis, moss protonemata were treated by 5 µM synthetic peptides for 2 and 4 h. All experiments were performed in three independent biological replicates.
Peptide pools extraction
Secreted peptides were extracted from 400 mL protonemata culture medium (Knop medium containing 500 mg/L ammonium tartrate). The culture medium was filtered through a 0.22 µm membrane filter (Millipore), lyophilized and resuspended in 700 µL 5 % acetonitrile (ACN) aqueous solution containing 0.1 % trifluoroacetic acid (TFA), followed by centrifugation at 10,000×g for 10 min, then the supernatant was transferred into clean tubes, cooled and centrifuged at 15,000×g for 30 minutes to remove any residual chitosan, and the pellet was discarded. For mass-spectrometry analysis, peptides were isolated from the culture medium by solid-phase extraction on reverse-phase DSC-18 cartridges (Discovery DSC-18, Supelco, USA) using a 50 % ACN/0.1 % TFA aqueous solution as eluent. The eluted peptides were concentrated in a SpeedVac and resuspended in 5 % ACN/0.1 % TFA. The evaporated precipitate was dissolved in aqueous solution containing chloracetomide (CAA), tris(2-carboxyethyl)phosphine (TCEP) and 8.5 pH Tris-HCl, heated to 90°C for 10 min and then cooled to restore S-S bridges in amino acids. The pool of peptides was isolated by solid-phase extraction on microtips with three SDB-RPS membranes using a 50 % ACN/0.1 % TFA aqueous solution as the eluent. Eluted peptides were concentrated in a SpeedVac and resuspended in 5 % ACN/0.1 % TFA, followed by mass spectrometry analysis of the prepared samples.
ROS detection and measurement
To detect intracellular ROS, we used the fluorescent dye 2ʹ,7ʹ-Dichlorofluorescin Diacetate (DCFH-DA, Sigma-Aldrich, USA). Protonema was treated with 5 µM synthetic peptides (Table 1) and incubated with 10 µM DCFH-DA for 15 min. 6 pmol retention time (RT) peptides mix (Biognosys, Switzerland) was used as negative control. 1 mg/mL chitosan was added up to 1 mL of total volume directly before the detection on the fluorescent microscope Axio Imager M2 (Zeiss) with an AxioCam 506 mono digital camera and filter units. The No. 44 filter (λex BP 475 nm/40 nm; λem BP 530 nm/50 nm) was used for DCFH-DA fluorescence detection. The exposure was set to 300 msec, FITC and Brightlight channels were used. Data on the fluorescence intensity were obtained from the related Zeiss software Zen.
To detect the accumulation of extracellular ROS a deesterified DCFH was obtained from DCFH-DA via hydrolysis in NaOH (10 mM) as described (Smirnova et al. 2009). 1 mL liquid medium containing protonemata filaments, 10 µM dye and elicitors were centrifuged at 15,000×g for 1 min, the precipitate then discarded. The level of extracellular ROS was detected in supernatant using a multifunction reader Varioskan Flash (Thermo Scientific, USA). The fluorescence intensity was measured at λ 485/535 nm (excitation/emission wavelength) at 25°C. The ROS accumulation intensity was finally reported as relative fluorescence units (RFU).
Luminol (Sigma-Aldrich, USA) was used to detect the accumulation of extracellular ROS. The measurements were made in 500 µL protonemata with 20 µL luminol solution (1-2 M stock solution), 5 µL horseradish peroxidase solution (10 mg/mL stock solution) and elicitors. The measurements were carried out on a Lum1200 chemiluminometer (MSU, Russia) in a single-channel mode with an interval of 1 s at room temperature for 30 min. The result was estimated as the area under the curve after adding a mixture of peroxidase with luminol.
At the end of the measurements, Н2О2 (5 µL from a 9 mM solution) was added and the chemiluminescence (CL) outbreak was recorded in the same mode as the maximum CL value. All measurements were made in three replicates. All control samples were treated with water.
LC-MS/MS analysis and peptide identification
Mass spectrometry analysis of endogenous peptides was performed in three biological repeats. LC-MS analysis was carried out on an Ultimate 3000 RSLCnano HPLC system connected to a Q Exactive Plus mass spectrometer (ThermoFisher Scientific). Samples were loaded to a home-made trap column 20 × 0.1 mm, packed with Inertsil ODS3 3 µm sorbent (GLSciences), in the loading buffer (2 % ACN, 98 % H2O, 0.1 % TFA) at 10 µL/min flow and separated at RT in a home-packed fused-silica column 500 × 0.1 mm packed with Reprosil PUR C18AQ 1.9 (Dr. Maisch) into the emitter prepared with P2000 Laser Puller (Sutter, USA) (Kovalchuk et al. 2019). Samples were eluted with a linear gradient of 80 % ACN, 19.9 % H2O, 0.1 % formic acid (FA) (buffer B) in 99.9 % H2O, 0.1 % FA (solvent A) from 4 to 36 % of solvent B in 1 h at 0.44 µL/min flow at RT.
MS data was collected in DDA mode. MS1 parameters were as follows: 140K resolution, 350–2000 scan range, max injection time 50 ms, AGC target 3 × 106. Ions were isolated with 1.4 m/z window and 0.2 m/z offset targeting the 10 highest intensity peaks of + 1 to + 6 charge, 8 × 103 minimum AGC, preferred peptide match and isotope exclusion. Dynamic exclusion was set to 40 s. MS2 fragmentation was carried out in HCD mode at 17.5K resolution with 27 % NCE. Ions were accumulated for max 50 ms with target AGC 1 × 105.
Mass-spectrometry data were searched with PEAKS Studio (version 8.0, Bioinfor Inc., CA, USA) against a database containing the protein sequences from Phytozome v12.0 merged with chloroplast and mitochondrial proteins (33,053 entries) and sequences predicted by SPADA. The search was done with the following parameters: precursor mass tolerance of 10 ppm and fragment mass tolerance of 0.05 Da; the “digest mode” was set as “unspecific”; fixed modifications - Carbamidomethylation (+ 57.02); variable modifications - Oxidation (M) - +15.99 and Acetylation (N-term) - +42.01. The instrument setting was set to „orbi-orbi‟ (orbitrap was used for both precursor and fragment ion m/z detection). A false discovery rate of 1 % was used.
Bioinformatic analysis
SSP genes prediction
SPADA bioinformatics approach (Small Peptide Alignment Discovery Application) was applied for the search of peptide signals in the genomes of P.patens (Lang et al. 2018), S.fallax (Shaw et al. 2016), M.polymorpha (Bowman et al. 2017), Anthoceros agrestis (Bonn and Oxford strains) and A.punctatus (Zhang et al. 2020a) (Zhou et al. 2013; Boschiero et al. 2020). The pipeline of the bioinformatics algorithm is publicly available at https://github.com/ZhaoBioinformaticsLab/PlantSSPProtocols. The genomes of P.patens, S.fallax and M.polymorpha in FASTA format were downloaded from Phytozome v12.1 (https://phytozome.jgi.doe.gov/pz/portal.html), the genomes of representatives of Anthoceros were downloaded from the database at https://www.hornworts.uzh.ch/en.html. The SPADA algorithm was launched using the Linux command line. The Augustus package within the SPADA pipeline was run using Arabidopsis thaliana as a reference genome. A docker image (CRP_PlantSSPv1_Noble) containing all currently known SSPs was used for the HMM search within the SPADA pipeline. For the gene model prediction, an E-value < 0.001 threshold was used to exclude false-positive results. As a result, new gene model prediction annotations in GFF3 format were obtained. For further functional annotation and classification of the predicted genes, potentially encoding SSPs proteins (< 250 aa) were generated. The potential SSP precursors were then classified as Known SSP, Likely Known SSP, Putative SSP and Non-SSP (Boschiero et al. 2020). The Known SSP (encoding known SSP) meets the following criteria: D-score SignalP > 0.45; HMM homology E-value < 0.01; Smith-Waterman homology E-value < 0.01 and precursor length < 200 aa. The Likely Known SSP genes have HMM homology value E < 0.01 and Smith-Waterman homology E-value < 0.01; precursor length < 250. Putative SSP genes have no or little sequence similarity with previous SSPs, precursor size < 230 aa; SignalP D-score > 0.25; and no presence of TM domains. Non-SSP genes did not fit the above categories. Predicted SSPs were BLASTed against the corresponding protein databases to obtain the protein ID.
Multiple sequence alignments and HMMER search
Overall, 43 angiosperms, 1 gymnosperm and 1 lycophyte species were selected to analyze SSPs diversity. Protein sequences of CAPE, TAX, thionin and hevein SSP families were downloaded from NCBI database (https://www.ncbi.nlm.nih.gov/) and RALF sequences were obtained from Phytozome database (https://phytozome.jgi.doe.gov/pz/portal.html) in FASTA format. The list of known PEPRs in angiosperms was obtained from the paper Lori et al. (2015). The ClustalW package was applied to generate multiple alignments with default parameters (Larkin et al. 2007). After each alignment representative sequences were manually selected. The visualization of multiple alignments was performed using the Jalview2 software package (Waterhouse et al. 2009). The HMMERv3.3 (http://hmmer.org) package was applied to search for PROPEP homologs in the genome of P.patens. HMM profiles of all known PROPEP in angiosperms were generated using hmmbuild. Both per-sequence and per-domain thresholds E-value < 0.001 was set for the console jackhmmer algorithm run. The console tool of the HMMER program, jackhmmer, searched for PROPEPs in the 6-frame translated P.patens genome using previously generated HMM profiles of 75 known amino acid sequences of PROPEPs identified in both monocots and dicots.
GO term analysis
GO terms list was obtained from the Phytozome tool PhytoMine (https://phytozome.jgi.doe.gov/phytomine/begin.do). The enrichment analysis was performed using online tool g:Profiler (https://biit.cs.ut.ee/gprofiler/gost).
Total RNA isolation and qRT-PCR
Total RNA from protonemata was isolated as previously described (Fesenko et al. 2019b). Quality and quantity were evaluated using electrophoresis on agarose gel with ethidium bromide staining. Total RNA concentration of samples was precisely measured using the Quant-iT™ RNA Assay Kit, 5–100 ng on a Qubit 3.0 (Invitrogen, US) fluorometer. cDNA was synthesized using the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, Russia) according to the manufacturer’s recommendations. OligoDT primers were used to prepare cDNA from 2 µg total RNA after DNase treatment. Primers were designed using the PrimerQuest Tool (http://eu.idtdna.com/Primerquest/Home/Index). Real-time PCR was performed using the qPCRmix-HS SYBR (5×) system fluorescent probes (Evrogen, Russia) on a LightCycler® 96 (Roche, Mannheim, Germany). qRT-PCR was carried out in three biological and three technical replicates. cDNA representation was normalized using stably transcribed reference gene actin 5 (Pp1s381_21V6.1). The 2-ddCT values were obtained using the LightCycler® 96 software. Control samples were used as a calibrator.
Statistical analysis
Statistical analysis was performed across at least three biological repeats. Statistical analyses and boxplots were made in Python v. 3.7.5 [G. van Rossum, Python tutorial, Technical Report CS-R9526, Centrum voor Wiskunde en Informatica (CWI, Amsterdam, May 1995)] using module scipy (Virtanen et al. 2020). The one-way analysis of variance (ANOVA) with a paired independent t-test were applied to determine which pairwise comparisons were statistically significant. Differences were considered to be significant at p-value < 0.05.
Peptide synthesis
P.patens endogenous signal peptides were chemically synthesized at Shanghai Ruifu Chemical Co., Ltd. (Shanghai, China), angiosperm plant elicitor peptides were chemically synthesized at the genetic engineering laboratory of the FRCC PCM (Moscow, Russia). The purity of the lyophilized peptides was > 95 %, their molecular weight was confirmed by mass spectrometric analysis. The synthesized peptides were dissolved in sterile water and stored at − 80 °C.
Results
Identification of established SSP families in bryophyte genomes
Comprehensive bioinformatic analysis of potential SSPs in non-vascular plants has not been provided to date. Therefore, we used the SPADA pipeline tool (Zhou et al. 2013), to predict genes encoding SSPs in the genomes of five bryophyte species (Supplementary Table S1, Supplementary Fig. S1). The general workflow of SSPs prediction is depicted in Fig. 1a. SPADA utilizes the in-built Hidden Markov Models of SSP families included in the PlantSSP database and scans any genome for them. However, the prediction of potential SSPs is limited to the availability of HMM models of known families identified in angiosperms. Therefore, one caveat of our approach is the inability to identify bryophyte-specific SSPs.

a A workflow of SSP prediction with SPADA tool. b Diagrams showing a distribution of SSP precursors groups in different plant taxons according to in silico prediction with SPADA. Percentage corresponds to: Physcomitrella patens «Signal» group − 31 precursors (23 %), «Peptidase inhibitor» group − 14 precursors (11 %), «Antimicrobial» group − 5 precursors (4 %), «Unknown» group − 82 precursors (62 %); Marchantia polymorpha - «Signal» group − 53 precursors (68 %), «Peptidase inhibitor» group − 10 precursors (13 %), «Antimicrobial» group − 12 precursors (15 %), «Unknown» group − 3 precursors (4 %); Medicago truncatula - «Signal» group − 694 precursors (35 %), «Peptidase inhibitor» group − 147 precursors (7 %), «Antimicrobial» group − 139 precursors (7 %), «Unknown» group − 994 precursors (50 %) - of the total number. c A heatmap showing a normalized distribution of SSP gene groups in different plant taxons according to in silico prediction with SPADA on a log2 scale. Colors in the heatmap correspond to a scaled number of SSP genes
Overall, we applied the SPADA pipeline to two moss species - Physcomitrella patens (v3.3) and Sphagnum fallax (v0.5); the liverwort Marchantia polymorpha (v3.1); three recently assembled hornwort genomes – Anthoceros agrestis (Bonn and Oxford strains) and A.punctatus (https://www.hornworts.uzh.ch/en.html).
At the first step, genes that potentially encode SSPs were predicted and classified (Table 2; Supplementary Table S5-7).
Additionally, to understand whether the predicted SSP precursor genes were previously annotated, we performed BLASTP analysis of the predicted precursors against annotated proteomes of studied species (E-value 0.00001 cut-off and at least 70 % hit coverage) (Supplementary Table S2-7).
We further focused on the detailed characterization of genes, which were predicted as ‘known SSP’ in each bryophyte species.
“Signal” group
Potential immune signaling peptides
CAPE
In tomato, the CAPE peptide is derived from the C-terminal end of the tomato PR1b preproprotein of the pathogenesis-related 1 proteins (CAP) (Chen et al. 2014). Among the potential SSP predicted by SPADA in P.patens, we found four proteins that are potentially precursors of CAPE peptides: Pp3c18_21170V3.1.p - CYSTEINE-RICH SECRETORY PROTEIN-RELATED; Pp3c18_21090V3.1.p - Defense-related protein containing SCP domain; Pp3c25_120V3.1.p - ALLERGEN V5/TPX-1-RELATED FAMILY PROTEIN-RELATED; Pp3c25_500V3.1.p - PR-1. We also found possible CAPE precursors in other examined species – nine genes in M.polymorpha (Supplementary Table S4); two proteins and one protein in A.agrestis Bonn and Oxford strains, respectively (Supplementary Table S5-6); one predicted precursor in S.fallax (Supplementary Table S3). There were no genes predicted as potential CAPE precursors in A.punctatus.
We manually performed multiple sequence alignments of predicted precursors of CAPE peptides from other plants (Supplementary Fig. S2). This analysis showed that the conserved motif of the CAPE peptide (P*GN*****PY) is present in almost all potential precursors (Fig. 2a, S2). We then analyzed publicly available data of moss P.patens gene expression (https://peatmoss.online.uni-marburg.de/) and revealed that the expression of four genes predicted as CAPE precursors is upregulated upon OPDA treatment (Supplementary Table S12).

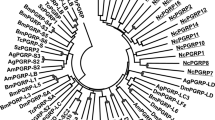
Logo for a a known motif of CAPE peptides; b a known motif of hevein peptides; c shared motifs of thionin peptide; d shared motifs of TAX peptide from some vascular plants and bryophytes candidates generated by the online tool MEME (http://memesuite.org/tools/meme). Representative sequences of CAPE peptides and candidates: DQ159948 from Solanum lycopersicum, AFK38989 from Medicago truncatula (downloaded from NCBI database), Pp3c18_21090V3.1 from Physcomitrella patens, Mapoly0097s0004.1 from Marchantia polymorpha, Sphfalx0162s0010.1 from Sphagnum fallax (downloaded from Phytozome database), AagrBONN_evm.model.Sc2ySwM_362.523.1 from Anthoceros agrestis Bonn strain and AagrOXF_evm.model.utg000076l.44.1 from Anthoceros agrestis Oxford strain (downloaded from https://www.hornworts.uzh.ch/en.html database). Representative sequences of hevein peptides and candidates: Q7Y238 from Euonymus europaeus, AAK96819.1 and NP_001321426.1 from Arabidopsis thaliana (downloaded from NCBI database), Pp3c16_13440V3.1 from Physcomitrella patens, Mapoly0057s0079.1 from Marchantia polymorpha, Sphfalx0020s0220.1 from Sphagnum fallax (downloaded from Phytozome database), AagrBONN_evm.model.Sc2ySwM_228.2301.1 from Anthoceros agrestis Bonn strain, AagrOXF_evm.model.utg000024l.73.1 from Anthoceros agrestis Oxford strain, Apun_evm.model.utg000049l.42.1 from Anthoceros punctatus (downloaded from https://www.hornworts.uzh.ch/en.html database). Representative sequences of thionin peptides and candidates: NP_001331704.1 from Arabidopsis thaliana (downloaded from NCBI database), Pp3c22_12110V3.1 from Physcomitrella patens, Sphfalx0248s0008.1 from Sphagnum fallax, Mapoly0031s0095.1 from Marchantia polymorpha (downloaded from Phytozome database), AagrBONN_evm.model.Sc2ySwM_344.1988.1 from Anthoceros agrestis Bonn strain, AagrOXF_evm.model.utg000006l.587.1 from Anthoceros agrestis Oxford strain, Apun_evm.model.utg000185l.148.1 from Anthoceros punctatus (downloaded from https://www.hornworts.uzh.ch/en.html database). Representative sequences of TAX peptides and candidates: AIW09141.1 from Taxus baccata, XP_013465154.1 from Medicago truncatula, XP_008650582.1 from Zea mays, XP_009141132.1 from Brassica rapa (downloaded from NCBI database), Pp3c4_17790V3.1.p from Physcomitrella patens, Sphfalx0071s0004.1 from Sphagnum fallax, Mapoly0016s0178.1 from Marchantia polymorpha (downloaded from Phytozome database), AagrBONN_evm.model.Sc2ySwM_368.1267.1 from Anthoceros agrestis Bonn strain, AagrOXF_evm.model.utg000010l.337.1 from Anthoceros agrestis Oxford strain, Apun_evm.model.utg000031l.724.1 from Anthoceros punctatus (downloaded from https://www.hornworts.uzh.ch/en.html database)
Peptides involved in developmental processes
The SPADA pipeline has enabled us to predict a handful of protein precursors of known peptide families involved in the regulation of plant development processes. This might indicate the importance of peptide regulation in adaptation to terrestrial environments (Whitewoods et al. 2018). Our analysis has revealed several SSPs (RALF, EPFL, CLE), which have been already identified in P.patens and also several SSPs (RALF), which have been annotated in M.polymorpha. We considered this result as a positive control of the SPADA prediction tool application.
RALF peptides
RALF peptides belong to the group of Cys-rich peptides and are identified in all angiosperms (Campbell and Turner 2017). Their primary function is connected with the regulation of root architecture; however, they have been also found in P.patens, M.polymorpha and S.fallax, which lack root systems (Campbell and Turner 2017; Bowman et al. 2017). Our analysis identified two RALF-like genes in P.patens, one of them was already annotated (Pp3c6_7200V3.1.p). There were also two genes predicted in M.polymorpha as RALF-like, which both were already annotated (Mapoly0076s0067.1, Mapoly0040s0047.1). One gene was predicted to be RALF-like in S.fallax (Supplementary Table S3) and one gene was predicted in A.punctatus, but none were found in both strains of A.agrestis (Supplementary Table S7, S5-6). Multiple sequence alignment also revealed the specific conserved motifs in predicted bryophyte precursors (Supplementary Fig. S3).
Plantcyanin/chemocyanin (PCY)
PCY peptides belong to the group of Cys-rich peptides and are involved in regulation of pollen tube growth in tracheophytes (Chae and Lord 2011). We identified 9 genes classified as Known or Likely Known SSP, which potentially encode protein precursors of plantcyanins in P.patens (Pp3c3_25110V3.1.p, Pp3c5_5180V3.1.p, Pp3c5_23940V3.1.p, Pp3c16_22330V3.1.p, Pp3c8_8380V3.1.p, Pp3c20_17730V3.1.p, Pp3c23_9820V3.1.p, pcy_Chr04_13M_1, Pp3c7_7010V3.1.p). PCY peptides were also predicted in all other examined species. The most number of precursors were identified in M.polymorpha (Supplementary Table S3-7). However, the functions of this family of secreted peptides require the further elucidation.
TAX peptides
TAX peptides belong to the group of Cys-rich peptides and are involved in the process of taxanes biosynthesis and synthesis of nicotinic alkaloids (Onrubia et al. 2014). Three genes (Pp3c5_22950V3.1.p, Pp3c3_16420V3.1.p, Pp3c4_17790V3.1.p) were predicted to encode the precursors of TAX peptides in P.patens. All of these protein precursors lacked any domain annotation in the current genome version v3.3 of P.patens and there was not any evidence of TAX presence in bryophytes. Two genes each were predicted to encode TAX peptides in M.polymorpha, A.punctatus and in both strains of A.agrestis (Supplementary Table S4-7). Four genes were predicted as TAX in S.fallax (Supplementary Table S3).
A multiple alignment of the bryophyte potential precursors with TAX protein precursors from other plants revealed strong sequences similarity (Fig. 2d, Supplementary Fig. S4).
Peptides involved in regulation of other processes
Non-specific lipid transfer protein (nsLTP)
NsLTP peptides represent a large group of Cys-rich peptides, which are involved in lipid transfer, defense responses and plant development in most vascular plants (Liu et al. 2015). Eleven genes were predicted to encode protein precursors of nsLTP in P.patens (Supplementary Table S2). Nine of these protein precursors were already annotated as Probable lipid transfer (LTP_2) and two were annotated de novo. In both strains of A.agrestis five nsLTP genes were identified (Supplementary Table S5-6). Four nsLTP genes were predicted in A.punctatus and M.polymorpha (Supplementary Table S7, S4). Two of predicted nsLTP were already annotated in M.polymorpha (Mapoly0120s0024.1.p; Mapoly0955s0001.1.p). Two proteins were predicted as precursors of nsLTP peptides in S.fallax and they were also already annotated (Sphfalx0002s0328.1.p; Sphfalx0005s0219.1.p).
The SPADA tool also predicted the already annotated “Signal” group peptides, such as Epidermal Patterning Factor-Like (EPFL) protein precursors and Clavata/Embryo Surrounding Region (CLE) protein precursors in P.patens (Supplementary Table S2) and potential precursors of EPFL peptides in the three Anthoceros genomes (Supplementary Table S5-7). We were also able to detect Root_Cap/Late_Embryogenesis protein precursors belonging to “Signal” group in P.patens and in both strains of A.agrestis (Supplementary Table S2, S5-6). Precursors of plant natriuretic peptides (PNPs) were predicted in M.polymorpha and both strains of A.agrestis, but were not identified in P.patens, S.fallax and A.punctatus (Supplementary Table S2-7).
“Peptidase inhibitor” group
Cytotoxic T-lymphocyte antigen-2 alpha (CTLA)
CTLA peptides are involved in regulation of Cys proteases activity and can inhibit protease activity (Sugita et al. 2011). Our analysis revealed 13 genes to encode potential CTLA peptides in P.patens (Pp3c2_30400V3.1.p and etc.; Supplementary Table S2). GO enrichment analysis showed that these protein precursors are involved in oxidation-reduction processes (Supplementary Table S13). CTLA peptide precursors were also predicted in all other examined species. The most number of precursors were identified in S. fallax (Supplementary Table S3-7).
“Antimicrobial” group
Antimicrobial peptides provide the first line of defense in pathogen attacks and have been found in all plant species (Czyzewicz et al. 2013; Oh et al. 2018).
Hevein-like peptides
Hevein-like peptides belong to the group of antimicrobial peptides and are identified in various monocot and dicot plants (Slavokhotova et al. 2017). In our data four genes were predicted to encode possible candidates of hevein-like peptide precursors in P.patens (Supplementary Table S2). All of the above-mentioned protein precursors participate in plant defense responses; they bind and degrade chitin of the fungal cell walls, according to the gene description in the publicly available annotations. However, some of them also encompass hevein-like peptide sequences according to the formula of known hevein peptides (C1 × 4 5 C 2 × 4C3C4 × 5 C 5 × 6C6, in which C stands for cysteine and X stands for any amino acid in between). The potential precursors of these peptides were also found in all examined bryophyte species (Tables S3-S7). Our manual multiple sequence alignment also revealed shared motifs among bryophytes and other plants protein precursors of known hevein peptides (Fig. 2b, Supplementary Fig. S5).
Thionin-like (THL)
Thionin-like peptides are cysteine-rich peptides that have both toxic and antimicrobial functions by disrupting the pathogenic membrane (Plattner et al. 2015). They have been identified in both monocots and eudicots. To date, there is no annotation of genes encoding thionin-like peptides in P.patens or other bryophytes. However, we predicted two genes as possible THL SSPs (Pp3c22_12110V3.1, Pp3c19_18370V3.1) in P.patens. These genes are non-annotated in Phytozome v12. One gene each was predicted as possible THL in M.polymorpha and three Anthoceros genomes (Supplementary Table S4-7). Two proteins were predicted as THL in S.fallax (Supplementary Table S3). We also performed a multiple alignment of those protein precursors with known THL precursors from other plants (Fig. 2c, Supplementary Fig. S6).
“Unknown” group
We identified Nodule-specific Glycine-rich Protein (NodGRP) precursors in all examined bryophyte species (Supplementary Table S2-7). The functions of this group of SSPs in bryophytes require further investigation. In addition, the SPADA pipeline predicted a number of low-molecular weight Cys-rich (LCR) peptides in P.patens, most of those were annotated de novo by SPADA (Supplementary Table S2). Precursors of LCRs were also predicted in S.fallax, A.punctatus and A.agrestis Oxford strain, but not in M.polymorpha and A.agrestis Bonn strain (Supplementary Table S3-7). The SPADA tool also predicted precursors of Nodule-specific Cysteine Rich Group (NCR) peptides in M.polymorpha, both strains of A.agrestis and in A.punctatus, but they were not identified in P.patens and S.fallax (Supplementary Table S2-7).
There were also precursors predicted as known SSP from different groups exclusively in several species: LEED.PEED (LP) precursors in A.agrestis Oxford strain and A.punctatus; Subtilisin inhibitor (SubIN) precursors in three Anthoceros genomes and in S.fallax; a Tapetum Determinant 1 (TPD) precursor in A.punctatus; and several others (Supplementary Table S2-7).
We found that the pattern of SSPs identified in S.fallax differed from P.patens and M.polymorpha, rather resembling that of hornworts (Supplementary Fig. S1, Supplementary Table S1). In the genome of S.fallax were identified several unique SSP precursors, such as MtSUBPEP, ProSCOOP, Kazal family inhibitors (Kaz) and Plant Defensin-like (PDL) (Supplementary Table S3).
In conclusion, the number of predicted genes encoding SSPs is higher in mosses than in both hornworts and liverworts but still lower than in angiosperms such as Medicago truncatula (Fig. 1b, c, Supplementary Fig. S1, Supplementary Table S1).
Identification of endogenous peptides induced by chitosan treatment
Taking into account that immune signalling peptides are often species-specific and cannot be predicted based on similarity search, we then performed mass-spectrometry analysis of the P.patens secretomes (Supplementary Table S8-11). Since the moss P.patens is unable to perceive the known bacterial elicitors such as flagellin or elf18, we induced the release of potential signalling peptides by chitosan (Supplementary Table S8, S9). First, we concentrated on the analysis of peptides and precursors from which peptides were identified in at least 2 biological repeats. In moss secretome treated by chitosan, 1187 unique peptides from 297 precursors were identified. A large portion of the precursors in chitosan-treated samples belong to chloroplastic, transport and proteolytic proteins, including proteins participating in amino acid transmembrane transport, and slightly fewer are translation factors, subunits of ribosomes and chaperones. We found that 6 % of precursors in chitosan-treated peptidomes were either uncharacterized or had an unclear function, with a half of them below 250 aa in length. Because of the huge variability of cell peptidomes, we discarded peptides derived from precursors also identified in control samples. This resulted in a filtered list of 52 peptides from 38 precursors, appearing only in chitosan-treated secretome. These precursors included stress-related proteins such as Pp3c2_12150 (molecular chaperone DnaK), Pp3c20_21140 (calcium binding protein); cell wall proteins such as Pp3c14_18450 (EXPANSIN-A5), Pp3c24_15950 (Leucine-rich repeat receptor-like protein kinase family protein), Pp3c5_23400 (Pectinesterase). Manual checking of the precursors revealed that 30 % of them were smaller than 250 aa, including Pp3c1_19210V3 (Protein of unknown function (DUF1118)), ribosomal proteins L28e and S19e and Pp3c26_5890V3 (peroxidase).
P.patens encodes four homologs of the Arabidopsis chitin receptor AtCERK1, but only one knock-out line (Δcerk1) showed insensitivity to chitin response (Bressendorff et al. 2016). To determine the peptides specifically induced by chitosan, we performed peptidome analysis of a Δcerk1 mutant secretome (Supplementary Table S9, S11). It should be noted that we cannot rule out the possibility that this mutant line senses chitin to some extent. Using peptides, identified in Δcerk1 as a filter, we identified 14 peptides, specifically appearing only in at least two chitosan-treated secretomes from wild type. Two peptides were derived from the C-terminus of the 165 aa peroxidase (Pp3c26_5890). Peroxidases are involved in numerous cellular processes such as development and stress responses.
The detection of bioactive peptides by mass-spectrometry might be difficult because of their low concentration and rapid degradation in cells. Therefore, we also looked at all peptides and precursors in chitosan-treated secretomes. First, we filtered off peptides found in control samples. Among the precursors of 5320 peptides from chitosan-treated samples were calmodulin (Pp3c10_21490), Chitinase-related (Pp3c1_30410), Probable lipid transfer (LTP_2) (Pp3c19_2080), Lactoperoxidase (Pp3c26_5890), cystatin-C (Pp3c9_24090) - inhibitor of cysteine proteinase, several actin proteins (Pp3c3_33410) and Cucumisins (Pp3c22_10010). Then, we selected 430 small (up to 250 aa) precursors appearing in chitosan-treated samples.
After discarding peptides that also appeared in ∆cerk mutants upon chitosan treatment, we identified 9 peptides from small precursors with unknown function specifically induced by chitosan.
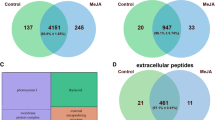
We then compared the identified peptides with our previously published moss secretomes, treated with salicylic acid (SA) (Filippova et al. 2019) and methyl jasmonate (MeJA) (Fesenko et al. 2019b). We found that peptidomes treated with methyl jasmonate and salicylic acid overlapped the most with each other (Supplementary Fig. S7). Peptides common to all three elicitor treatments were cleaved from precursors, most of which take part in photosynthesis; in addition, peptides were cleaved from Cucumisin (Pp3c2_4520) as well as from several uncharacterized proteins. Among the common precursors of induced peptides, we also found precursors of our signaling peptide candidates selected and synthesized in previous studies (INIINAPLQGFKIA and EAAPAPVAEVEAPKAEE) (Filippova et al. 2019; Fesenko et al. 2019b). Several peptides induced only in wild type upon chitosan treatment and found previously in peptidomes treated with other stress factors were synthesized for further study. These were peptides derived from precursors Pp3c14_22870V3.1.p, Pp3c21_4350V3.1.p (already selected in our previous works (Filippova et al. 2019; Fesenko et al. 2019b)) and also from Pp3c13_3880V3.1.p and Pp3c17_15750V3.1.p (Table 1). However, the role of all these peptides as DAMP signals requires further elucidation.
Next, we searched for endogenous peptides derived from protein precursors, which were predicted as Known or Likely Known SSP in extracellular peptidomes. We detected hevein-like peptides in cerk-mutant lines upon treatment with chitosan (Pp3c1_30410V3.1.p). Three unique peptides contained a motif of Cys-rich peptides, which form disulfide bridges between the following cysteine residues: C1–C4, C2–C5 and C3–C6. Thus, these peptides are very likely to be hevein peptides. Although we were not able to detect such peptides in wild type samples treated with chitosan. The low concentration and the rapid degradation of the endogenous peptides in cells and extracellular space can make the isolation and identification of certain peptides very difficult. Thus, we cannot confirm that certain peptides are not produced in the plants using only LC-MS analysis.
Also, we detected a short sequence of the RALF peptide in MeJA-treated samples. In each stress-treated and control sample we identified potential PCY peptides derived from Pp3c5_23940V3.1.p, Pp3c16_22330V3.1.p, pcy_Chr04_13M_1 (SPADA-predicted gene). Interestingly, chitosan upregulated the potential Cys-rich PCY peptides in extracellular peptidomes.
Treatment with known peptide elicitors resulted in ROS accumulation
PEPs and their receptors—plant elicitor peptide receptors (PEPRs)—are found in almost all angiosperms, but not in bryophytes. PEPRs amino acid sequences show high interfamily diversity and are mostly represented by one form in angiosperms (Lori et al. 2015). Using the sequences list of known PEPRs in monocots and dicots, we performed a BLASTp similarity search for PEPR homologs in Physcomitrella patens. As a result, we identified 14 potential PEPR-like homologs and performed a multiple alignment using the ClustalW algorithm (Supplementary Fig. S8). According to the analysis of key amino acid residues involved in the interaction between ligand and its receptors, 7 sequences were excluded from the analysis (Supplementary Fig. S9). Surprisingly, the key amino acid residues essential for interaction between PEP and PEPRs, such as R487, D441, F419, were the most conserved ones in PERP sequences in both angiosperms and P.patens. The highest level of similarity between angiosperm PEPRs and P.patens PEPR-like homologs was shown by genes Pp3c3_12560V3.1 and Pp3c2_18570V3.1, as they seem to have the key amino acid residues in the exact positions required for efficient interaction between the PEP-PEPR-BAK complex (Supplementary Fig. S9). Thus, we suggest that P.patens might sense peptide elicitors derived from angiosperms due to similar structures in potential PEPR-like homologs receptor-like kinases. We then used HMMER and its in-built jackhmmer search tool to search for PROPEP homologs in P.patens. We identified 12 potential protein transcripts, which might encompass PEP sequences. We then performed multiple sequence alignments of known PROPEP sequences and our potential candidates, however we did not find any shared motifs. We next treated moss P.patens with synthetic PEPs - AtPEP1 and StPEP1 (plant elicitor peptide from Solanum tuberosum), and analyze whether the treatment lead to ROS accumulation and changes at the transcriptional level of selected genes. We choose these PEPs because AtPEP1 is a commonly use in studies of plant immunity (Kadota et al. 2014; Huffaker et al. 2006; Roux et al. 2011) and StPEP1 comes from agriculturally important plant (Solanum tuberosum).
The release of ROS is a common indicator of stress reaction in plants (Shapiguzov et al. 2012). Therefore, we measured intra- and extracellular ROS accumulation in P.patens protonemata treated with synthetic PEPs and chitosan as a control (Fig. 3a and b; Supplementary Fig. S10). As expected, the chitosan treatment induced the release of ROS in wild-type cells, whereas a∆cerk mutant line did not show significant changes in ROS accumulation (Fig. 3a). In wild-type plants, fluorescent signal was mainly detected in the cytoplasm and near the cell walls (Fig. 3b). Synthetic PEPs treatment also resulted in an increase of intracellular ROS in chloroplasts and in the cytoplasm (Fig. 3b). In addition, AtPEP1 and chitosan induced extracellular ROS accumulation, whereas StPEP1 did not (Supplementary Fig. S10). We also analyzed the expression of some pathogenesis-related genes, such as allene oxide synthase (AOS), OPDA reductase (OPR), chalcone synthase (CHS) and phenylalanine ammonia lyase (PAL4) in P.patens protonemata. The quantitative RT-PCR analysis (qRT-PCR) showed that the transcriptional level of CHS and PAL4 significantly increased upon treatment with 5 µM AtPEP1 (Supplementary Fig. S11a).

A comparison between ROS accumulation after treatment with 5 µM AtPEP1 and StPEP and with 1 mg/mL chitosan of wild type and cerk-mutant P.patens showed in a a boxplot and b fluorescence microscopy. All experiments had at least three biological repeats (a paired t-test: *P-value < 0.05; **P-value < 0.005; ***P-value < 0.0005). Control samples were treated with water
Then, we investigated peptide candidates identified in our previous experiments (Filippova et al. 2019; Fesenko et al. 2019b) and confirmed by mass-spectrometry analysis in this study. We found a significant increase in ROS accumulation upon treatment with synthetic candidate peptides - pep7(EAA) EAAPAPVAEVEAPKAEE and pep8 INIINAPLQGFKIA (Fig. 4a). These peptides were also found in peptidomes treated with stress hormones salicylic acid (Filippova et al. 2019) and methyl jasmonate (Fesenko et al. 2019b). The level of intracellular ROS accumulation was comparable with chitosan, suggesting the probable potential of the selected peptides as immune signals. The treatment with synthetic pep7 also resulted in an increase of extracellular ROS accumulation (Supplementary Fig. S10).

A comparison between ROS accumulation after treatment with 5 µM synthesized peptides and 6 pmol retention time (RT) peptides mix (Biognosys, Switzerland; https://biognosys.com/shop/irt-kit) of wild type P.patens in a a boxplot and b fluorescence microscopy. All experiment had at least three biological repeats (a paired t-test: *P-value < 0.05; **P-value < 0.005; ***P-value < 0.0005). Control samples were treated with water
We used qRT-PCR to check the ability of synthetic candidate peptides to induce the transcription of known pathogenesis-related genes as well. In our previous study we reported the ability of pep8 to alter the expression of OPR and AOS genes in a similar way to MeJA (Fesenko et al. 2019b). In this study, the qRT-PCR analysis showed that the expression of some pathogenesis-related genes was influenced in the time-dependent manner upon treatment with 5 µM pep7 peptide (Supplementary Fig. S11b). However, whether these peptides play a role as DAMPs requires further elucidation.
Our results suggest that mosses might be able to detect PEPs and the downstream signals are similar to those in angiosperms. Also moss itself may secrete a number of peptide signals that are able to trigger general stress reactions such as accumulation of ROS.
Discussion
Because of their specific features such as small size or specific patterns of expression, SSPs often remain unannotated. Our study showed that the number of SSPs in annotated genomes is underestimated and further re-annotation efforts are needed.
It is unclear whether SSPs from angiosperms are similar to those from non-vascular plants. To shed light on this, we conducted a bioinformatic screening of potential homologs of well-known plant SSPs in the genome of the moss Physcomitrella patens as well as in the genomes of some other bryophytes, such as a liverwort M.polymorpha, moss S.fallax and three species of hornworts Anthoceros ssp. It is worth noting that the branching order of non-vascular plants is still uncertain (Delaux et al. 2019). Three main hypotheses support the branching order of the bryophytes: mosses and liverworts are monophyletic, whereas hornworts sister to tracheophytes (Puttick et al. 2018; Qiu et al. 2006); mosses and liverworts monophyletic with hornworts sister to all land plants (Wickett et al. 2014) and mosses, liverworts and hornworts form a monophyletic group (Wickett et al. 2014; Frangedakis et al. 2021). The recent analysis of 1KP plant transcriptomes recovered extant bryophytes as monophyletic, with hornworts sister to a moss and liverwort clade (Leebens-Mack et al. 2019). The hypothesis that liverworts are sister to all other extant land plant lineages was rejected (Leebens-Mack et al. 2019). Until very recently, the genomes of Anthoceros spp. had not been analyzed, but now the availability of three high-quality genomes of Anthoceros hornworts make it possible to perform comparative studies among other lineages of extant bryophytes (Zhang et al. 2020a). Hornworts are organisms of special interest as they share common traits with both green algae and other land plants (Zhang et al. 2020a). Therefore, understanding whether they share peptide signals among other lineages of Bryophytes seems a promising approach in evolutionary studies.
We observed a contrast pattern of predicted SSPs among studied bryophyte species. SSPs were grouped by mode of action as in MtSSPdb, in which the “Signal” group includes peptides involved in development and growth processes and immune signaling, such as CLE, PEP, RALF, CAPE and SCOOP; the “Peptidase inhibitor” group includes peptides involved in regulation of proteolysis, such as phytocystatin and CTLA; the “Antimicrobial” group includes Cys-rich peptides such as thionin and plant defensin and also CTLA; the “Unknown” group refers to peptides with unidentified mode of action, such as LCR and NCR. Our analysis showed that the “Peptidase Inhibitors” group was the most abundant group of SSPs in S.fallax and Anthoceros spp., whereas in other analyzed species it is not as pronounced. The “Signal” SSPs were the most abundant in M.polymorpha in comparison to other bryophytes. The “Signal” group in three Anthoceros species had a similar size, suggesting low variability of these SSPs in related species. A substantial portion of predicted SSPs belongs to the “Unknown” group in M.truncatula and P.patens, whereas in all other species they constitute only a small part. The smallest group is the “Antimicrobial” group of peptides in all species. However, the largest number of them are represented in M.polymorpha.
Surprisingly, the pattern of SSP activity groups distribution was very similar for P.patens and M.truncatula. The patterns of SSPs in the liverwort M.polymorpha was quite unique compared with the rest of the analyzed species. For example, M.polymorpha has the largest number of signaling SSPs PCY, STIG-GRI and CAPE genes and antimicrobial Hevein-like peptide genes compared with other bryophyte species. The functions of these peptides in non-vascular plants are still unknown. Plastocyanin (PCY) and STIG-GRI have been reported to affect pollen tube growth in angiosperms (Chae and Lord 2011; Huang et al. 2014). At the same time, the number of predicted SSPs having a similarity to Cytotoxic T-lymphocyte antigen-2 alpha (CTLA) peptides was lower in M.polymorpha, as well as in P.patens and M.truncatula, than in hornworts and S.fallax. Cytotoxic T-lymphocyte antigen-2 alpha (CTLA) peptides were first found in mammals. This peptide class inhibits Cathepsin Cys proteases (Sugita et al. 2011). Taken together, our findings suggest that small peptides, probably controlling the proteolysis, form the largest group of SSPs in studied species. As expected, the number of NCR genes, which are associated with selection of symbiotic partners in nodules (Mergaert et al. 2003; Nallu et al. 2013), was much greater in M.truncatula then in the bryophytes, which seems logical as bryophytes do not have structures such as nodules for which these peptides are specific.
Surprisingly, the SSP patterns significantly differ in two studied mosses—P.patens and S.fallax. The reasons for this difference might be related to the conditions of their growth and external threats (Beike et al. 2015) as well as the quality of genome assembly and annotation.
We also found that P.patens has the largest number of nsLTP genes and LCR in comparison with other bryophyte species. NsLTPs participate in plant defense responses and various development pathways (Liu et al. 2015). P.patens also has SSP genes with high homology to known angiosperm SSPs, for example, antimicrobial Hevein-like and Thionin-like peptides, signaling CAPE and TAX peptides (Figs. 1c and 2; Supplementary Fig. S2-6). The other moss from our study—S.fallax—also has a homolog of signaling peptide SUBPEP, cleaved out of subtilisin protease in M.truncatula, and also a homolog of the precursor of the recently discovered peptide SCOOP. These findings raise a question about the origin of functional SSPs from non-specific genes during evolution.
It is known that upon the transition from water to land and from unicellular to multicellular organisms, plants have acquired new signaling mechanisms that have allowed them to adapt to new environmental conditions (Bowles et al. 2020). For example, CLE peptides have been identified in bryophytes and in green alga Chlamydomonas reinhardtii, which lack vasculature (Oelkers et al. 2008). This raises a question whether the ancestral function of the CLE peptide was initially involved in developmental processes and why the peptide started to act as a regulator of vascular development in seed plants. In this context, our knowledge about peptide signaling in extant land plants as well as the origin of most phytocytokines is even more limited. It have been shown that immunity-related receptors evolved before the transition to land, but only the genomes of bryophytes encode a large repertoire of PRR proteins with all associated domains (> 320 in Physcomitrella patens, > 600 in A.thaliana and > 1100 in Oryza sativa) (Shiu and Bleecker 2003; Shiu et al. 2004; Han 2019). In this regard, the question remains as to when the peptide signaling system appeared, and also what should appear earlier: a peptide signal or a receptor recognizing it?
The study of immune signaling peptides evolution is a challenging task because of their species-specific or even family-specific nature (Lori et al. 2015). Therefore, we can only hypothesize on the existence of functional homologs of immune signaling peptides in different plant taxa. For example, PEPs are thought to be the missing functional homologs of systemin outside of the Solanaceae (Huffaker et al. 2013).
The PEP recognition induces phosphorylation of RBOHD (Respiratory Burst Oxidase Protein D), the NADPH oxidase, which is responsible for the production of ROS in ETI. It has also been shown in soybean that GmPEP treatment enhances RBOHD gene expression. In addition, it also induced expression of GmPEP precursor gene together with other genes associated with a defense response against nematodes (Lee et al. 2018). In our study, we detected the response of moss protonemata to exogenously applied AtPEP1 and StPEP1. Treatment with AtPEP and StPEP induced the production of intracellular ROS and change the transcription of selected pathogenesis-related genes, which suggested that moss genomes encode a receptor that can perceive PEP. Recent studies also showed that taxonomically distant species can effectively perceive non-self-danger peptides. There are a few studies that support the perception of systemin (SYS) in plant species lacking its orthologs. For example, constitutive expression of the tomato prosystemin gene in tobacco affected the host proteome, with the up-regulation of proteins involved in defense responses against pathogens in leaves (Rocco et al. 2008; Zhang and Hu 2017). It was also reported that external application of 5 and 10 µM tomato systemin led to inhibition of Arabidopsis seedling root growth and the expression of the plant defensin PDF1.2 (Zhang et al. 2018). Finally, Arabidopsis thaliana perceives signals upon exogenous treatment with systemins and HypSyS, which are specific for solanaceous species. Moreover, these treatments induced resistance to the necrotrophic fungus Plectosphaerella cucumerina (Pastor-Fernández et al. 2020). These findings support the hypothesis that plants may have a common receptor’s repertoire and receptor-mediated intracellular signaling pathway in response to peptides. It is interesting to note that signaling cascades are shared among different species, while the main domain of the PEPRs have evolved with the PEPs resulting in interfamily incompatibility but high intrafamily compatibility (Huffaker et al. 2013; Lori et al. 2015; Ruiz et al. 2018).
The number of studies used fluorescent dye DCFH-DA to detected intracellular ROS accumulation under biotic stress in moss P.patens (Oliver et al. 2009; Ponce de León 2011; Ponce de León et al. 2012; Ponce de León and Montesano 2013). However, the mechanisms of extracellular ROS generation in bryophytes are poorly studied. In our study, we detected both intracellular and extracellular ROS in P.patens using DCFH-DA and DCFH as well as luminol. Despite its wide use, luminol is not suitable for the detection of NO, ONOO−, H2O2 or OH· in living systems (Pavelescu 2015; Halliwell and Whiteman, 2004). Moreover, because luminol can interfere with different phenolic compounds (Cui et al. 2004), the analysis of extracellular ROS in mosses may be difficult (Klavina et al. 2015). Therefore, we additionally measured the extracellular ROS accumulation by DCFH. This dye is a non-selective probe for specific types of ROS, as it can interact with H2O2, OH ·− and even ONOO− (Halliwell and Whiteman 2004; Gomes et al. 2005).
It is interesting to note that, in contrast to the ROS level that was observed after treatment with flg22 and elf18, the ROS level after treatment with chitin and PEP was significantly lower (approximately two times), whereas it was comparable between chitin and PEP (slightly higher for chitin) (Ma et al. 2013). In our analysis, the levels of activated ROS in response to treatment with AtPEP1 and chitosan were comparable. Taking into account that chitin is an established immune elicitor, this may indicate quite strong response to PEP treatment in P.patens protonemata. The level of extracellular and intracellular ROS in response to StPEP treatment were contrasted. This result could be due to the release of compounds with antioxidant activity into the extracellular space.
Data Availability
All data generated or analyzed during this study are included in the manuscript. The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al. 2019) partner repository with the dataset identifier PXD021721.
References
Beike AK, Spagnuolo V, Lüth V, Steinhart F, Ramos-Gómez J, Krebs M, Adamo P, Rey-Asensio AI, Angel Fernández J, Giordano S, Decker EL, Reski R (2015) Clonal in vitro propagation of peat mosses (Sphagnum L.) as novel green resources for basic and applied research. Plant Cell Tissue Organ Cult PCTOC 120:1037–1049. https://doi.org/10.1007/s11240-014-0658-2
Bigeard J, Colcombet J, Hirt H (2015) Signaling mechanisms in pattern-triggered immunity (PTI). Mol Plant 8:521–539. https://doi.org/10.1016/j.molp.2014.12.022
Boschiero C, Dai X, Lundquist PK, Roy S, de Bang TC, Zhang S, Zhuang Z, Torres-Jerez I, Udvardi MK, Scheible W-R, Zhao PX (2020) MtSSPdb: the medicago truncatula small secreted peptide database. Plant Physiol 183:399–413. https://doi.org/10.1104/pp.19.01088
Boutrot F, Zipfel C (2017) Function, discovery, and exploitation of plant pattern recognition receptors for broad-spectrum disease resistance. Annu Rev Phytopathol 55:257–286. https://doi.org/10.1146/annurev-phyto-080614-120106
Bowles AMC, Bechtold U, Paps J (2020) The origin of land plants is rooted in two bursts of genomic novelty. Curr Biol 30:530-536.e2. https://doi.org/10.1016/j.cub.2019.11.090
Bowman JL, Kohchi T, Yamato KT, Jenkins J, Shu S, Ishizaki K, Yamaoka S, Nishihama R, Nakamura Y, Berger F, Adam C, Aki SS, Althoff F, Araki T, Arteaga-Vazquez MA, Balasubrmanian S, Barry K, Bauer D, Boehm CR, Briginshaw L, Caballero-Perez J, Catarino B, Chen F, Chiyoda S, Chovatia M, Davies KM, Delmans M, Demura T, Dierschke T, Dolan L, Dorantes-Acosta AE, Eklund DM, Florent SN, Flores-Sandoval E, Fujiyama A, Fukuzawa H, Galik B, Grimanelli D, Grimwood J, Grossniklaus U, Hamada T, Haseloff J, Hetherington AJ, Higo A, Hirakawa Y, Hundley HN, Ikeda Y, Inoue K, Inoue S, Ishida S, Jia Q, Kakita M, Kanazawa T, Kawai Y, Kawashima T, Kennedy M, Kinose K, Kinoshita T, Kohara Y, Koide E, Komatsu K, Kopischke S, Kubo M, Kyozuka J, Lagercrantz U, Lin S-S, Lindquist E, Lipzen AM, Lu C-W, Luna ED, Martienssen RA, Minamino, N., Mizutani, Masaharu, Mizutani, Miya, Mochizuki, Monte N, Mosher I, Nagasaki R, Nakagami H, Naramoto H, Nishitani S, Ohtani K, Okamoto M, Okumura T, Phillips M, Pollak J, Reinders B, Rövekamp A, Sano M, Sawa R, Schmid S, Shirakawa MW, Solano M, Spunde R, Suetsugu A, Sugano N, Sugiyama S, Sun A, Suzuki R, Takenaka Y, Takezawa M, Tomogane D, Tsuzuki H, Ueda M, Umeda T, Ward M, Watanabe JM, Yazaki Y, Yokoyama K, Yoshitake R, Yotsui Y, Zachgo I, Schmutz S, J (2017) . Cell 171:287–304. https://doi.org/10.1016/j.cell.2017.09.030
Breiden M, Simon R (2016) Q&A: How does peptide signaling direct plant development? BMC Biol 14:58. https://doi.org/10.1186/s12915-016-0280-3
Bressendorff S, Rasmussen MW, Petersen M, Mundy J (2017) Chitin-induced responses in the moss physcomitrella patens. In: Shan L, He P (eds) Plant pattern recognition receptors: methods and protocols. methods in molecular biology. Springer, New York, pp 317–324. https://doi.org/10.1007/978-1-4939-6859-6_27
Bressendorff S, Azevedo R, Kenchappa CS, Ponce de León I, Olsen JV, Rasmussen MW, Erbs G, Newman M-A, Petersen M, Mundy J (2016) An innate immunity pathway in the moss Physcomitrella patens. Plant Cell 28:1328–1342. https://doi.org/10.1105/tpc.15.00774
Butenko MA, Vie AK, Brembu T, Aalen RB, Bones AM (2009) Plant peptides in signalling: looking for new partners. Trends Plant Sci 14:255–263. https://doi.org/10.1016/j.tplants.2009.02.002
Carella P, Gogleva A, Hoey DJ, Bridgen AJ, Stolze SC, Nakagami H, Schornack S (2019) Conserved biochemical defenses underpin host responses to oomycete infection in an early-divergent land plant lineage. Curr Biol 29:2282-2294.e5. https://doi.org/10.1016/j.cub.2019.05.078
Campbell L, Turner SR (2017) A comprehensive analysis of RALF proteins in green plants suggests there are two distinct functional groups. Front Plant Sci. https://doi.org/10.3389/fpls.2017.000372017.00037
Chae K, Lord EM (2011) Pollen tube growth and guidance: roles of small, secreted proteins. Ann Bot 108:627–636. https://doi.org/10.1093/aob/mcr015
Chen Y-L, Fan K-T, Hung S-C, Chen Y-R (2020) The role of peptides cleaved from protein precursors in eliciting plant stress reactions. New Phytol 225:2267–2282. https://doi.org/10.1111/nph.16241
Chen Y-L, Lee C-Y, Cheng K-T, Chang W-H, Huang R-N, Nam HG, Chen Y-R (2014) Quantitative peptidomics study reveals that a wound-induced peptide from PR-1 regulates immune signaling in tomato. Plant Cell 26:4135–4148. https://doi.org/10.1105/tpc.114.131185
Couto D, Zipfel C (2016) Regulation of pattern recognition receptor signalling in plants. Nat Rev Immunol 16:537–552. https://doi.org/10.1038/nri.2016.77
Cui H, Shi M-J, Meng R, Zhou J, Lai C-Z, Lin X-Q (2004) Effect of pH on inhibition and enhancement of luminol–H2O2–Co2 chemiluminescence by phenolic compounds and amino acids. Photochem Photobiol 79:233–241. https://doi.org/10.1562/BE-03-28.1
Czyzewicz N, Yue K, Beeckman T, De Smet I (2013) Message in a bottle: small signalling peptide outputs during growth and development. J Exp Bot 64:5281–5296. https://doi.org/10.1093/jxb/ert283
de Bang TC, Lundquist PK, Dai X, Boschiero C, Zhuang Z, Pant P, Torres-Jerez I, Roy S, Nogales J, Veerappan V, Dickstein R, Udvardi MK, Zhao PX, Scheible W-R (2017) Genome-wide identification of medicago peptides involved in macronutrient responses and nodulation. Plant Physiol 175:1669–1689. https://doi.org/10.1104/pp.17.01096
de Vries J, de Vries S, Slamovits CH, Rose LE, Archibald JM (2017) How embryophytic is the biosynthesis of phenylpropanoids and their derivatives in streptophyte algae? Plant Cell Physiol 58:934–945. https://doi.org/10.1093/pcp/pcx037
De la Concepcion JC, Franceschetti M, MacLean D, Terauchi R, Kamoun S, Banfield MJ (2019) Protein engineering expands the effector recognition profile of a rice NLR immune receptor. eLife. https://doi.org/10.7554/eLife.47713
Delaux P-M, Radhakrishnan GV, Jayaraman D, Cheema J, Malbreil M, Volkening JD, Sekimoto H, Nishiyama T, Melkonian M, Pokorny L, Rothfels CJ, Sederoff HW, Stevenson DW, Surek B, Zhang Y, Sussman MR, Dunand C, Morris RJ, Roux C, Wong GK-S, Oldroyd GED, Ané J-M (2015) Algal ancestor of land plants was preadapted for symbiosis. Proc Natl Acad Sci 112:13390–13395. https://doi.org/10.1073/pnas.1515426112
Delaux P-M, Hetherington AJ, Coudert Y, Delwiche C, Dunand C, Gould S, Kenrick P, Li F-W, Philippe H, Rensing SA, Rich M, Strullu-Derrien C, Vries J de (2019) Reconstructing trait evolution in plant evo–devo studies. Curr Biol 29:R1110–R1118. https://doi.org/10.1016/j.cub.2019.09.044
Dufayard J-F, Bettembourg M, Fischer I, Droc G, Guiderdoni E, Périn C, Chantret N, Diévart A (2017) New insights on leucine-rich repeats receptor-like kinase orthologous relationships in angiosperms. Front Plant Sci. https://doi.org/10.3389/fpls.2017.003812017.00381
Felix G, Duran JD, Volko S, Boller T (1999) Plants have a sensitive perception system for the most conserved domain of bacterial flagellin. Plant J 18:265–276. https://doi.org/10.1046/j.1365-313X.1999.00265.x
Fesenko I, Kirov I, Knyazev A, Khazigaleeva R, Lazarev V, Kharlampieva D, Grafskaia E, Zgoda V, Butenko I, Arapidi G, Mamaeva A, Ivanov V, Govorun V (2019) Distinct types of short open reading frames are translated in plant cells. Genome Res. https://doi.org/10.1101/gr.253302.119
Fesenko I, Azarkina R, Kirov I, Kniazev A, Filippova A, Grafskaia E, Lazarev V, Zgoda V, Butenko I, Bukato O, Lyapina I, Nazarenko D, Elansky S, Mamaeva A, Ivanov V, Govorun V (2019) Phytohormone treatment induces generation of cryptic peptides with antimicrobial activity in the moss physcomitrella patens. BMC Plant Biol 19:9. https://doi.org/10.1186/s12870-018-1611-z
Filippova A, Lyapina I, Kirov I, Zgoda V, Belogurov A, Kudriaeva A, Ivanov V, Fesenko I (2019) Salicylic acid influences the protease activity and posttranslation modifications of the secreted peptides in the moss physcomitrella patens. J Pept Sci 25:e3138. https://doi.org/10.1002/psc.3138
Frangedakis E, Shimamura M, Villarreal JC, Li F-W, Tomaselli M, Waller M, Sakakibara K, Renzaglia KS, Szövényi P (2021) The hornworts: morphology, evolution and development. New Phytol 229:735–754. https://doi.org/10.1111/nph.16874
Fürst-Jansen JMR, de Vries S, de Vries J (2020) Evo-physio: on stress responses and the earliest land plants. J Exp Bot 71:3254–3269. https://doi.org/10.1093/jxb/eraa007
Galotto G, Abreu I, Sherman C, Liu B, Gonzalez-Guerrero M, Vidali L (2020) Chitin triggers calcium-mediated immune response in the plant model physcomitrella patens. Mol Plant-Microbe Interact 33:911–920. https://doi.org/10.1094/MPMI-03-20-0064-R
Gao Y, Wang W, Zhang T, Gong Z, Zhao H, Han G-Z (2018) Out of water: the origin and early diversification of plant R-genes. Plant Physiol 177:82–89. https://doi.org/10.1104/pp.18.00185
Ghorbani S, Lin Y-C, Parizot B, Fernandez A, Njo MF, Van de Peer Y, Beeckman T, Hilson P (2015) Expanding the repertoire of secretory peptides controlling root development with comparative genome analysis and functional assays. J Exp Bot 66:5257–5269. https://doi.org/10.1093/jxb/erv346
Gomes A, Fernandes E, Lima JLFC (2005) Fluorescence probes used for detection of reactive oxygen species. J Biochem Biophys Methods 65:45–80. https://doi.org/10.1016/j.jbbm.2005.10.003
Halliwell B, Whiteman M (2004) Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol 142:231–255. https://doi.org/10.1038/sj.bjp.0705776
Han G-Z (2019) Origin and evolution of the plant immune system. New Phytol 222:70–83. https://doi.org/10.1111/nph.15596
Hao G, Pitino M, Duan Y, Stover E (2016) Reduced susceptibility to xanthomonas citri in transgenic citrus expressing the FLS2 Receptor From Nicotiana benthamiana. Mol Plant-Microbe Interact MPMI 29:132–142. https://doi.org/10.1094/MPMI-09-15-0211-R
Huang W-J, Liu H-K, McCormick S, Tang W-H (2014) Tomato pistil factor STIG1 promotes in vivo pollen tube growth by binding to phosphatidylinositol 3-phosphate and the extracellular domain of the pollen receptor kinase LePRK2. Plant Cell 26:2505–2523. https://doi.org/10.1105/tpc.114.123281
Huffaker A, Pearce G, Ryan CA (2006) An endogenous peptide signal in Arabidopsis activates components of the innate immune response. Proc Natl Acad Sci 103:10098–10103. https://doi.org/10.1073/pnas.0603727103
Huffaker A, Pearce G, Veyrat N, Erb M, Turlings TCJ, Sartor R, Shen Z, Briggs SP, Vaughan MM, Alborn HT, Teal PEA, Schmelz EA (2013) Plant elicitor peptides are conserved signals regulating direct and indirect antiherbivore defense. Proc Natl Acad Sci 110:5707–5712. https://doi.org/10.1073/pnas.1214668110
Kadota Y, Sklenar J, Derbyshire P, Stransfeld L, Asai S, Ntoukakis V, Jones JD, Shirasu K, Menke F, Jones A, Zipfel C (2014) Direct regulation of the NADPH oxidase RBOHD by the PRR-associated kinase BIK1 during plant immunity. Mol Cell 54:43–55. https://doi.org/10.1016/j.molcel.2014.02.021
Klavina L, Springe G, Nikolajeva V, Martsinkevich I, Nakurte I, Dzabijeva D, Steinberga I (2015) Chemical composition analysis, antimicrobial activity and cytotoxicity screening of moss extracts (moss phytochemistry). Molecules 20:17221–17243. https://doi.org/10.3390/molecules200917221
Kovalchuk SI, Jensen ON, Rogowska-Wrzesinska A (2019) FlashPack: fast and simple preparation of ultrahigh-performance capillary columns for LC-MS. Mol Cell Proteomics: MCP 18:383–390. https://doi.org/10.1074/mcp.TIR118.000953
Kunze G, Zipfel C, Robatzek S, Niehaus K, Boller T, Felix G (2004) The N terminus of bacterial elongation factor Tu elicits innate immunity in arabidopsis plants. Plant Cell 16:3496–3507. https://doi.org/10.1105/tpc.104.026765
Lacombe S, Rougon-Cardoso A, Sherwood E, Peeters N, Dahlbeck D, van Esse HP, Smoker M, Rallapalli G, Thomma BPHJ, Staskawicz B, Jones JDG, Zipfel C (2010) Interfamily transfer of a plant pattern-recognition receptor confers broad-spectrum bacterial resistance. Nat Biotechnol 28:365–369. https://doi.org/10.1038/nbt.1613
Lang D, Ullrich KK, Murat F, Fuchs J, Jenkins J, Haas FB, Piednoel M, Gundlach H, Bel MV, Meyberg R, Vives C, Morata J, Symeonidi A, Hiss M, Muchero W, Kamisugi Y, Saleh O, Blanc G, Decker EL, Gessel N van, Grimwood J, Hayes RD, Graham SW, Gunter LE, McDaniel SF, Hoernstein SNW, Larsson A, Li F-W, Perroud P-F, Phillips J, Ranjan P, Rokshar DS, Rothfels CJ, Schneider L, Shu S, Stevenson DW, Thümmler F, Tillich M, Aguilar JCV, Widiez T, Wong GK-S, Wymore A, Zhang Y, Zimmer AD, Quatrano RS, Mayer KFX, Goodstein D, Casacuberta JM, Vandepoele K, Reski R, Cuming AC, Tuskan GA, Maumus F, Salse J, Schmutz J, Rensing SA (2018) The Physcomitrella patens chromosome-scale assembly reveals moss genome structure and evolution. Plant J 93:515–533. https://doi.org/10.1111/tpj.13801
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinforma. Oxf Engl 23:2947–2948. https://doi.org/10.1093/bioinformatics/btm404
Lease KA, Walker JC (2006) The arabidopsis unannotated secreted peptide database, a resource for plant peptidomics. Plant Physiol 142:831. https://doi.org/10.1104/pp.106.086041
Lee MW, Huffaker A, Crippen D, Robbins RT, Goggin FL (2018) Plant elicitor peptides promote plant defences against nematodes in soybean. Mol Plant Pathol 19:858–869. https://doi.org/10.1111/mpp.12570
Leebens-Mack JH, Barker MS, Carpenter EJ, Deyholos MK, Gitzendanner MA,Graham SW, Grosse I, Li Z, Melkonian M, Mirarab S, Porsch M, Quint M, Rensing SA, Soltis DE, Soltis PS, Stevenson DW, Ullrich KK, Wickett NJ, DeGironimo L, Edger PP, Jordon-Thaden IE, Joya S, Liu T, Melkonian B, Miles NW, Pokorny L, Quigley C, Thomas P, Villarreal JC, Augustin MM, Barrett MD, Baucom RS, Beerling DJ, Benstein RM, Biffin E, Brockington SF, Burge DO, Burris JN, Burris KP, Burtet-Sarramegna V, Caicedo AL, Cannon SB, Çebi Z, Chang Y, Chater C, Cheeseman JM, Chen T, Clarke ND, Clayton H, Covshoff S, Crandall-Stotler BJ, Cross H, dePamphilis CW, Der JP, Determann R, Dickson RC, Di Stilio VS, Ellis S, Fast E, Feja N, Field KJ, Filatov DA, Finnegan PM, Floyd SK, Fogliani B, García N, Gâteblé G, Godden GT, Goh F, Qi Y, Greiner S, Harkess A, Heaney JM, Helliwell KE, Heyduk K, Hibberd JM, Hodel RGJ, Hollingsworth PM, Johnson MTJ, Jost R, Joyce B, Kapralov MV, Kazamia E, Kellogg EA, Koch MA, VonKonrat M, Könyves K, Kutchan TM, Lam V, Larsson A, Leitch AR, Lentz R, Li F-W, Lowe AJ, Ludwig M, Manos PS, Mavrodiev E, McCormick MK, McKain M, McLellan T, McNeal JR, Miller RE, Nelson MN, Peng Y, Ralph P, Real D, Riggins CW, Ruhsam M, Sage RF, Sakai AK, Scascitella M, Schilling EE, Schlösser E-M, Sederoff H, Servick S, Sessa EB, Shaw AJ, Shaw SW, Sigel EM, Skema C, Smith AG, Smithson A, Stewart CN, Stinchcombe JR, Szövényi P, Tate JA, Tiebel H, Trapnell D, Villegente M, Wang C-N, Weller SG, Wenzel M, Weststrand S, Westwood JH, Whigham DF, Wu S, Wulff AS, Yang Y, Zhu D, Zhuang C, Zuidof J, Chase MW, Pires JC, Rothfels CJ, Yu J, Chen C, Chen L, Cheng S, Li J, Li R, Li X, Lu H, Ou Y, Sun X, Tan X, Tang J, Tian Z, Wang F, Wang J, Wei X, Xu X, Yan Z, Yang F, Zhong X, Zhou F, Zhu Y, Zhang Y, Ayyampalayam S, Barkman TJ, Nguyen N, Matasci N, Nelson DR, Sayyari E, Wafula EK, Walls RL, Warnow T, An H, Arrigo N, Baniaga AE, Galuska S, Jorgensen SA, Kidder TI, Kong H, Lu-Irving P, Marx HE, Qi X, Reardon CR, Sutherland BL, Tiley GP, Welles SR, Yu R, Zhan S, Gramzow L, Theißen G, Wong GK-S. (2019) One thousand plant transcriptomes initiative. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574:679–685. https://doi.org/10.1038/s41586-019-1693-2
Lehtonen MT, Akita M, Frank W, Reski R, Valkonen JPT (2012) Involvement of a class III peroxidase and the mitochondrial protein TSPO in oxidative burst upon treatment of moss plants with a fungal elicitor. Mol Plant-Microbe Interact MPMI 25:363–371. https://doi.org/10.1094/MPMI-10-11-0265
Lehtonen MT, Akita M, Kalkkinen N, Ahola-Iivarinen E, Rönnholm G, Somervuo P, Thelander M, Valkonen JPT (2009) Quickly-released peroxidase of moss in defense against fungal invaders. N Phytologist 183:432–443. https://doi.org/10.1111/j.1469-8137.2009.02864.x
Li YL, Dai XR, Yue X, Gao X-Q, Zhang XS (2014) Identification of small secreted peptides (SSPs) in maize and expression analysis of partial SSP genes in reproductive tissues. Planta 240:713–728. https://doi.org/10.1007/s00425-014-2123-1
Liu F, Zhang X, Lu C, Zeng X, Li Y, Fu D, Wu G (2015) Non-specific lipid transfer proteins in plants: presenting new advances and an integrated functional analysis. J Exp Bot 66:5663–5681. https://doi.org/10.1093/jxb/erv313
Lori M, van Verk MC, Hander T, Schatowitz H, Klauser D, Flury P, Gehring CA, Boller T, Bartels S (2015) Evolutionary divergence of the plant elicitor peptides (Peps) and their receptors: interfamily incompatibility of perception but compatibility of downstream signalling. J Exp Bot 66:5315–5325. https://doi.org/10.1093/jxb/erv236
Luo W, Xiao Y, Liang Q, Su Y, Xiao L (2019) Identification of potential auxin-responsive small signaling peptides through a peptidomics approach in Arabidopsis thaliana. Molecules 24:3146. https://doi.org/10.3390/molecules24173146
Luu DD, Joe A, Chen Y, Parys K, Bahar O, Pruitt R, Chan LJG, Petzold CJ, Long K, Adamchak C, Stewart V, Belkhadir Y, Ronald PC (2019) Biosynthesis and secretion of the microbial sulfated peptide RaxX and binding to the rice XA21 immune receptor. Proc Natl Acad Sci USA 116:8525–8534. https://doi.org/10.1073/pnas.1818275116
Ma Y, Zhao Y, Walker RK, Berkowitz GA (2013) Molecular steps in the immune signaling pathway evoked by plant elicitor peptides: Ca2+-dependent protein kinases, nitric oxide, and reactive oxygen species are downstream from the early Ca2 + signal1[OPEN]. Plant Physiol 163:1459–1471. https://doi.org/10.1104/pp.113.226068
Matsui H, Iwakawa H, Hyon G-S, Yotsui I, Katou S, Monte I, Nishihama R, Franzen R, Solano R, Nakagami H (2020) Isolation of natural fungal pathogens from marchantia polymorpha reveals antagonism between salicylic acid and jasmonate during liverwort–fungus interactions. Plant Cell Physiol 61:265–275. https://doi.org/10.1093/pcp/pcz187
Mergaert P, Nikovics K, Kelemen Z, Maunoury N, Vaubert D, Kondorosi A, Kondorosi E (2003) A novel family in Medicago truncatula consisting of more than 300 nodule-specific genes coding for small, secreted polypeptides with conserved cysteine motifs. Plant Physiol 132:161–173. https://doi.org/10.1104/pp.102.018192
Miya A, Albert P, Shinya T, Desaki Y, Ichimura K, Shirasu K, Narusaka Y, Kawakami N, Kaku H, Shibuya N (2007) CERK1, a LysM receptor kinase, is essential for chitin elicitor signaling in Arabidopsis. Proc Natl Acad Sci USA 104:19613–19618. https://doi.org/10.1073/pnas.0705147104
Monte I, Franco-Zorrilla JM, García-Casado G, Zamarreño AM, García-Mina JM, Nishihama R, Kohchi T, Solano R, (2019) A single JAZ repressor controls the Jasmonate pathway in Marchantia polymorpha. Mol. Plant 12:185–198. https://doi.org/10.1016/j.molp.2018.12.0172018.12.017
Monte I, Ishida S, Zamarreño AM, Hamberg M, Franco-Zorrilla JM, García-Casado G, Gouhier-Darimont C, Reymond P, Takahashi K, García-Mina JM, Nishihama R, Kohchi T, Solano R (2018) Ligand-receptor co-evolution shaped the jasmonate pathway in land plants. Nat Chem Biol 14:480–488. https://doi.org/10.1038/s41589-018-0033-4
Murphy E, Smith S, Smet ID (2012) Small signaling peptides in arabidopsis development: how cells communicate over a short distance. Plant Cell 24:3198. https://doi.org/10.1105/tpc.112.099010
Nallu S, Silverstein KAT, Samac DA, Bucciarelli B, Vance CP, VandenBosch KA (2013) Regulatory patterns of a large family of defensin-like genes expressed in nodules of Medicago truncatula. PLoS ONE 8:e60355. https://doi.org/10.1371/journal.pone.0060355
Oelkers K, Goffard N, Weiller GF, Gresshoff PM, Mathesius U, Frickey T (2008) Bioinformatic analysis of the CLE signaling peptide family. BMC Plant Biol 8:1. https://doi.org/10.1186/1471-2229-8-1
Oh E, Seo PJ, Kim J (2018) Signaling peptides and receptors coordinating plant root development. Trends Plant Sci 23:337–351. https://doi.org/10.1016/j.tplants.2017.12.007
Ohyama K, Ogawa M, Matsubayashi Y (2008) Identification of a biologically active, small, secreted peptide in Arabidopsis by in silico gene screening, followed by LC-MS-based structure analysis. Plant J 55:152–160. https://doi.org/10.1111/j.1365-313X.2008.03464.x
Oliver JP, Castro A, Gaggero C, Cascón T, Schmelz EA, Castresana C, Ponce de León I (2009) Pythium infection activates conserved plant defense responses in mosses. Planta 230:569–579. https://doi.org/10.1007/s00425-009-0969-4
Onrubia M, Pollier J, Bossche RV, Goethals M, Gevaert K, Moyano E, Vidal-Limon H, Cusidó RM, Palazón J, Goossens A (2014) Taximin, a conserved plant-specific peptide is involved in the modulation of plant-specialized metabolism. Plant Biotechnol J 12:971–983. https://doi.org/10.1111/pbi.12205
Overdijk EJR, Keijzer JD, Groot DD, Schoina C, Bouwmeester K, Ketelaar T, Govers F (2016) Interaction between the moss Physcomitrella patens and Phytophthora: a novel pathosystem for live-cell imaging of subcellular defence. J Microsc 263:171–180. https://doi.org/10.1111/jmi.12395
Pan B, Sheng J, Sun W, Zhao Y, Hao P, Li X (2013) OrysPSSP: a comparative platform for small secreted proteins from rice and other plants. Nucleic Acids Res 41:D1192–D1198. https://doi.org/10.1093/nar/gks1090
Pastor-Fernández J, Gamir J, Pastor V, Sanchez-Bel P, Sanmartín N, Cerezo M, Flors V (2020) Arabidopsis plants sense non-self peptides to promote resistance against Plectosphaerella cucumerina. Front Plant Sci. https://doi.org/10.3389/fpls.2020.005292020.00529
Pavelescu L (2015) On reactive oxygen species measurement in living systems. J Med Life 8:38–42
Peñuelas M, Monte I, Schweizer F, Vallat A, Reymond P, García-Casado G, Franco-Zorrilla JM, Solano R (2019) Jasmonate-related MYC transcription factors are functionally conserved in Marchantia polymorpha. Plant Cell 31:2491–2509. https://doi.org/10.1105/tpc.18.00974
Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Pérez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilmaz S, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, Vizcaíno JA (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47:D442–D450. https://doi.org/10.1093/nar/gky1106
Plattner S, Gruber C, Stadlmann J, Widmann S, Gruber CW, Altmann F, Bohlmann H (2015) Isolation and characterization of a thionin proprotein-processing enzyme from barley. J Biol Chem 290:18056–18067. https://doi.org/10.1074/jbc.M115.647859
Ponce de León I, Montesano M (2013) Activation of defense mechanisms against pathogens in mosses and flowering plants. Int J Mol Sci 14:3178–3200. https://doi.org/10.3390/ijms14023178
Ponce de León I, Oliver JP, Castro A, Gaggero C, Bentancor M, Vidal S (2007) Erwinia carotovora elicitors and Botrytis cinerea activate defense responses in Physcomitrella patens. BMC Plant Biol 7:52. https://doi.org/10.1186/1471-2229-7-52
Ponce de León I, Schmelz EA, Gaggero C, Castro A, Alvarez A, Montesano M (2012) Physcomitrella patens activates reinforcement of the cell wall, programmed cell death and accumulation of evolutionary conserved defence signals, such as salicylic acid and 12-oxo‐phytodienoic acid, but not jasmonic acid, upon Botrytis cinerea infection. Mol Plant Pathol 13:960–974. https://doi.org/10.1111/j.1364-3703.2012.00806.x
Ponce de León I (2011) The moss physcomitrella patens as a model system to study interactions between plants and phytopathogenic fungi and oomycetes. J. Pathog. 2011:719873. https://doi.org/10.4061/2011/7198732011/719873
Puttick MN, Morris JL, Williams TA, Cox CJ, Edwards D, Kenrick P, Pressel S, Wellman CH, Schneider H, Pisani D, Donoghue PCJ (2018) The interrelationships of land plants and the nature of the ancestral embryophyte. Curr Biol 28:733-745.e2. https://doi.org/10.1016/j.cub.2018.01.063
Qi J, Wang J, Gong Z, Zhou J-M (2017) Apoplastic ROS signaling in plant immunity. Curr. Opin Plant Biol Biotic interactions 38:92–100. https://doi.org/10.1016/j.pbi.2017.04.0222017.04.022
Qiu Y-L, Li L, Wang B, Chen Z, Knoop V, Groth-Malonek M, Dombrovska O, Lee J, Kent L, Rest J, Estabrook GF, Hendry TA, Taylor DW, Testa CM, Ambros M, Crandall-Stotler B, Duff RJ, Stech M, Frey W, Quandt D, Davis CC (2006) The deepest divergences in land plants inferred from phylogenomic evidence. Proc Natl Acad Sci 103:15511–15516. https://doi.org/10.1073/pnas.0603335103
Reboledo G, del Campo R, Alvarez A, Montesano M, Mara H, Ponce de León I (2015) Physcomitrella patens activates defense responses against the pathogen Colletotrichum gloeosporioides. Int J Mol Sci 16:22280–22298. https://doi.org/10.3390/ijms160922280
Rocco M, Corrado G, Arena S, D’Ambrosio C, Tortiglione C, Sellaroli S, Marra M, Rao R, Scaloni A (2008) The expression of tomato prosystemin gene in tobacco plants highly affects host proteomic repertoire. J Proteom 71:176–185. https://doi.org/10.1016/j.jprot.2008.04.003
Roux M, Schwessinger B, Albrecht C, Chinchilla D, Jones A, Holton N, Malinovsky FG, Tör M, de Vries S, Zipfel C (2011) The Arabidopsis leucine-rich repeat receptor–like kinases BAK1/SERK3 and BKK1/SERK4 are required for innate immunity to hemibiotrophic and biotrophic pathogens[W]. Plant Cell 23:2440–2455. https://doi.org/10.1105/tpc.111.084301
Ruiz C, Nadal A, Foix L, Montesinos L, Montesinos E, Pla M (2018) Diversity of plant defense elicitor peptides within the Rosaceae. BMC Genet 19:11. https://doi.org/10.1186/s12863-017-0593-4
Saur IML, Kadota Y, Sklenar J, Holton NJ, Smakowska E, Belkhadir Y, Zipfel C, Rathjen JP (2016) NbCSPR underlies age-dependent immune responses to bacterial cold shock protein in Nicotiana benthamiana. Proc Natl Acad Sci 113:3389–3394. https://doi.org/10.1073/pnas.1511847113
Schoonbeek H, Wang H-H, Stefanato FL, Craze M, Bowden S, Wallington E, Zipfel C, Ridout CJ (2015) Arabidopsis EF-Tu receptor enhances bacterial disease resistance in transgenic wheat. New Phytol 206:606–613. https://doi.org/10.1111/nph.13356
Schwessinger B, Bahar O, Thomas N, Holton N, Nekrasov V, Ruan D, Canlas PE, Daudi A, Petzold CJ, Singan VR, Kuo R, Chovatia M, Daum C, Heazlewood JL, Zipfel C, Ronald PC (2015) Transgenic expression of the dicotyledonous pattern recognition receptor EFR in rice leads to ligand-dependent activation of defense responses. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1004809
Shapiguzov A, Vainonen J, Wrzaczek M, Kangasjärvi J (2012) ROS-talk – how the apoplast, the chloroplast, and the nucleus get the message through. Front Plant Sci. https://doi.org/10.3389/fpls.2012.002922012.00292
Shaw AJ, Schmutz J, Devos N, Shu S, Carrell AA, Weston DJ (2016) Chapter five---the sphagnum genome project: a new model for ecological and evolutionary genomics. In: Rensing SA (Ed) Advances in botanical research, genomes and evolution of charophytes, bryophytes, lycophytes and ferns. Academic Press, London, pp 167–187. https://doi.org/10.1016/bs.abr.2016.01.0032016.01.003
Shiu SH, Bleecker AB (2003) Expansion of the receptor-like kinase/Pelle gene family and receptor-like proteins in Arabidopsis. Plant Physiol 132:530–543. https://doi.org/10.1104/pp.103.021964
Shiu S-H, Karlowski WM, Pan R, Tzeng Y-H, Mayer KFX, Li W-H (2004) Comparative analysis of the receptor-like kinase family in arabidopsis and rice. Plant Cell 16:1220–1234. https://doi.org/10.1105/tpc.020834
Slavokhotova AA, Shelenkov AA, Andreev YaA, Odintsova TI (2017) Hevein-like antimicrobial peptides of plants. Biochem Mosc 82:1659–1674. https://doi.org/10.1134/S0006297917130065
Smakowska-Luzan E, Mott GA, Parys K, Stegmann M, Howton TC, Layeghifard M, Neuhold J, Lehner A, Kong J, Grünwald K, Weinberger N, Satbhai SB, Mayer D, Busch W, Madalinski M, Stolt-Bergner P, Provart NJ, Mukhtar MS, Zipfel C, Desveaux D, Guttman DS, Belkhadir Y (2018) An extracellular network of Arabidopsis leucine-rich repeat receptor kinases. Nature 553:342–346. https://doi.org/10.1038/nature25184
Smirnova AV, Matveyeva NP, Polesskaya OG, Yermakov IP (2009) Generation of reactive oxygen species during pollen grain germination. Russ J Dev Biol 40:345. https://doi.org/10.1134/S1062360409060034
Song WY, Wang GL, Chen LL, Kim HS, Pi LY, Holsten T, Gardner J, Wang B, Zhai WX, Zhu LH, Fauquet C, Ronald P (1995) A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 270: 1804–1806. https://doi.org/10.1126/science.270.5243.18041804
Sugita S, Yamada Y, Horie S, Nakamura O, Ishidoh K, Yamamoto Y, Yamagami S, Mochizuki M (2011) Induction of T regulatory cells by cytotoxic T-lymphocyte antigen-2α on corneal endothelial cells. Invest Ophthalmol Vis Sci 52:2598–2605. https://doi.org/10.1167/iovs.10-6322
Vellosillo T, Vicente J, Kulasekaran S, Hamberg M, Castresana C (2010) Emerging complexity in reactive oxygen species production and signaling during the response of plants to pathogens. Plant Physiol 154:444–448. https://doi.org/10.1104/pp.110.161273
Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, van der Walt SJ, Brett M, Wilson J, Millman KJ, Mayorov N, Nelson ARJ, Jones E, Kern R, Larson E, Carey CJ, Polat İ, Feng Y, Moore EW, VanderPlas J, Laxalde D, Perktold J, Cimrman R, Henriksen I, Quintero EA, Harris CR, Archibald AM, Ribeiro AH, Pedregosa F, van Mulbregt P (2020) SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17:261–272. https://doi.org/10.1038/s41592-019-0686-2
Vries S de, Vries J de, Dahlen JK von, Gould SB, Archibald JM, Rose LE, Slamovits CH (2018) On plant defense signaling networks and early land plant evolution. Commun Integr Biol 11:1–14. https://doi.org/10.1080/19420889.2018.1486168
Wan J, Zhang X-C, Neece D, Ramonell KM, Clough S, Kim S-Y, Stacey MG, Stacey G (2008) A LysM receptor-like kinase plays a critical role in chitin signaling and fungal resistance in Arabidopsis. Plant Cell 20:471–481. https://doi.org/10.1105/tpc.107.056754
Wang P, Yao S, Kosami K, Guo T, Li J, Zhang Y, Fukao Y, Kaneko-Kawano T, Zhang H, She Y-M, Wang P, Xing W, Hanada K, Liu R, Kawano Y (2020) Identification of endogenous small peptides involved in rice immunity through transcriptomics- and proteomics-based screening. Plant Biotechnol J 18:415–428. https://doi.org/10.1111/pbi.13208
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. https://doi.org/10.1093/bioinformatics/btp033
Whitewoods CD, Cammarata J, Nemec Venza Z, Sang S, Crook AD, Aoyama T, Wang XY, Waller M, Kamisugi Y, Cuming AC, Szövényi P, Nimchuk ZL, Roeder AHK, Scanlon MJ, Harrison CJ (2018) CLAVATA was a genetic novelty for the morphological innovation of 3d growth in land plants. Curr Biol 28:2365-2376.e5. https://doi.org/10.1016/j.cub.2018.05.068
Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E, Matasci N, Ayyampalayam S, Barker MS, Burleigh JG, Gitzendanner MA, Ruhfel BR, Wafula E, Der JP, Graham SW, Mathews S, Melkonian M, Soltis DE, Soltis PS, Miles NW, Rothfels CJ, Pokorny L, Shaw AJ, DeGironimo L, Stevenson DW, Surek B, Villarreal JC, Roure B, Philippe H, dePamphilis CW, Chen T, Deyholos MK, Baucom RS, Kutchan TM, Augustin MM, Wang J, Zhang Y, Tian Z, Yan Z, Wu X, Sun X, Wong GK-S, Leebens-Mack J (2014) Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc Natl Acad Sci 111:E4859–E4868. https://doi.org/10.1073/pnas.1323926111
Yamaguchi Y, Huffaker A (2011) Endogenous peptide elicitors in higher plants. Curr Opin Plant Biol 14:351–357. https://doi.org/10.1016/j.pbi.2011.05.001
Yamaguchi Y, Huffaker A, Bryan AC, Tax FE, Ryan CA (2010) PEPR2 Is a second receptor for the Pep1 and Pep2 peptides and contributes to defense responses in arabidopsis. Plant Cell 22:508–522. https://doi.org/10.1105/tpc.109.068874
Zhang H, Hu Y (2017) Long-distance transport of prosystemin messenger RNA in tomato. Front Plant Sci. https://doi.org/10.3389/fpls.2017.018942017.01894
Zhang H, Yu P, Zhao J, Jiang H, Wang H, Zhu Y, Botella MA, Šamaj J, Li C, Lin J (2018) Expression of tomato prosystemin gene in Arabidopsis reveals systemic translocation of its mRNA and confers necrotrophic fungal resistance. New Phytol 217:799–812. https://doi.org/10.1111/nph.14858
Zhang J, Fu X-X, Li R-Q, Zhao X, Liu Y, Li M-H, Zwaenepoel A, Ma H, Goffinet B, Guan Y-L, Xue J-Y, Liao Y-Y, Wang Q-F, Wang Q-H, Wang J-Y, Zhang G-Q, Wang Z-W, Jia Y, Wang M-Z, Dong S-S, Yang J-F, Jiao Y-N, Guo Y-L, Kong H-Z, Lu A-M, Yang H-M, Zhang S-Z, Van de Peer Y, Liu Z-J, Chen Z-D (2020a) The hornwort genome and early land plant evolution. Nature Plants 6:107–118. https://doi.org/10.1038/s41477-019-0588-4
Zhang Z, Liu L, Kucukoglu M, Tian D, Larkin RM, Shi X, Zheng B (2020) Predicting and clustering plant CLE genes with a new method developed specifically for short amino acid sequences. BMC Genom. https://doi.org/10.21203/rs.2.21530/v2
Zhou B, Benbow HR, Brennan CJ, Arunachalam C, Karki SJ, Mullins E, Feechan A, Burke JI, Doohan FM (2020) Wheat encodes small, secreted proteins that contribute to resistance to septoria tritici blotch. Front Genet. https://doi.org/10.3389/fgene.2020.004692020.00469
Zhou P, Silverstein KA, Gao L, Walton JD, Nallu S, Guhlin J, Young ND (2013) Detecting small plant peptides using SPADA (small peptide alignment discovery application). BMC Bioinform 14:335. https://doi.org/10.1186/1471-2105-14-335
Funding
This work was funded by the Russian Science Foundation (Project No. 17-14-01189).
Author information
Authors and Affiliations
Contributions
IL, AF, IF wrote the manuscript. IL, AF conducted all experiments, performed bioinformatic search for SSP candidates and contributed to data analysis. AM, EM and OP contributed to ROS accumulation analysis. OI performed bioinformatic search for homologes of receptor-like kinases. SK, RZ conducted mass-spectrometry analysis. VL and IL conducted peptide synthesis. IF performed data analysis. IF and VI designed and supervised the study.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Consent for publication
All authors have approved the manuscript and agreed with its submission.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lyapina, I., Filippova, A., Kovalchuk, S. et al. Possible role of small secreted peptides (SSPs) in immune signaling in bryophytes. Plant Mol Biol 106, 123–143 (2021). https://doi.org/10.1007/s11103-021-01133-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-021-01133-z