
Abstract
Purpose
Pituitary adenomas commonly arise in patients with MEN1 syndrome, an autosomal dominant condition predisposing to neuroendocrine tumor formation, and typically diagnosed in patients with a relevant family cancer history. In these patients with existing germline loss of MEN1 on one allele, somatic loss of the second MEN1 allele leads to complete loss of the MEN1 protein, menin, and subsequent tumor formation.
Methods
Whole exome sequencing was performed on the tumor and matching blood under an institutional board approved protocol. DNA extraction and analysis was conducted according to previously described methods.
Results
We describe a 23 year-old patient with no significant past medical history or relevant family history who underwent surgical resection of a symptomatic and medically resistant prolactinoma. Whole exome sequencing of tumor and blood samples revealed somatic loss of MEN1 at both alleles, suggesting a double hit mechanism, with no underlying germline MEN1 mutation.
Conclusion
To our knowledge, this is the first case of pituitary adenoma to arise from somatic loss of MEN1 and in the absence of an underlying germline MEN1 mutation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant inherited disorder predisposing to multiple tumors, most commonly those of the parathyroid, pancreas, and pituitary gland, with the latter affecting 30–40% of patients [1]. 90% of patients with MEN1 syndrome are diagnosed with a relevant family history, while the remaining 10% present with a de novo germline mutation [2]. MEN1 mutated pituitary adenomas, like other MEN1 related tumors, form after a second change on the other MEN1 allele at the somatic level, resulting in complete loss of the MEN1 protein, menin.
We describe the first case to our knowledge of a pituitary adenoma, arising from somatic loss of both alleles of MEN1, and notably in the absence of an underlying germline MEN1 mutation. Interestingly, whole exome sequencing also detected a pathogenic germline mutation in the Parkin RBR E3 ubiquitin protein ligase (PRKN) gene, which encodes the multifunctional ubiquitin ligase Parkin and has been linked to development of autosomal recessive juvenile parkinsonism [3]. Notably, PRKN mutations have been increasingly reported in various human cancers whereby under-expression or loss of Parkin carries clinical prognostic significance [4]. We review the relevant literature and hypothesize on potential mechanisms linking loss of PRKN and a predisposition to mutations of hallmark oncogenes, such as MEN1.
Methods
Whole exome sequencing (WES) was performed on the tumor and matching blood in accordance with our previously described methods [5]. DNA extraction, exome capture (IDT xGen Exome Research Panel Version 1, with additional spike-ins), and sequencing was carried out at Yale Cancer for Genome Analysis (YCGA) with Illumina NovaSeq 600 WES system, resulting in 2 × 101-bp reads. High mean coverage of 301.8X and 141.8X was achieved for tumor and blood respectively. Downstream analysis including alignment to reference genome (Grch37), marking duplicates, and local realignment was performed using GATK (v3.4, Grch37). Germline single-nucleotide variations (SNVs), insertions/deletions (INDELs) were identified using GATK HaplotypeCaller (v3.4). Annotations of the variants were performed using ANNOVAR (version 2019-10-24) and VEP (v95). Rare germline variants were identified by filtering out variants with allele-frequency > 1% in the control databases, such as gnomAD-genome and gnomAD-exome (release 170,228), for all the subpopulations. For somatic variant discovery, MuTect (v2.7) and Indelocator (IndelGenotyperV2) were used to call SNVs and INDELs, respectively. Somatic calls were further filtered based on the variant allele frequency (VAF) in the matching normal sample and tumor tissue, as well as the frequency of these variants in control databases such as 1000Genomes, ExAC and NHLBI (< 1%). Copy number variations (CNV) were identified using GATK (v4)’s corresponding CNV workflow for both somatic and germline analysis. Somatic CNVs and loss-of-heterozygosity (LOH) was assessed using the matching normal sample to normalize and denoise the CNV events for the tumor. Germline CNV analysis was carried out in cohort mode with default parameters for the workflow using a panel of 114 blood samples obtained from other sequencing projects carried out by our group and sequenced with the same capture method.
Case presentation
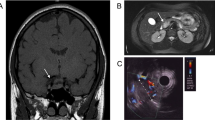
A 23-year-old male with a known prolactinoma was referred for neurosurgical consultation. His oncologic history was as follows. His prolactinoma had been diagnosed 10 years earlier when he presented with several months of headache and polydipsia. Magnetic resonance imaging (MRI) at the time revealed a 5 cm sellar mass and serum testing identified elevated prolactin (PRL) levels (6730 ng/mL). There was no relevant family history of neuroendocrine tumors to suggest an underlying diagnosis of any of the multiple endocrine neoplasia syndromes. Likewise, results from a comprehensive metabolic panel were all within normal limits, and he had no gastrointestinal, urinary, or hypoglycemic symptoms to suggest the presence of an additional neuroendocrine tumor. On visual field testing, the patient exhibited bitemporal hemianopsia (Fig. 1A, B). The patient was started on cabergoline and repeat imaging obtained four months later demonstrated reduction in tumor size, decreased prolactin levels (PRL 176 ng/mL), as well as clinical improvement of his visual deficits (Fig. 1C, D). Unfortunately, he was then lost to follow-up for the next two years, after which he re-presented with worsening vision and admitted to non-compliance with cabergoline over the past year. Interval imaging revealed regrowth of the lesion to 3 cm, prompting increasing his cabergoline dosing. The patient was then lost to follow-up for another 1.5 years until he presented to the emergency department after a seizure. MRI demonstrated a large 3.6 cm lobulated sellar mass with suprasellar extension, causing effacement of the left lateral ventricle (Fig. 2A, B). Pre-operative labs were notable for a PRL > 4700 ng/mL. On examination, the patient demonstrated severe gynecomastia without galactorrhea, as well as bitemporal hemianopsia. Given concern for development of tumor resistance to cabergoline as well as his history of inconsistent medication compliance, he was referred for surgical resection.

Serial MRI of the patient’s tumor leading up to surgery. Representative T1-weighted post-contrast (A) coronal and (B) sagittal images demonstrate a 3.5 cm sellar lesion at time of original diagnosis. (C, D) Repeat imaging obtained four months after cabergoline treatment demonstrated interval reduction in tumor size

Pre- and post-operative MRI of the patient’s tumor. Representative T1-weighted post-contrast (A) coronal and (B) sagittal images obtained at time of seizure presentation and preceding surgery showed tumor regrowth, concerning for development of tumor resistance to medical therapy. (C, D) Post-operative imaging obtained one day after surgery showed interval tumor removal and decompression of the optic chiasm
After obtaining informed consent, the patient subsequently underwent endoscopic endonasal resection of his tumor. The majority of the tumor was resected, and the optic chiasm was adequately decompressed (Fig. 2C, D). A small residual portion of tumor was left anterolaterally where it was significantly adhered to the carotid bifurcation. Post-operatively, he experienced transient diabetes insipidus and maintained a normal hypothalamus-pituitary axis, precluding any need for steroids. At last follow-up, he had been weaned to a lower dose of 0.5 mg cabergoline three times weekly. He reported improvement in his vision, energy levels, and denied any breast tenderness or galactorrhea, polyuria or polydipsia.
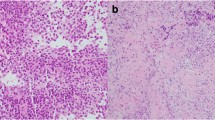
Final pathology revealed an adenohypophyseal neoplasm with round to oval nuclei, stippled chromatin, variably distinct nucleoli, and eosinophilic cytoplasm (Fig. 3A). Sheet-like growth was highlighted by reticulin staining (Fig. 3B). Mitotic activity was rare (Ki-67 index < 1%) and necrosis was absent. Immunohistochemical staining was positive for synaptophysin, PIT-1, Cam5.2, and prolactin (Fig. 3C), taken together consistent with a diagnosis of prolactinoma.

Pathology. Representative photomicrographs of resected tissue with (A) H&E stain, (B) reticulin staining, and (C) immunohistochemical staining for prolactin depict histologic features consistent with prolactinoma. Magnification x200
Genomics findings
We conducted WES on both the tumor and matching blood samples in order to detect somatic alterations specific to the tumor, as well as to uncover any rare germline alterations that could be pertinent to the presented pathology. Our analysis revealed 11 somatic SNV/INDELs with variant allele frequency (VAF) greater than 10% in the tumor. Only one of these 11 identified somatic SNV/INDELs, MEN1 (NM_000244, p.G161R), was previously reported to be cancer related. MEN1 (NM_000244, p.G161R), was reported to be “Likely-pathogenic” by ClinVar [6] (RCV001269563.1) and predicted to be deleterious by SIFT [7], Polyphen2 [8], FATHMM [9], and MetaSVM [10]. Our somatic CNV analysis yielded an intriguing result, as only 4.2% of the genome showed alterations due to a somatic CNV event. The analysis revealed the deletion of the entire chromosome 11, which overlaps with the MEN1 locus. Additionally, the variant allele frequency (VAF) of the MEN1-p.G161R missense mutation was 71.5% in the tumor sample, suggesting a somatic double-hit mechanism (Fig. 4).

Somatic bi-allelic loss of MEN1. (A) A somatic deleterious MEN1 missense mutation with increased VAF, overlapping with (B) a somatic deletion of chr11 indicates somatic bi-allelic loss of MEN1.
Rare germline variant analysis revealed 1194 coding region and splicing variants, with only three “Pathogenic” (PRKN: p.R126W: NM_013988, CASP14: p.D154fs: NM_012114 ), or “Likely pathogenic” (DHCR7: p.G147D: NM_001163817) variants per ClinVar. Interestingly, germline analysis did not reveal any SNV/INDELs or CNVs on MEN1 gene. None of these pathogenic/likely pathogenic rare variants were previously reported to be associated with familial or early-onset pituitary adenoma, or any other neuroendocrine tumor syndromes. The pathogenic PRKN: p.R12W mutation identified in this study is noteworthy as it affects the RING finger 1 domain of the Parkin protein, which impairs mitochondrial ubiquitination by disrupting the catalytic site and proper protein localization [12, 13]. This mutation has also been found in cases of familial early-onset Parkinson’s disease [14,15,16], as well as in somatic and germline forms of lung cancer [11, 17]. Furthermore, a recent study that examined the germline genomic profiles of children, adolescents, and young adults with solid tumors found a higher frequency of PRKN loss-of-function mutations [4].
Discussion
Despite the genetically unremarkable profile of the vast majority of pituitary tumors, the common molecular mechanisms that drive them remain yet to be discovered. However, in recent years, a subset of functional pituitary tumors have been associated with recurrent somatic gain-of-function mutations, including mutations of GNAS in growth hormone (GH)-secreting tumors causing acromegaly [18] and mutations of USP8 in cortisol-secreting tumors causing Cushing disease [19]. On the other hand, a minority of pituitary tumors (~ 5%) are hereditary, presenting with a relevant family history and/or de novo germline mutations. Examples of such tumor-predisposing syndromes include germline PRKAR1A mutations in Carney complex and germline AIP mutations among others [20, 21]. That said, syndromic pituitary tumors have been most commonly and classically associated with MEN1 syndrome [22].
The MEN1 gene has been identified as the primary genetic contributor to multiple endocrine neoplasia type 1, a disorder characterized by the development of tumors in multiple endocrine glands. To date, over 1,300 distinct mutations in the MEN1 gene have been identified as causative factors for MEN1. The most commonly affected endocrine glands in MEN1 include the parathyroid, pancreas, and pituitary, the latter of which leads to pituitary adenoma formation in approximately 40% of patients [23, 24]. The majority of MEN1 gene mutations result in the production of a truncated, non-functional form of the protein menin, or a protein that is rapidly degraded. This leads to the absence of functional menin in affected cells, as a result of the loss of function of one copy of the MEN1 gene. In instances where both copies of the MEN1 gene are altered, no functional menin is produced. Menin is a tumor suppressor protein that is involved in transcriptional regulation of genomic stability and transcription, as well as cell cycle control during cell division [25]. The specific mechanism by which the loss of menin leads to tumor formation in endocrine glands remains to be fully understood.
Interestingly, germline MEN1 mutations have been diagnosed in young patients with sporadic pituitary adenomas and no relevant family history. Cuny et al. screened 174 such patients, all under age 30, and detected germline MEN1 mutations in 3.4% of patients [26]. However, to our knowledge, this is the first report of a pituitary adenoma, arising from a somatic loss of both alleles, resulting in complete loss of functional menin. Sequencing of germline DNA did not reveal an underlying MEN1 germline mutation, supported by the fact that our patient had no relevant family history or clinical findings of MEN1 syndrome. Interestingly, somatic double hit mutations have been identified in other neuroendocrine tumors in the absence of a germline MEN1 mutation, including those of the pancreas and parathyroid gland [27,28,29]. However, similar studies in pituitary adenomas have failed to find a case where somatic loss of both MEN1 alleles was identified [30,31,32]. As such, our case, while rare, demonstrates that somatic double hit mutations in MEN1 can occur and likely contributes to the pathogenesis of sporadic pituitary adenoma.
Although we have not found any other germline mutations previously linked to familial or early-onset pituitary adenomas or neuroendocrine tumor syndromes, the discovery of a pathogenic PRKN mutation is intriguing. Mutations in PRKN have been linked to autosomal recessive juvenile parkinsonism, an inherited form of Parkinson’s disease [3]. The gene encodes Parkin, which is a ubiquitin ligase that has been most notably studied in neuroprotection. Its functions include maintaining protein and mitochondrial homeostasis through regulation of cellular autophagy and mitophagy [33,34,35]. PRKN also plays a role in regulation of the cell cycle, apoptosis, and reactive oxygen species, and, interestingly, there are increasing data to suggest a role for PRKN in various human cancers via its role as a potential tumor suppressor gene [36,37,38]. Numerous mechanisms for PRKN in tumorigenesis have been described, including regulation of the G1/S-phase cell cycle transition[39], stabilization of the anaphase promoting complex during mitosis [40], and maintenance of cellular metabolism and suppression of glycolysis via interaction with TP53 [41]. Additionally, a recent review of clinical findings of PRKN abnormalities in human patients described downregulation of PRKN across many different cancers, including a role for predicting poor clinical prognosis in patients with cancers of the brain, nasopharynx, breast, and lung among others [42]. Notably, several studies have also shown germline mutations to predispose patients to development of lung cancer [43, 44], including one group who detected the same germline variant seen in our patient in a family with eight cases of lung cancer [11]. This gene is known to cause juvenile Parkinson’s disease and has recently been linked to a higher frequency of loss-of-function germline mutations in children, adolescents, and young adults with solid tumors [4]. However, it remains unclear whether germline loss-of-function PRKN mutations have clinical significance in terms of cancer predisposition. Interestingly, this variant is also one of the most common variants detected in patients with autosomal recessive juvenile parkinsonism [45]. PRKN promotes apoptosis in cancer cells via ubiquitination and degradation of the Bcl-2 family proteins [46]. That said, there is no established mechanistic link between the germline PRKN and somatic MEN1 mutations, detected in our case. Lastly, several studies have demonstrated PRKN deficiency results in increased inflammation and genomic instability [11, 40, 47]. As such, one hypothesis could be that a combination of loss of normal apoptosis and regulation of reactive oxygen species may predispose to genomic instability, leading to mutations of hallmark oncogenes, such as MEN1. Clearly, further studies are needed to answer this question.
In conclusion, we describe a case of a sporadic pituitary adenoma with somatic loss of both MEN1 alleles in the absence of an underlying germline mutation. Additionally, we detected a pathogenic germline mutation in PRKN, for which there is increasing evidence to suggest a role as a tumor suppressor gene. Further genomic reports are needed to establish whether there is a mechanistic link between germline PRKN mutations and increased cancer predisposition risk.
Data Availability
Additional data may be requested from the corresponding author.
References
Al-Salameh A, Cadiot G, Calender A, Goudet P, Chanson P (2021) Clinical aspects of multiple endocrine neoplasia type 1. Nat Rev Endocrinol 17(4):207–224
Iacovazzo D, Hernandez-Ramirez LC, Korbonits M (2017) Sporadic pituitary adenomas: the role of germline mutations and recommendations for genetic screening. Expert Rev Endocrinol Metab 12(2):143–153
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392(6676):605–608
Akhavanfard S, Padmanabhan R, Yehia L, Cheng F, Eng C (2020) Comprehensive germline genomic profiles of children, adolescents and young adults with solid tumors. Nat Commun 11(1):2206
Fomchenko EI, Erson-Omay EZ, Kundishora AJ, Hong CS, Daniel AA, Allocco A et al (2019) Genomic alterations underlying spinal metastases in pediatric H3K27M-mutant pineal parenchymal tumor of intermediate differentiation: case report. J Neurosurg Pediatr. :1–10
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S et al (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46(D1):D1062–D7
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC (2016) SIFT missense predictions for genomes. Nat Protoc 11(1):1–9
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ et al (2013) Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 34(1):57–65
Liu X, Wu C, Li C, Boerwinkle E (2016) dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat 37(3):235–241
Xiong D, Wang Y, Kupert E, Simpson C, Pinney SM, Gaba CR et al (2015) A recurrent mutation in PARK2 is associated with familial lung cancer. Am J Hum Genet 96(2):301–308
Cookson MR, Lockhart PJ, McLendon C, O’Farrell C, Schlossmacher M, Farrer MJ (2003) RING finger 1 mutations in parkin produce altered localization of the protein. Hum Mol Genet 12(22):2957–2965
Kazlauskaite A, Kelly V, Johnson C, Baillie C, Hastie CJ, Peggie M et al (2014) Phosphorylation of parkin at Serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of parkin E3 ligase activity. Open Biol 4(3):130213
Li H, Yusufujiang A, Naser S, Zhu Y, Maimaiti M, He X et al (2014) Mutation analysis of PARK2 in a Uyghur family with early-onset Parkinson’s disease in Xinjiang, China. J Neurol Sci 342(1–2):21–24
Lohmann E, Periquet M, Bonifati V, Wood NW, De Michele G, Bonnet AM et al (2003) How much phenotypic variation can be attributed to parkin genotype? Ann Neurol 54(2):176–185
Ruffmann C, Zini M, Goldwurm S, Bramerio M, Spinello S, Rusconi D et al (2012) Lewy body pathology and typical Parkinson disease in a patient with a heterozygous (R275W) mutation in the parkin gene (PARK2). Acta Neuropathol 123(6):901–903
Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS et al (2016) Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48(6):607–616
Vallar L, Spada A, Giannattasio G (1987) Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature 330(6148):566–568
Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF et al (2015) Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res 25(3):306–317
Boikos SA, Stratakis CA (2007) Molecular genetics of the cAMP-dependent protein kinase pathway and of sporadic pituitary tumorigenesis. Hum Mol Genet 16:1:R80–R87 Spec No
Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A et al (2006) Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 312(5777):1228–1230
Zhuang Z, Ezzat SZ, Vortmeyer AO, Weil R, Oldfield EH, Park WS et al (1997) Mutations of the MEN1 tumor suppressor gene in pituitary tumors. Cancer Res 57(24):5446–5451
Scheithauer BW, Laws ER Jr, Kovacs K, Horvath E, Randall RV, Carney JA (1987) Pituitary adenomas of the multiple endocrine neoplasia type I syndrome. Semin Diagn Pathol 4(3):205–211
Verges B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G et al (2002) Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab 87(2):457–465
Agarwal SK (2017) The future: genetics advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer 24(10):T119–T34
Cuny T, Pertuit M, Sahnoun-Fathallah M, Daly A, Occhi G, Odou MF et al (2013) Genetic analysis in young patients with sporadic pituitary macroadenomas: besides AIP don’t forget MEN1 genetic analysis. Eur J Endocrinol 168(4):533–541
Bergman L, Boothroyd C, Palmer J, Grimmond S, Walters M, Teh B et al (2000) Identification of somatic mutations of the MEN1 gene in sporadic endocrine tumours. Br J Cancer 83(8):1003–1008
Heppner C, Kester MB, Agarwal SK, Debelenko LV, Emmert-Buck MR, Guru SC et al (1997) Somatic mutation of the MEN1 gene in parathyroid tumours. Nat Genet 16(4):375–378
Qi C, Duan J, Shi Q, Wang M, Yan C (2018) Two nonsense somatic mutations in MEN1 identified in sporadic insulinomas. FEBS Open Bio 8(2):295–301
Asteria C, Anagni M, Persani L, Beck-Peccoz P (2001) Loss of heterozygosity of the MEN1 gene in a large series of TSH-secreting pituitary adenomas. J Endocrinol Invest 24(10):796–801
Farrell WE, Simpson DJ, Bicknell J, Magnay JL, Kyrodimou E, Thakker RV et al (1999) Sequence analysis and transcript expression of the MEN1 gene in sporadic pituitary tumours. Br J Cancer 80(1–2):44–50
Poncin J, Stevenaert A, Beckers A (1999) Somatic MEN1 gene mutation does not contribute significantly to sporadic pituitary tumorigenesis. Eur J Endocrinol 140(6):573–576
Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y et al (2005) Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci 25(8):2002–2009
Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD et al (2007) Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J Cell Biol 178(6):1025–1038
Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP et al (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 17(3):431–439
Bernardini JP, Brouwer JM, Tan IK, Sandow JJ, Huang S, Stafford CA et al (2019) Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J. ;38(2)
Gong Y, Zack TI, Morris LG, Lin K, Hukkelhoven E, Raheja R et al (2014) Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat Genet 46(6):588–594
Mowers EE, Sharifi MN, Macleod KF (2018) Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J 285(10):1751–1766
Ikeuchi K, Marusawa H, Fujiwara M, Matsumoto Y, Endo Y, Watanabe T et al (2009) Attenuation of proteolysis-mediated cyclin E regulation by alternatively spliced parkin in human colorectal cancers. Int J Cancer 125(9):2029–2035
Lee SB, Kim JJ, Nam HJ, Gao B, Yin P, Qin B et al (2015) Parkin regulates mitosis and genomic Stability through Cdc20/Cdh1. Mol Cell 60(1):21–34
Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ et al (2011) Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci U S A 108(39):16259–16264
Perwez A, Wahabi K, Rizvi MA, Parkin (2021) A targetable linchpin in human malignancies. Biochim Biophys Acta Rev Cancer 1876(1):188533
Iwakawa R, Okayama H, Kohno T, Sato-Otsubo A, Ogawa S, Yokota J (2012) Contribution of germline mutations to PARK2 gene inactivation in lung adenocarcinoma. Genes Chromosomes Cancer 51(5):462–472
Parry EM, Gable DL, Stanley SE, Khalil SE, Antonescu V, Florea L et al (2017) Germline mutations in DNA repair genes in Lung Adenocarcinoma. J Thorac Oncol 12(11):1673–1678
Castelo Rueda MP, Raftopoulou A, Gogele M, Borsche M, Emmert D, Fuchsberger C et al (2021) Frequency of heterozygous parkin (PRKN) Variants and Penetrance of Parkinson’s disease risk markers in the Population-Based CHRIS Cohort. Front Neurol 12:706145
Carroll RG, Hollville E, Martin SJ (2014) Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep 9(4):1538–1553
Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I et al (2010) Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 42(1):77–82
Funding
None.
Author information
Authors and Affiliations
Contributions
C.S.H. and H.A. wrote the main manuscript text. C.S.H., H.A., and M.D. prepared the figures. C.S.H., S.B.O., and E.Z.E. conceptualized the study and provided supervision. All authors were involved in the clinical care of the patient and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was approved under an institutional board approved protocol. Written informed consent was obtained.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hong, C.S., Alanya, H., DiStasio, M. et al. Sporadic pituitary adenoma with somatic double-hit loss of MEN1. Pituitary 26, 488–494 (2023). https://doi.org/10.1007/s11102-023-01336-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11102-023-01336-1