
Abstract
Context
Regional variation in prevalence of genetic mutations in growth hormone deficiency (GHD) is known.
Aim
Study phenotype and prevalence of mutations in GH1, GHRHR, POU1F1, PROP1 genes in GHD cohort.
Methods
One hundred and two patients {Isolated GHD (IGHD): 79; combined pituitary hormone deficiency (CPHD): 23} with orthotopic posterior pituitary were included. Auxologic, hormonal and radiological details were studied. All four genes were analysed in IGHD patients. POU1F1 and PROP1 were studied in CPHD patients.
Results
Of 102, 19.6% were familial cases. Height SDS, mean (SD) was − 5.14 (1.63). Peak GH, median (range) was 0.47 ng/ml (0–6.59), 72.5% patients had anterior pituitary hypoplasia (APH). Twenty mutations (novel: 11) were found in 43.1% patients (n = 44, IGHD-36, CPHD-8). GHRHR mutations (n = 32, p.Glu72* = 24) were more common than GH1 mutations (n = 4) in IGHD cohort. POU1F1 mutations (n = 6) were more common than PROP1 mutations (n = 2) in CPHD cohort. With few exceptions, this prevalence pattern is contrary to most studies in world-literature. No patients with peak GH > 4 ng/ml had mutations, signifying it as negative predictor. While many parameters were significant on univariate analysis, only positive family history and lower median peak GH levels were significant predictors of mutations on multivariate analysis in IGHD patients.
Conclusion
At variance with world literature, we found reverse predominance of GHRHR over GH1 mutations, POU1F1 over PROP1 mutations and predominance of GHRHR p.Glu72* mutations thus re-affirming the regional diversity in GHD genetics. We report positive and negative predictors of mutations in GHD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although influenced by multiple environmental factors, human growth remains a highly heritable trait [1]. Growth hormone deficiency (GHD) is the commonest congenital pituitary hormone deficiency presenting as isolated GHD (IGHD) or as component of combined pituitary hormone deficiency (CPHD) [2, 3]. Generally, different genes are implicated in pathogenesis of IGHD and CPHD, though GHD can be the first manifestation of CPHD [2, 3]. Common genes implicated in IGHD include GH1 (growth hormone 1) and GHRHR (growth hormone-releasing hormone receptor), while those in CPHD include PROP1 (PROP paired-like homeobox 1), POU1F1 (POU class 1 homeobox 1), HESX1, LHX3, LHX4, SOX2, SOX3, OTX2 and GLI2 [1,2,3,4]. With use of next-generation sequencing, this list continues to grow. Up-to 3–30% of GHD patients are familial [1,2,3]. Study of genetics helps in enhancing patient care by enabling opportunities for genetic counselling, early diagnosis, and timely initiation of hormone replacement therapy. Knowing certain consistent genotype–phenotype associations like rarity of mutations in later-acting transcription factor genes (POU1F1, PROP1) in patients with ectopic posterior pituitary (EPP) or pituitary stalk interruption syndrome (PSIS) may direct study of specific genes [5]. Similarly, the absence of other hormone deficiencies in patients with GHRHR mutations, and that of corticotropin or gonadotropin deficiency in those with POU1F1 mutations may influence clinical follow-up [2, 5]. Moreover, ethnic-specific differences in prevalence of specific genetic mutations are known [4, 6,7,8]. This information might help in prioritising genetic testing in specific populations, thus, emphasising need for genetic characterisation of regional cohorts of GHD patients. In this sense, comprehensive genetic studies on Asian-Indian patient cohorts are limited [9, 10]. We aim to study phenotypic characteristics and prevalence of mutations in four common genes (GH1, GHRHR, POU1FI1, and PROP1) in a cohort of consecutive GHD patients from western India.
Patients and methods
After approval from Institutional Ethics Committee II, Seth GS medical college and KEM hospital, Mumbai, 145 consecutive, unrelated probands with idiopathic GHD were evaluated. Patients having EPP/PSIS (n = 30) and septo-optic dysplasia (n = 7) were excluded as these features are rarely reported in patients with GH1, GHRHR, POU1F1, and PROP1 mutations [2, 5]. Six patients were excluded for inadequate phenotypic data. Thus, final cohort consisted of 102 patients. Written informed consent was taken from the patients and/or their parents.
Diagnosis of GHD was based on peak GH value < 7 ng/ml for those less than 18 years of age, or < 3 ng/ml for those with ≥ 18 years of age, on at-least one GH stimulation test (clonidine stimulation test, insulin tolerance test or glucagon stimulation test) and low serum insulin-like growth factor 1 (IGF1) level. Sex steroid priming was done in children ≥ 8 years old and tanner staging ≤ 2 with estradiol valerate tablets (1–2 mg OD) for three days prior to testing [11]. Absence of acquired causes (e.g. systemic illness, intracranial masses, cranio-spinal radiation) was ascertained.
Following phenotypic details were recorded: age at presentation, gender, family history of consanguinity and that of similar affection of other members. Auxological parameters like height-SDS, weight-SDS, mid-parental height, sexual maturity by Tanner staging and bone-age were recorded. Following hormonal parameters were recorded: peak GH on any of the GH stimulation tests (as mentioned above), serum levels of IGF1, free/total T3, free/total T4, TSH, prolactin, 8.00 am cortisol, FSH, LH and total testosterone. Central hypothyroidism was defined as low free/total T4 with low or inappropriately normal TSH levels. Central hypo-cortisolism was defined as 8.00 am serum cortisol < 5 µg/dl, and/or serum cortisol < 18 µg/dl during insulin tolerance test (whenever available). Central hypogonadism was defined as absence of pubertal onset/progression with low or inappropriately normal serum FSH and LH levels with bone age > 13 years in females or > 14 years in males. Serum prolactin level < 5 ng/ml was indicative of prolactin deficiency. ACTH and TRH stimulation tests were not performed due to limited availability of these drugs at our centre. GHD patients developing one additional pituitary hormone deficiency (thyroid, cortisol or gonadal axes) till last available follow up were considered to have CPHD. All hormonal measurements were done by chemiluminescence assay (Advia Centaur CP) with intra and inter-assay coefficients of variation less than 8 and 10% respectively.
MRI (1.5 T) of brain and pituitary was done with gadolinium contrast and read by single radiologist. Following parameters were recorded: anterior pituitary height (mm), location of posterior pituitary (ectopically placed or normal), morphology of pituitary stalk (interrupted or continuous), optic nerves (normal or hypoplastic) and midline brain structures (corpus callosum and septum pellucidum abnormalities). Maximum height of pituitary was measured perpendicular to sella turcica and considered hypoplastic when less than − 2 SD of normal [12].
Genotyping
We studied four genes (GH1 (ENSG00000259384), GHRHR (ENSG00000106128), PROP1 (ENSG00000175325) and POU1F1 (ENSG00000064835)) in IGHD patients. Only PROP1 and POU1F1 were studied in CPHD patients. Genomic DNA was isolated from peripheral blood leukocytes by standard techniques. PCR primers were designed to amplify exons, intron–exon boundaries, 5′/3′ untranslated regions and promoter regions. PCR reactions were standardized using GoTaq Green Master mix (Promega). Capillary DNA sequencing was carried out using BigDye® Terminator v3.1 cycle sequencing kit chemistry on ABI PRISM® 3100 Genetic Analyzer. The sequence obtained was aligned against primary assembly of human genome (GRCh37.p10) using Basic Local Alignment Search Tool (BLAST). ExAC, 1000 Genomes and gnomAD databases were used to find frequency of novel variations, which were reported to ClinVar databases to obtain accession numbers. In patients who were found to be mutation negative, multiplex ligation-dependent probe amplification (MLPA) was done to assess large deletions. Whenever possible, first degree relatives were screened for variations observed in index cases. Novel variants were considered pathogenic/likely pathogenic if in-silico tools (human splice site finder, Mutation Taster, Polyphen-2 and Sort Intolerant From Tolerant) predicted them to be damaging and minor allele frequency was < 1% on the above databases [3].
Statistical analysis
Categorical variables were represented as actual numbers/percentages and differences between them were compared using chi-square test or Fisher exact t test. Continuous variables were expressed as mean ± SD or median and compared using independent “t” test or Mann–Whitney U test. P value < 0.05 was considered significant. Multiple regression analysis was done for predictors of mutation positivity. Data were analysed using software SPSS version 23.0 (SPSS software, IL, Chicago, SA).
Results
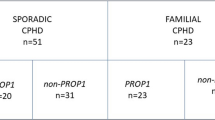
Study cohort included 102 index patients (males: 57, females: 45); 79 patients had IGHD (males: 46, females: 33) and 23 patients had CPHD (males-11, females-12). Eighty-two patients (IGHD: 64, CPHD: 18) were apparently sporadic (AS) while family history of similar affection was present in 20 patients (IGHD: 15, CPHD: 5). Twenty-six patients (25.4%) had history of consanguinity. Few patients had significant perinatal history in the form of documented hypoglycaemic events (n = 5, IGHD-3, CPHD-2). Amongst the 57 male patients, 2 had history of micropenis at birth (IGHD-1, CPHD-1). Table 1 summarises baseline characteristics of patients.
With mean height-SDS of − 5.14 and 86% patients having peak GH < 3 ng/ml, our cohort predominantly had patients with severe GHD. In CPHD cohort, other than GH axis, thyroid (n = 17, 73.9%) was the commonest axis involved followed by prolactin (n = 10, 43.4%), gonadotropin (n = 9, 39.1%), and cortisol (n = 4, 17.4%) axes. The commonest deficiency pattern was that of GH + TSH + prolactin deficiency (n = 7, 30.4%) followed by GH + gonadotropin deficiency (n = 5, 21.7%). MRI was available in all patients. Anterior pituitary hypoplasia (APH) was observed in 72.5% patients (n = 74, IGHD-56, CPHD-18).
Twenty different mutations were found in four genes (GH1, GHRHR, PROP1, POU1F1) in 44 patients (43.1%), out of which 11 were novel (Tables 2, 3). Mutation yield was higher in familial cases (19/20, 95%) than in sporadic patients (25/82, 30.4%).
IGHD cohort
Thirty-six IGHD patients (36/79, 45.5%) had mutations (GH1: 4 patients, GHRHR: 32 patients). Expectedly, mutation positivity was higher in familial (93%, 14/15) than in sporadic cases (34.3%, 22/64). On univariate analysis, mutation positive patients had significantly higher prevalence of familial cases and APH, lower peak GH and IGF1 levels than mutation negative patients (Table 4). Rate of mutation positivity declined with increasing peak GH values (63.2% in patients with peak GH < 1 ng/ml, 24% in patients with peak GH 1–4 ng/ml and none in those with peak GH > 4 ng/ml). However, in multivariate analysis, positive family history and lower peak GH levels were the only significant predictors for mutation positivity.
GH1 gene
One patient had splice-site mutation while three had deletions in GH1 gene. The patient with splice site mutation had presented at 14.5 years of age with severe growth failure (height SDS: − 7.8) and very low GH/ IGF1 levels (Table 2). He was born to normal statured parents having consanguineous marriage and had younger brother similarly affected with IGHD. Due to financial constraints, he received intermittent GH therapy, and showed good response. Both siblings were homozygous for novel intron 1 splicing acceptor site mutation (c.11-2A > G). Their mother was heterozygous for same mutation, while father’s sample was un-available for analysis (Supplementary Fig. S1 Pedigree 1).
Three patients had same deletion involving exons 3, 4 and 5 in GH1 gene (2: homozygous, 1: heterozygous) All three were sporadic cases, had presented early (by 5 years of age) with severe growth failure and almost undetectable IGF1/GH levels (Table 2). Two of them had APH. One patient couldn’t afford GH treatment, while other two patients showed good response to GH therapy over follow up of 6 and 10 years respectively.
GHRHR gene
Nine different mutations (novel: 5) were found in 32 patients (29: homozygous, 3: compound heterozygous). Thirteen patients (40%) were familial cases. Of 19 sporadic patients, 7 (36.8%) had history of consanguinity. Previously reported p.Glu72* was the commonest mutation, found in 24 patients (22: homozygous, 2: compound heterozygous) thus accounting for its remarkable prevalence of 30% in overall IGHD cohort, 22% in sporadic and 66% in familial IGHD patients. Three other previously reported mutations were: p.Arg161Trp (n = 2, 1: homozygous, 1: compound heterozygous), p.Arg94Trp (n = 1) and p.Arg94Leu (n = 2) (Table 2). We found five novel mutations in 6 patients (Table 3). Out of them, we have separately published one novel gross indel g.30999250_31006943delinsAGAGATCCA in two non-consanguineous families [13]. Other four novel mutations were: a. p.His165Gln in compound heterozygous state with previously reported p.Arg161Trp in one apparently sporadic patient (Supplementary Fig. S1 Pedigree 2) b. p.Arg94Gln in compound heterozygous with p.Glu72* in a familial case (Supplementary Fig. S1 Pedigree 3) c. homozygous p.Ser140Pro in an apparently sporadic patient (Supplementary Fig. S1 Pedigree 4) and d. homozygous p.Cys55Phe in a consanguineous family (Supplementary Fig. S1 Pedigree 5).
CPHD cohort
Eight patients (34.7%) had pathogenic mutations (POU1F1: 6, PROP1: 2). Mutation positivity was higher in familial patients (5/5, 100%) than in sporadic cases (3/18, 16.6%). On univariate analysis, mutation positive patients had significantly higher prevalence of familial cases, earlier presentation with lower Ht SDS and more delayed bone age than mutation negative patients (Table 4). Due to small sample size, multivariate analysis could not be performed in CPHD patients’ cohort.
POU1F1 gene
We found six different mutations in POU1F1 gene (3: novel, 3: previously reported) in six patients (6/23, 26%) (Table 3). All patients had presented with central hypothyroidism early in life (less than 2 years of age), except one patient who presented at 5 years of age. All patients had severe deficiency of GH and prolactin (Table 2). Three novel mutations include a. Homozygous c.665 + 1G > T intron 5 splice-site mutation in a consanguineous familial case (Supplementary Fig. S1 Pedigree 6) b. Homozygous p.Arg213Lysfs*12 (c.634_638delGAAAG) exon 5 mutation found in an apparently sporadic patient (Supplementary Fig. S1 Pedigree 7) c. Homozygous c.215-3C > G intron 2 mutation causing aberrant splicing in an another apparently sporadic patient (Supplementary Fig. S1 Pedigree 8) (Table 3). Apart from these novel mutations, three affected siblings from a consanguineous family were homozygous for a reported p.Glu250* mutation in exon 6, while in another consanguineous family two affected siblings were homozygous for reported p.Arg265Trp mutation in exon 6. Additionally, in another family, two cousins were homozygous for reported splice-site mutation, c.605-1G > A in intron 4.
PROP1 gene
Two patients had three pathogenic PROP1 mutations (2 novel, 1 previously described) (Tables 2, 3). Both had GH, thyroid and gonadotrophin deficiency, while prolactin was low in one patient. These patients include: a. One familial case with novel p.Gln92* mutation in exon 2 in a compound heterozygous state with previously described p.Arg125Trp exon 3 mutation (Supplementary Fig. S1. Pedigree 9) b. One apparently sporadic patient (Supplementary Fig. S1 Pedigree 10) was homozygous for a novel 13 bp deletion c.110_122delACTCGAGTCCTCC (p.S38Pfs*123) in exon 2 gene resulting in frameshift with a premature stop codon at position 480.
Discussion
Genetics of GHD has been evaluated in worldwide IGHD/CPHD cohorts and wide variation in mutation prevalence has been reported in different studies [4, 5, 8,9,10, 14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. From time to time, various authors have described predominance of specific mutations in certain populations. Notable instances of such reports include IVSI + 1G > A splice site GHRHR mutation in Brazilian Itabaianinha cohort by Salvatori et al., p.Glu72* GHRHR mutation in Sindh province by Maheshwari et al., and c.296delGA PROP1 mutation in Lithuanian CPHD cohort by Navardauskaite et al. [6, 8, 35]. Presence of a founder effect or mutational hotspots are the common reasons proposed for such recurrent mutations [7, 36]. Interestingly, independent occurrence of same mutation in patients from different continents without a common ancestor (as proven on haplotype analysis) has also been described [37]. Such evident geographic and/or ethnic character of mutations signify importance of studying genetics of regional cohorts of GHD patients. From Indian subcontinent, most previous studies had some limitations [17, 20, 38]. In their respective western-Indian and Sri- Lankan cohorts of IGHD patients, Desai et al. [17] and deSilva et al. [38] studied only specific mutations (deletions in GH1 gene and/or p.Glu72* mutation in GHRHR gene). Similarly, Khadilkar et al. studied deletions alone in western-Indian cohort [20]. Hence, we aimed to study the complete coding sequences in four common genes in our cohort of consecutive GHD patients along with MLPA.
We found 43.1% prevalence of mutations in four selected genes. The fact that our cohort consisted predominantly of severe GHD patients, may be one of the factors contributing to such high mutation yield [14, 15, 21, 39]. Understandably, prevalence was higher in familial (95%) than in sporadic patients (30.4%), which is a consistent finding in most studies [5, 15,16,17, 25, 30].
We found 45.5% mutation positivity in IGHD cohort which is within the wide range of prevalence (0–52%) reported in different genes in worldwide IGHD cohorts [5, 9, 10, 14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. We found GHRHR mutations were more common than GH1 mutations. While this distribution has been reported in few other studies, especially from Indian subcontinent [9, 17, 40], most of the cohorts of other ethnicities have GH1 mutations more common than GHRHR mutations (Table 5) [14,15,16, 18, 21]. This may be due to predominance of p.Glu72* GHRHR mutation, which has been reported to have founder effect in patients from Indian sub-continent [7]. In our cohort, 75% patients with GHRHR mutations had p.Glu72* mutation. We had four patients with GH1 variations, three of them had deletions. Congruous with the phenotype described for severe GH1 mutations, all four of our patients had presented with severe growth failure and undetectable GH/IGF1 levels. All treated patients continued to show good response to GH therapy over entire period of treatment. This substantiates previous observation that phenomenon of immune intolerance to GH and the consequent treatment failure among patients with GH1 deletion is not universal [2]. Intriguing feature in our cohort is a patient who was heterozygous for GH1 deletion. GH1 deletions are not reported to be pathogenic in heterozygous state, and hence, the phenotype seen in our patient remains un-explained [2]. Her parents’ samples were not available for segregation analysis. Mutations in regulatory and other non-coding sequences of second GH1 allele that escaped our current detection methods can be a plausible explanation for this phenomenon.
On univariate analysis, our mutation positive IGHD patients had significantly more prevalence of positive family history, APH and lower IGF1 and peak GH levels than mutations negative patients. However, on multivariate analysis, only significant factors were positive family history and lower peak GH levels. In this regard, our findings are similar to previous studies [14, 15, 21]. In 89 Dutch IGHD patients, de Graaff found positive family history, lower height-SDS, peak GH levels and IGF1 SDS as significant predictors of GH1 mutation [14]. Alatzoglou found significant difference in auxologic parameters (lower height SDS in mutation positive patients) but not in endocrine or MRI features between mutation positive and negative patients in their multi-ethnic IGHD cohort [15]. Recently, in a large multinational prospective observational study of GeNeSIS cohort (Genetics and Neuroendocrinology of Short Stature International Study), Blum et al. reported younger age at presentation and lower peak GH levels as indicators of mutations in GH1 and GHRHR genes in IGHD patients [21]. In our cohort, none of patients with peak GH level > 4 ng/ml had mutations, suggesting this feature to have negative predictability for mutation positivity. Similar to our observation, Lido et al. reported that GH1 mutations (n = 9) were found only in the sub-group of patients having peak GH < 3.3 μg/l in 135 Brazilian children [39]. However, Blum et al. caution against this notion. In their IGHD sub-cohort, 4 out of 23 patients with GH1 mutations and one of two patients with SOX3 mutations had peak GH levels between 3–6 μg/l [21]. With similar findings in CPHD cohort, they cautioned against precluding genetic analysis in patients with such ‘measurable’ GH levels. In our cohort, considering the fact that p.Glu72* is the commonest mutation accounting for one third of IGHD patients, it remains intuitive to speculate whether direct testing for this mutation with a simpler technique like PCR based restriction digestion (using BfaI endonuclease) in patients with suggestive phenotype of idiopathic severe growth failure and very low IGF1 levels, can obviate the need for GH stimulation tests in at least one-third of patients [17].
In our CPHD cohort, 34.7% patients had mutations, with POU1F1 mutations more common (26%) than PROP1 mutations (8.6%). Our prevalence of POU1F1 mutations (26%) is one of the highest reported worldwide (Table 5). With 4 out of 6 mutations being novel, no recurring mutations and only 21% familial cases in our CPHD patients, we cannot speculate any definite cause for this high prevalence. Notably, we have excluded patients with EPP/PSIS from our analysis due to rarity of mutations in POU1F1/PROP1 genes in such patients. Most reported studies analysing prevalence of POU1F1/PROP1 mutations have included varying proportions of patients with these abnormalities (2–80%) in their cohorts [5, 18, 21, 24, 27,28,29, 31, 32, 34]. Since not all authors have excluded these patients from analysis, there is possible under-estimation in reported prevalence figures of mutations in these genes.
Unlike POU1F1 mutations, our rate of PROP1 mutations (8.6%) is well within that described in world literature (Table 5). In an exhaustive systematic review of 21 published studies, De Rienzo reported global prevalence of 11.2% for PROP1 mutations, clearly higher than that of POU1F1 mutations (2.8%) [5]. Importantly, they observed wide regional variation (0–65%) in prevalence of PROP1 mutations, which was largely accounted by uneven geographic concentration of two common variations, c.296delGA (25–100%) and c.150delA (12–50%) in certain regions. Our cohort did not have these mutations. Importantly, Turton et al. reported founder effect for 13 bp deletion in PROP1 (112–124Δ) gene in Indian patients [41]. Intriguingly, in our cohort, we found a novel 13 bp deletion in same region of PROP1 gene, but 2 bp upstream to this founder mutation. We don’t know significance of this finding. Our contrasting observation of relative predominance of POU1F1 over PROP1 mutations might be partly attributed by absence of common dominating PROP1 mutations in our cohort. While at contrast with world literature, our observation is similar to that of other Indian cohorts like that of Birla et al. who reported 14% POU1F1 mutations and 6% PROP1 mutations in their North Indian cohort of 51 CPHD children [10].
Our mutation positive patients had significantly higher prevalence of positive family history, early presentation, lower height SDS and delayed bone age as compared to mutation negative patients. Very few studies have reported predictors of mutations in CPHD cohorts. In GeNeSIS cohort, Blum et al. reported lower height-SDS minus target height-SDS as the only significant indicator of mutations in CPHD patients [21].
We have included all consecutive GHD patients from single centre. Hence, we believe our cohort is representative of GHD patients in a routine growth clinic. However, ours being tertiary care centre, risk of referral bias cannot be discounted completely. We have considered pituitary height alone as a parameter of APH in the current study. While calculation of pituitary volume could have represented a more sensitive parameter of assessment of APH, this remains an important limitation of our study. Unlike previous Indian studies, we have comprehensively evaluated complete coding sequences of four genes along with MLPA. We report 11 novel variations, adding to the genetic literature of GHD. However, we couldn’t do functional studies or splicing assays for these novel variations, which is an important limitation of our study. We were able to study only four genes, while number of implicated genes in GHD is increasing [1]. However, the contribution of other genes to known genetic prevalence worldwide is reported to be less than 1% [5].
To conclude, we present a cohort of consecutive GHD patients from western India and report mutation prevalence of 45.5% in IGHD patients and 34.7% in CPHD patients. At variance with world literature, we report higher prevalence of GHRHR than GH1 mutations, with predominance of the founder mutation p.Glu72*, in IGHD patients and predominance of POU1F1 over PROP1 mutations in CPHD patients. In addition to re-affirming some of the previously reported predictors of mutation positivity, our study also provides few novel predictors of mutation positivity, especially in CPHD patients. Summarising, we emphasise the importance of studying genetics of regional cohorts of GHD patients, as subtle cohort specific differences in genetic prevalence can be established. This might help in establishing individualised region-specific prioritisation of genetic study in GHD patients.
Abbreviations
- GHD:
-
Growth hormone deficiency
- IGHD:
-
Isolated growth hormone deficiency
- CPHD:
-
Combined pituitary hormone deficiency
- GH1:
-
Growth hormone 1
- GHRHR:
-
Growth hormone releasing hormone receptor
- PROP1:
-
PROP paired-like homeobox 1
- POU1F1:
-
POU class 1 homeobox 1
- EPP:
-
Ectopic posterior pituitary
- PSIS:
-
Pituitary stalk interruption syndrome
- IGF-1:
-
Insulin like growth factor 1
- SDS:
-
Standard deviation score
- GH:
-
Growth hormone
- TSH:
-
Thyroid stimulating hormone
- FSH:
-
Follicle stimulating hormone
- LH:
-
Luteinizing hormone
- ACTH:
-
Adrenocorticotropic hormone
- TRH:
-
Thyrotropin releasing hormone
- BLAST:
-
Basic local alignment search tool
- ExAC:
-
Exome aggregation consortium
- APH:
-
Anterior pituitary hypoplasia
References
Giordano M (2016) Genetic causes of isolated and combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab 30(6):679–691
Alatzoglou KS, Dattani MT (2010) Genetic causes and treatment of isolated growth hormone deficiency – an update. Nat Rev Endocrinol 6:562–576
Fang Q, George AS, Brinkmeier ML et al (2016) Genetics of combined pituitary hormone deficiency: roadmap into the genome era. Endocr Rev 6:636–675
Madeira J, Nishi M, Nakaguma M et al (2017) Molecular analysis of Brazilian patients with combined pituitary hormone deficiency and orthotopic posterior pituitary lobe reveals eight different PROP1 alterations with three novel mutations. Clin Endocrinol (Oxf) 87(6):725–732
De Rienzo F, Mellone S, Bellone S et al (2015) Frequency of genetic defects in combined pituitary hormone deficiency: a systematic review and analysis of a multicentre Italian cohort. Clin Endocrinol 83:849–860
Salvatori R, Hayashida CY, Aguiar-Oliveira MH et al (1999) Familial dwarfism due to a novel mutation of the growth hormone-releasing hormone receptor gene. J Clin Endocrinol Metab 84:917–923
Kamijo T, Hayashi Y, Seo H et al (2004) A nonsense mutation (E72X) in growth hormone releasing hormone receptor (GHRHR) gene is the major cause of familial isolated growth hormone deficiency in Western region of India: founder effect suggested by analysis of dinucleotide repeat polymorphism close to GHRHR gene. Growth Horm IGF Res 14:394–401
Navardauskaite R, Dusatkova P, Obermannova B et al (2014) High prevalence of PROP1 defects in Lithuania: phenotypic findings in an ethnically homogenous cohort of patients with multiple pituitary hormone deficiency. J Clin Endocrinol Metab 99(1):299–306
Birla S, Khadgawat R, Jyotsna VP et al (2016) Identification of novel GHRHR and GH1 mutations in patients with isolated growth hormone deficiency. Growth Horm IGF Res 29:50–56
Birla S, Khadgawat R, Jyotsna VP et al (2016) Identification of Novel PROP1 and POU1F1 mutations in patients with combined pituitary hormone deficiency. Horm Metab Res 48(12):822–827
Martinez AS, Domene HM, Ropelato G et al (2000) Estrogen priming effect on growth hormone (GH) provocative test: a useful tool for the diagnosis of GH deficiency. J Clin Endocrinol Metab 11(1):4168–4172
Argyropoulou M, Perignon F, Brunelle F, Brauner R, Rappaport R (1991) Height of normal pituitary gland as a function of age evaluated by magnetic resonance imaging in children. Pediatr Radiol 21:247–249
Kale S, Budyal S, Kasaliwal R et al (2014) A novel gross indel in the growth hormone releasing hormone receptor gene of Indian IGHD patients. Growth Horm IGF Res 24(6):227–232
De Graaff LC, Argente J, Veenma DCM et al (2009) Genetic screening of a Dutch population with isolated GH deficiency (IGHD). Clin Endocrinol (Oxf) 70:742–750
Alatzoglou KS, Turton JP, Kelberman D et al (2009) Expanding the spectrum of mutations in GH1 and GHRHR: genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab 94:3191–3199
Juanes M, Marino R, Ciaccio M et al (2014) Presence of GH1 and absence of GHRHR gene mutations in a large cohort of Argentinian patients with severe short stature and isolated GH deficiency. Clin Endocrinol (Oxf) 80(4):618–620
Desai MP, Mithbawkar SM, Upadhye PS, Rao SC, Bhatia V, Vijaykumar M (2013) Molecular genetic studies in isolated growth hormone deficiency (IGHD). Indian J Pediatr 80(8):623–630
Fritez N, Sobrier ML, Iraqi H et al (2015) Molecular screening of a large cohort of Moroccan patients with congenital hypopituitarism. Clin Endocrinol 82(6):876–884
Sundralingam T, Tennekoon KH, de Silva S, De Silva S, Hewage AS (2017) Pathogenic and likely pathogenic genetic alterations and polymorphisms in growth hormone gene (GH1) and growth hormone releasing hormone receptor gene (GHRHR) in a cohort of isolated growth hormone deficient (IGHD) children in Sri Lanka. Growth Horm IGF Res 36:22–29
Khadilkar V, Phadke N, Khatod K et al (2017) Molecular genetics of growth hormone deficient children: correlation with auxology and response to first year of growth hormone therapy. J Pediatr Endocrinol Metab 30(6):669–675
Blum W, Klammt J, Amselemc S et al (2018) Screening a large pediatric cohort with GH deficiency for mutations in genes regulating pituitary development and GH secretion: frequencies, phenotypes and growth outcomes. EBioMedicine 36:390–400
McLennan K, Jeske Y, Cotterill A et al (2003) Combined pituitary hormone deficiency in Australian children: clinical and genetic correlates. Clin Endocrinol 58:785–794
Lebl J, Vosahlo J, Pfaeffle RW et al (2005) Auxological and endocrine phenotype in a population-based cohort of patients with PROP1 gene defects. Eur J Endocrinol 153:389–396
Rainbow LA, Rees SA, Shaikh MG et al (2005) Mutation analysis of POUF-1, PROP-1 and HESX-1 show low frequency of mutations in children with sporadic forms of combined pituitary hormone deficiency and septo-optic dysplasia. Clin Endocrinol 62:163–168
Reynaud R, Gueydan M, Saveanu A et al (2006) Genetic screening of combined pituitary hormone deficiency: experience in 195 patients. J Clin Endocrinol Metab 91:3329–3336
Vieira TC, Boldarine VT, Abucham J (2007) Molecular analysis of PROP1, PIT1, HESX1, LHX3 and LHX4 shows high frequency of PROP1 mutations in patients with familial forms of combined pituitary hormone deficiency. Arq Bras Endocrinol Metab 51(7):1097–1103
De Graaff LCG, Argente J, Veenma DCM et al (2010) PROP1, HESX1, POU1F!, LHX3 and LHX4 mutation and deletion screening and GH1 P89L and IVS3+1/+2 mutation screening in a dutch nationwide cohort of patients with combined pituitary hormone deficiency. Horm Res Paediatr 73:363–371
Nyström HF, Saveanu A, Barbosa EJ et al (2011) Detection of genetic hypopituitarism in an adult population of idiopathic pituitary insufficiency patients with growth hormone deficiency. Pituitary 14:208–216
Takagi M, Ishii T, Inokuchi M et al (2012) Gradual loss of ACTH due to a novel mutation in LHX4: comprehensive mutation screening in Japanese patients with congenital hypopituitarism. PLoS ONE 7(9):e46008
Bas F, Uyguner O, Darendeliler F et al (2015) Molecular Analysis of PROP1, POU1F1, LHX3, and HESX1 in Turkish patients with combined pituitary hormone deficiency: a multicentre study. Endocrine 49(2):479–491
Kim S, Kim Y, Shin Y et al (2003) Clinical characteristics and molecular analysis of PIT1, PROP1, LHX3 and HESX1 in combined pituitary hormone deficiency patients with abnormal pituitary MR imaging. Horm Res 60:277–283
Coya R, Vela A, de Nanclares GP et al (2007) Panhypopituitarism: genetic versus acquired etiological factors. J Pediatr Endocrinol Metab 20:27–36
Dateki S, Fukami M, Uematsu A et al (2010) Mutation and gene copy number analyses of six pituitary transcription factor genes in 71 patients with combined pituitary hormone deficiency: identification of a single patient with LHX4 deletion. J Clin Endocrinol Metab 95:4043–4047
Choi J, Jung C, Kang E et al (2017) Rare frequency of mutations in pituitary transcription factor genes in combined pituitary hormone or isolated growth hormone deficiencies in Korea. Yonsei Med J 58(3):527–532
Maheshwari H, Silverman B, Dupui J et al (1998) Phenotype and genetic analysis of a syndrome caused by an inactivating mutation in the growth hormone-releasing hormone receptor: Dwarfism of Sindh. J Clin Endocrinol Metab 83(11):4065–4074
Deladoëy J, Flück C, Büyükgebiz A et al (1999) Hot spot in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab 84(5):1645–1650
Salvatori R, Aguiar-Oliveira MH, Monte L et al (2002) Detection of a recurring mutation in the human growth hormone-releasing hormone receptor gene. Clin Endocrinol 57:77–80
de Silva KSH, Tennekoon KH, Sundralingam T et al (2016) Growth hormone releasing hormone receptor codon 72 mutation in a cohort of Sri Lankan patients with growth hormone deficiency. Ceylon Med J 61:18–21
Lido AC, Franca MM, Correa FA et al (2014) Autosomal recessive form of isolated growth hormone deficiency is more frequent than the autosomal dominant form in a Brazilian cohort. Growth Horm IGF Res 24(5):180–186
Salvatori R, Fan X, Phillips J III et al (2001) Three new mutations in the gene for the growth hormone -releasing hormone receptor in familial isolated GH deficiency type IB. J Clin Endocrinol Metab 86:273–279
Turton J, Mehta A, Raza J et al (2005) Mutations within the transcription factor PROP1 are rare in a cohort of patients with sporadic combined pituitary hormone deficiency (CPHD). Clin Endocrinol 63:10–18
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The study has been approved by the Institutional Ethics Committee II, Seth GS medical college and KEM hospital, Mumbai, India.
Informed consent
All the patients/their parents have given written informed consent for participation in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kale, S., Gada, J.V., Jadhav, S. et al. Genetic spectrum and predictors of mutations in four known genes in Asian Indian patients with growth hormone deficiency and orthotopic posterior pituitary: an emphasis on regional genetic diversity. Pituitary 23, 701–715 (2020). https://doi.org/10.1007/s11102-020-01078-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11102-020-01078-4