
Abstract
Purpose
This study aimed to incorporate ondansetron hydrochloride (ODS), a water-soluble drug into nanostructured lipid carriers (NLCs) to improve the pharmacokinetic properties of the drug.
Methods
NLCs were produced by solvent injection method. Various parameters of formulation and process were assessed to enhance the drug incorporation into NLCs. Physicochemical analyses, in vitro drug release, and pharmacokinetic studies were performed.
Results
Entrapment efficiency (EE) of ODS was considerably improved (>90%) by increasing pH of the aqueous phase. The use of an appropriate level of liquid lipid resulted in small, monodispersed NLCs with the enhanced EE and drug loading (DL). The optimized NLCs formulation exhibited particle size of 185.2 ± 1.9 nm, polydispersity index of 0.214 ± 0.006, EE of 93.2 ± 0.5%, and DL of 10.43 ± 0.05% as well as an in vitro sustained-release profile of ODS. Differential scanning calorimetry and X-ray powder diffraction suggested the amorphous state of ODS in the NLCs. The pharmacokinetic study in rats exhibited the sustained-release characteristic of the optimized ODS-loaded NLCs following subcutaneous administration with an extended Tmax and mean residence time as well as the enhanced systemic exposure compared to the ODS solution.
Conclusions
The ODS-loaded NLCs appear potential for prolongation of drug action and reduction in dosing frequency.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) are alternative carrier systems to liposomes, emulsions and polymeric nanoparticles with a potential for controlled release of the drug (1,2). They have found wide applications in parenteral (3), topical (4), and oral delivery systems (5). By altering the drug physicochemical properties, SLNs and NLCs could control the biodistribution and pharmacokinetics of the drug and thereby improve the drug therapeutic performance (6,7). NLCs are regarded as the second generation of lipid nanoparticles developed by modifications of SLNs to overcome their limitations (8). NLCs are prepared from mixtures of solid and liquid lipids, which are still solid at room and body temperature. Lipids and surfactants used in NLCs are generally biodegradable with low toxicity (9). The use of solid and liquid lipids blends creates an imperfect structure in the NLCs, which enhances the drug loading capacity and minimizes the drug expulsion during storage (10).
Various methods to prepare SLNs and NLCs have been studied including high-pressure homogenization, emulsification sonification, microemulsion, emulsion/solvent evaporation, solvent diffusion, and phase inversion (2,11). These methods relate to several limitations such as the utilization of sophisticated equipment (e.g., high-shear mixer and high-pressure homogenizer), high surfactant concentrations, toxic solvent residue, and substantial dilutions of suspension. Solvent injection, an alternative method for SLNs and NLCs preparation, was first reported in 2003 (12) and further investigated in several studies (13,14,15). In this method, lipids and drugs are dissolved in water-miscible solvents such as ethanol, acetone, isopropanol, and methanol. The organic phase is rapidly injected into an aqueous phase with surfactants under continuous mechanical stirring, leading to the solvents diffusion and SLNs/NLCs formation (16). The advantages of solvent injection method include easiness of handling, quick production process, and avoidance of complicated instruments (11).
Ondansetron hydrochloride (ODS) is a 5-hydroxytryptamine subtype 3 receptor antagonist used for the prevention of chemotherapy-induced nausea and vomiting (CINV), common side effects of cancer treatment (17). ODS has a short plasma half-life (3–6 h) and a low oral bioavailability (60%) due to the first pass metabolism (18,19). When ODS is used for CINV management in cancer patients, the current formulations might show some limitations. After oral administration, the drug has a tendency to be discharged by vomiting, which considerably reduces the therapeutic effects (20). Following intravenous administration, side effects such as headache, constipation, and diarrhea are prominent as a result of the rapidly high drug level in the blood (21). In addition, patients have to repeat the administration 2–3 times a day for several days until the end of chemotherapy (18). Although ODS does not belong to the drugs with a narrow therapeutic window and the side effects caused by IV administration may be not serious, the repetition of the administration together with the side effects could cause discomforts and tiredness to the patients, who are under chemotherapy. The sustained-release of the drug could reduce the side effects caused by the conventional IV injection as well as the dosing frequency, which help to improve patient compliance. Therefore, sustained-release parenteral formulations could also be an attractive candidate to solve the recent limitations.
Previously, to overcome these drawbacks, ODS was formulated into an intranasal drug delivery system with an increased permeation rate and a prolonged nasal residence time (22). Transdermal patch containing ODS was prepared to reduce side effects and provide drug sustained-release (23). ODS-loaded SLNs/NLCs were also prepared as sustained-release formulations for nose-to-brain delivery (10,24). Nasal delivery is a potential approach for certain drugs like ODS. However, it remains some limitations, including the mucociliary clearance and the irritation of the nasal mucosa (25). The low drug retention time was solved to some extent in the previous studies, but the drug was still needed the frequent administration (10,22). ODS-loaded SLNs/NLCs that previously fabricated (10,24) could be used as sustained-release parenteral formulations. They would be potential alternatives to the intranasal formulations. However, SLNs/NLCs produced in these studies showed a low entrapment efficiency (EE) of 16.0–56.6% and a limited drug loading (DL) due to the hydrophilicity of ODS. Generally, SLNs loaded with water-soluble molecules were associated with the drug leakage to the outer phase during the fabrication process, resulted in a low DL and EE (16,26). In the present study, ODS-loaded NLCs were developed with the improved EE and DL by assessing various variables of the preparation process and formulation. Solvent injection method was selected for the NLCs production. The optimized formulation was aimed to parenteral delivery for prolonging blood circulation of the drug, which could reduce side effects and improve patient compliance.
Materials and Methods
Materials
ODS was obtained from Cadila Pharmaceuticals (India). Tripalmitin was supplied by Tokyo Chemical Industry (Japan). Phosal® 53MCT (Soybean lecithin 53% and medium-chain triglycerides) was received from Phospholipid GmbH (Germany). Polysorbate 80 was purchased from Sigma-Aldrich (MO, USA). The HPLC grade water and acetonitrile were purchased from Avantor (PA, USA). The Milli-Q® purification system (Millipore, MA, USA) was used to de-ionize and purify water. Other analytical grade reagents were used without further purifications.
Preparation of NLCs
ODS-loaded NLCs were prepared using the solvent injection method as reported previously (12,16). Briefly, the organic phase was prepared by dissolving tripalmitin, Phosal® 53 MCT, and the drug (ODS) in ethanol at 70 ± 2°C. The aqueous phase consisting of polysorbate 80 in different pH buffers was pre-heated to a predetermined temperature. The organic phase was rapidly injected into the aqueous phase using a hypodermic needle (26-gauge) under magnetic stirring at 600 rpm. The mixture was continuously stirred at 4 ± 2°C (30 min) for solidification of NLCs followed by sonication for varying time (2–6 min). Suspension NLCs was lyophilized using a freeze-drier (8508, Ilshin, South Korea) and stored in refrigerator for further investigations.
Evaluation of Parameters of Formulation and Process on ODS-Loaded NLCs Properties
The effects of various parameters of formulation and process on NLCs properties were evaluated. Each parameter was varied, whereas others were kept constant. The NLCs were characterized for the particle size, PDI, EE, and DL. The preparation process and formulation variables included the pH of the aqueous phase (2.0 to 12.0), the temperature of the aqueous phase (20 to 70°C) the ratio of aqueous phase volume (Va) and organic phase volume (Vo) (Va/Vo = 5/1 to 20/1), the total lipid concentration in organic phase (20–80 mg/ml), the emulsifier concentration (0–1% w/v), the initial drug added (6.3–16.7% of the total lipid added), and the sonication time (0–6 min). Effect of liquid lipid on NLCs was also investigated by altering the liquid lipid level in the solid lipid-liquid lipid mixture (0–50%).
Analysis of ODS Using HPLC Method
An HPLC method for ODS quantification was developed with some modifications from a previous report (23) and validated according to the Q2(R1) ICH guideline. The HPLC system (1260 Infinity, Agilent Technologies, CA, USA) consisted of a quaternary pump, an autosampler, and a UV detector. The Phenomenex® C18 column (5 μm particle size, 250 × 4.6 mm, Phenomenex, CA, USA) was used with the mobile phase of acetonitrile/0.02 M acetate buffer pH 4.8 (40/60, V/V). The system was operated with a flow rate of 1.0 ml/min, an injection volume of 10 μl, and a detection wavelength of 310 nm. The chromatographic peaks were quantified using the OpenLAB Chromatography Data System version A.02.10 (Agilent Technologies, CA, USA).
Drug Solubility in Water and Buffer Solutions
ODS solubility in water and buffer solutions (pH 2.0–12.0, 0.05 M) was determined using a previously reported method (27). An excessive amount of ODS was added to water and buffer solutions in glass vials, which were sealed and shaken at 25 ± 0.5°C under a speed of 50 rpm for 48 h using a shaking water bath (HST 205 SW, Hanbaek ST, South Korea). The suspensions were filtered through 0.45 μm membranes and adequately diluted with the mobile phase prior to the HPLC analysis.
Characterization of NLCs
Particle Size, PDI, and Zeta Potential
The z-average particle size and PDI of NLCs were determined by dynamic light scattering (DLS) using a particle size analyzer (Zetasizer Nano-S90, Malvern Instruments, UK). The NLCs suspension was diluted with distilled water (10 times) and the size measurement was performed at 25°C using 90-degree scattering optics. Zeta potential of NLCs was measured using the zeta potential and particle size analyzer (ELSZ-1000, Photal Otsuka Electronics, Japan) based on the DLS method at 25°C and a fixed detector angle of 90°.
Determination of EE and DL
The total drug amount in the suspension was determined by dissolving the sample in methanol at 60°C. It was appropriately diluted with the mobile phase and filtered through 0.45 μm membranes prior to the HPLC analysis. For the determination of the free drug amount, the NLCs suspension was added into a centrifugal filter unit (MWCO 10,000, Amicon® Ultra, Millipore, MA, USA) and centrifuged for 1 h at 14,000 rpm. After centrifugation, the aqueous phase containing free drug was collected and appropriately diluted with the mobile phase prior to the HPLC analysis. The drug loaded amount was determined indirectly by subtracting the free drug amount from the total drug amount in the suspension (16,28). The EE and DL were calculated using the following equations:
Differential Scanning Calorimetry (DSC)
DSC analyses were performed using the DSC 8000 (PerkinElmer, MA, USA). An exact amount of samples (1–5 mg) were placed in aluminum pans with covers and heated over a temperature range of 30°C to 250°C with a rate of 20°C/min using the Pyris software 11.1.1. The nitrogen purge was 20 ml/min.
X-Ray Powder Diffraction (XRD)
XRD patterns were obtained using an X-ray diffractometer (MiniFlex600, Rigaku, Japan) at room temperature with Ni-filtered CuKα radiation at 15 mA and 40 kV. The 2θ diffraction angle was 3° - 90° with a scanning rate of 2°/min and a step width of 0.02°.
Fourier-Transform Infrared Spectroscopy (FTIR)
FTIR spectra were recorded using an FTIR spectrophotometer (Tensor 27, Bruker, MA, USA). The samples were scanned 16 times in the range of 500–4000 cm−1 with a resolution of 4 cm−1.
Transmission Electron Microscopy (TEM)
After preparation, approximately 20 μL of the NLCs suspension was dropped onto copper grids. The grids were left for drying overnight at room temperature. TEM images were obtained using a Tecnai G2 F30 transmission electron microscope (FEI, OR, USA) at an accelerating voltage of 300 kV.
In Vitro Drug Release
In vitro drug release studies were carried out using the dialysis bag method as reported previously (6,13) with some modifications. Lyophilized NLCs were re-suspended in phosphate saline buffer (pH 7.4) at a concentration equivalent to ODS 4 mg/ml. NLCs suspension (1 ml) was added into a dialysis bag (MWCO 3.5 kDa, Spectra/Por, Spectrum Laboratories, CA, USA), which was then fixed to a paddle of a dissolution tester (DST-810, Labfine, South Korea). The dissolution vessels were filled with 200 ml phosphate buffer (pH 7.4, 0.02 M) containing 0.2% polysorbate 80. The drug release studies were performed at 37 ± 0.5°C with the paddles’ rotating speed of 50 rpm. Samples were withdrawn at predetermined intervals, appropriately diluted with the mobile phase and quantified using the HPLC method. The drug release was expressed as a percentage of cumulative ODS released.
Pharmacokinetic Study
The animal study was approved by the Animal Care and Use Committee and carried out following the standard operating procedures of the Animal Care and Use Guidelines (Gachon University). Sprague-Dawley male rats (8–9 weeks, 250–300 g, Nara Biotech, South Korea) were allowed to adjust to the laboratory environment for 1 week before the experiment and maintained under 12-h light/dark cycles. All the rats were randomly assigned into two groups (n = 5). The control group received ODS solution in saline at a dose of 2 mg/kg, prepared aseptically by dissolving ODS in saline solution. The test group received the optimized ODS-loaded NLCs formulation at a dose of 4 mg/kg, which was prepared by re-suspending the lyophilized NLCs in phosphate saline buffer (pH 7.4). The drug was administered by SC injection using 26-gauge needles. At 0, 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, and 96 h after SC administration, blood samples (0.25 ml) were collected from tail veins and centrifuged at 14,000 rpm for 15 min at 4°C. The plasma was collected and stored at −20°C until analysis.
For the drug analysis, granisetron in acetonitrile solution (20 ng/ml) was used as internal standard (IS). Plasma samples were left at 4°C for liquefying. After that, 100 μL IS were added to a microtube containing 50 μL plasma samples for deproteinization. The tubes were then vortex-mixed in 1 min and centrifuged at 14,000 rpm for 15 min at 4°C. The supernatants were collected and injected for LC-MS/MS analysis.
ODS concentration in rat plasma was determined using a developed LC-MS/MS method with some modifications from previously reported methods (29,30). The LC-MS/MS system (Agilent Technologies, CA, USA) consisted of an electrospray tandem triple quadrupole mass spectrometer (Agilent 6490 QQQ) coupled with an HPLC system (Agilent 1290 Infinity). The plasma samples were separated on a reverse-phase column (Synergi 4 μm polar-RP 80A, 150 × 2.0 mm, Phenomenex, CA, USA) equipped with a guard-column (SecurityGuard 4.0 × 3.0 mm, Phenomenex, CA, USA). The column temperature was maintained at 30°C. The mobile phase was a mixture of 0.1% formic acid and acetonitrile (40:60, V/V) with a flow rate of 0.2 ml/min under the isocratic condition for 6-min. The temperature of the autosampler was 4°C and the injection volume was 2 μL. Quantification was performed using multiple reaction monitoring (MRM). The electrospray ionization (ESI) source was operated in positive mode. The MRM transitions of m/z 294.1 → 170.0 and m/z 313.1 → 138.1 were applied for ODS and IS, respectively. Optimization of collision energy for ODS and IS was 28 and 22 eV, respectively. The cell accelerator voltages were 7 and 5 V for ODS and IS, respectively. Data acquisition and integration were performed using the Mass Hunter version A.06.00 software (Agilent Technologies, CA, USA).
For descriptive pharmacokinetic analysis, pharmacokinetic parameters including the peak plasma concentration (Cmax) and the time to reach Cmax (Tmax) were directly defined from individual ODS plasma concentration-time profiles. Other parameters including the half-life (t1/2), the mean residence time (MRT), the area under the plasma concentration-time curve from time zero to the last time point (AUClast) and to infinity (AUCinf) were calculated by the non-compartmental analysis using WinNonlin 5.0.1 (Pharsight, NC, USA).
Statistics
All experiments were carried out in triplicate except the pharmacokinetic studies. The data were presented as the mean values with standard deviations (SDs). Student’s t test was performed to analyze the data and a value of p < 0.05 was considered statistically significant.
Results and Discussion
Formulation of NLCs Using Solvent Injection Method
ODS is a water-soluble drug with the water solubility of 29.5 ± 1.0 mg/ml. The most popular procedure to incorporate water-soluble compounds like ODS to NLCs was double-emulsion method (31). Besides, other techniques such as cold high-pressure homogenization, solvent emulsification-evaporation, and solvent emulsification-diffusion were also used (32). Our study employed the solvent injection method with ethanol as the water-miscible solvent to prepare ODS-loaded NLCs. In this method, following the rapid injection of the organic phase containing ODS and lipids, the solvent quickly diffused across the interface into the aqueous phase, resulting in the formation of droplets. The original droplets were broken into smaller ones as a result of the interfacial pulsation and turbulence during solvent diffusion (12). Consequently, the lipid precipitated, and NLCs were formed and stabilized with an emulsifier. The diffusion rate of the organic solvent into the aqueous phase is a critical factor affecting the particle size and the size distribution (12). During the process, the leakage of ODS to the aqueous phase may occur due to its good water solubility, resulting in the low EE and DL. Certain studies have evaluated the effects of several parameters of the solvent injection method on particle size and polydispersity index (PDI) of NLCs (12,13). However, their effects on EE and DL were not considerably studied. Therefore, parameters of formulation and preparation process were evaluated in this study to improve EE and DL.
Effect of pH of the Aqueous Phase on NLCs
As shown in Table I, the particle size of the NLCs ranged between 168 and 188 nm with PDI of 0.185–0.214 at pH from 2.0 to 7.4. The NLCs prepared using distilled water as the aqueous phase showed a similar particle size (171.4 ± 0.8 nm) and PDI (0.219 ± 0.04). As alkaline buffer solutions (pH from 8.0 to 12.0) were used as the aqueous phase, the size and PDI increased significantly. Increasing pH of the aqueous phase from 2.0 to 7.4 resulted in significant increases in EE and DL, particularly EE reached 93.2 ± 0.5% at pH 7.4. Further increases in the pH of the aqueous phase did not show any significant improvement in EE and DL. These findings were most likely due to the ODS solubility. As shown in Table I, ODS has a pH-dependent solubility. The decrease in solubility of ODS upon the increase in pH of the aqueous phase resulted in the reduction of ODS leakage and consequently improved EE and DL. These findings were consistent with a previous report, which pointed that EE of an anti-proliferative agent increased from 28.2% to 84.3% in SLNs preparation when the aqueous pH increased from 6.5 to 8.4 (33). Another study also reported that by adjusting the pH of the aqueous phase from 5.8 to 9.3, EE of procaine hydrochloride increased from 11.0 to 58.2% (34). The low solubility of ODS in pH >7.4 resulted in similar EE and DL among the corresponding NLCs. Therefore, ODS solubility in the aqueous phase reversely affected EE and DL and was the key to improve them. The aqueous phase with pH 7.4 was chosen for further investigations.
Effect of Temperature of the Aqueous Phase on NLCs
Effect of temperature of the aqueous phase on ODS-loaded NLCs was shown in Table II. The appropriate temperature for NLCs preparation was 30–40°C, which resulted in NLCs with the lowest particle size and PDI. The higher particle size and PDI at a lower temperature (20°C) could be due to the rapid precipitation of lipids. At higher temperatures (50–70°C), lipids in the semisolid or melting state could easily coalesce together to create larger droplets, which explained for the large particle size and the high PDI. EE and DL decreased as the temperature increased. It could be attributed to the increase in drug solubility at high temperature leading to an increase in the drug leakage to the aqueous phase. In addition, at low temperatures, the lipids rapidly precipitated at the interface between the droplets and the aqueous medium, which prevented the leakage of the drug (35). Further studies were conducted with the aqueous phase maintained at 30°C.
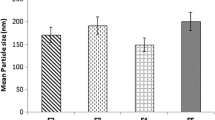
Effect of Va/Vo Ratio on NLCs
As shown in Fig. 1(a), decreasing Va/Vo ratio resulted in increases in particle size and PDI, which could be due to the reduction in the diffusion rate of the organic solvent. In addition, a low Va/Vo ratio decreased drug migration to the aqueous phase, leading to increases in EE and DL (Table S1). A high Va/Vo ratio was favorable to NLCs with respect to the particle size and distribution, but it was associated with the low EE and DL as well as the use of a high amount of water and surfactant (2). Hence, the Va/Vo ratio was a crucial factor affecting the NLCs. It should be small to improve EE and DL but not lower than a critical value to achieve small and narrow-distributed NLCs (12). Considering particle size, PDI, EE, and DL, the Va/Vo ratio of 10/1 was chosen for further investigations.

Effect of volume of aqueous phase/ volume of organic phase (Va/Vo) ratio (a) and total lipid concentration (b) on NLCs.
Effect of Total Lipid Concentration in Organic Phase on NLCs
As shown in Fig. 1(b), the particle size increased as total lipid concentration in the organic phase increased, which was consistent with previous reports (24,36). As the total lipid concentration varied from 20 to 60 mg/ml, the PDI was not considerably affected, but its further rise to 80 mg/ml significantly increased the PDI. These results could be attributed to the increase in viscosity of the organic phase at higher lipid concentrations, which consequently reduced the breaking of droplets into smaller ones (12). The rapid precipitation of lipid at higher concentrations also prevented drug leakage. In addition, since the drug/total lipid ratio was constant (12.5%), the initial drug input increased along with the total lipid, which consequently led to an increase in EE and DL (Table S2). Therefore, the total lipid concentration played a critical role in NLCs preparation. In this study, compared with the highest lipid concentration (80 mg/ml), the concentration of 60 mg/ml showed a slight reduction in EE and DL, whereas the particle size and the PDI significantly decreased. Hence, the lipid concentration of 60 mg/ml was chosen for further studies.
Effect of Emulsifier Concentration on NLCs
NLCs prepared without emulsifier exhibited a considerable large size and a high PDI (Table III). The addition of polysorbate 80 resulted in significant decreases in particle size and PDI, indicating the critical role of the emulsifier in the preparation of NLCs. It reduced the interfacial surface tension, resulting in the formation of initially smaller solvent droplets at the injection site (12,13). In addition, the emulsifier contributed to the stability of the droplets and prevented them from coalescing (24). Increasing polysorbate 80 concentration from 0.1% to 0.5% did not significantly affect NLCs particle size and PDI. However, its further increase to 1% resulted in a significant rise in PDI, which was possibly due to the irregular size reduction of the droplets (13,15,37). The emulsifier concentration did not considerably affect EE and DL. Therefore, in this study, polysorbate 80 could be used at a concentration of 0.1–0.5% to achieve small particles with a narrow size distribution. Further studies were conducted using the lowest concentration of polysorbate 80 (0.1%).
Effect of Initial Drug Added on NLCs
Increasing the initial drug from 6.3% to 12.5% resulted in negligible increases in particle size and PDI as shown in Table S3. However, its further increase to 16.7% led to the larger particle size and the higher PDI. These findings suggested the saturation of the drug in the lipid matrix that may cause the particle aggregation (13). More drug molecules were entrapped into the lipid matrix upon increasing the initial drug, which could contribute to the increase in DL. The EE also increased when the initial drug varied from 6.3% to 12.5%. However, a further rise in the initial drug resulted in a decrease in EE, which could be due to the higher amount of drug escaped to the aqueous phase (28). The initial drug of 12.5% was selected for further studies.
Effect of Sonication Time on NLCs
As shown in Table IV, slight decreases in particle size and PDI were observed when sonication was used. Upon increasing the sonication time from 2 to 6 min, the particle size was unchanged. However, long sonication time (6 min) resulted in a slight increase in the PDI, possibly due to the irregular size decrease (13). It was found that EE and DL were not affected by the sonication. Therefore, the sonication time was a minor factor influencing NLCs with respect to particle size, size distribution, EE, and DL.
Effect of the Liquid Lipid on NLCs
The optimized parameters of process and formulation were fixed based on the above results: the aqueous phase (pH 7.4) with 0.1% polysorbate 80 preheated to 30°C, the Va/Vo ratio of 10/1, the total lipid concentration of 60 mg/ml, the initial drug added of 12.5%, and the sonication time of 4 min. To investigate the effect of the liquid lipid on NLCs, four NLCs formulations were prepared (Table V). As the liquid lipid amount increased from 0 to 40%, significant decreases in particle size and PDI were observed. It could be attributed to the surfactant action of soybean lecithin (a component of the liquid lipid) during NLCs preparation (10). In addition, the presence of medium-chain triglycerides in the liquid lipid may reduce the viscosity of the lipid blend, increase the diffusion rate of ethanol, and consequently contribute to the formation of smaller droplets (1,38). However, a further increase in liquid lipid to 50% resulted in the larger particle size and the higher PDI. It was possibly due to the formation of mixed micelles, liposomes, and other structures when an excess of the liquid lipid leaked to the aqueous phase (13,28). EE and DL increased with the liquid lipid level, which could be attributed to the presence of a lipid mixture with different chain lengths and structures. This lipid matrix was possibly a disorganized structure with many imperfections, which could offer more space for drug molecules (1,39). The optimized ODS-loaded NLCs was F3 with 40% of liquid lipid.
Characterization of NLCs
The zeta potential of F1, F2, and F3 was approximate −27 mV (Table V), which provided repulsive interaction among the nanoparticles. The negative charge could be attributed to the ionization of tripalmitin molecules (31). Zeta potential of nanoparticles provides information about their surface properties. In general, colloidal dispersions with a high zeta potential (absolute value >30 mV) are considered physically stable. However, the presence of stabilizers could improve the stability of the dispersion although the zeta potential value was not high (1,37). The TEM image of F3 (Fig. 2a) showed the spherical particle with a diameter around 200 nm, which was in agreement with the particle size analysis. The size distribution (Fig. 2b) indicated the monodispersity of the ODS-loaded NLCs.

Characterization of NLCs. (a) Representative TEM image of F3, (b) Size distribution of F3, (c) FTIR spectra of ODS, tripalmitin, Phosal 53® MCT, and lyophilized F3, (d) DSC thermograms of ODS, tripalmitin, and lyophilized F3, (e) XRD patterns of tripalmitin, ODS, and lyophilized F3.
Figure 2c showed the FTIR spectrum of ODS with peaks at 3481, 3409 cm−1 (N–H stretching), and 1636 cm−1 (C=O stretching) (40). Absorption bands of tripalmitin were found at 2914 cm−1 (C-H stretching), 1735 cm−1 (C=O stretching), and 1177 cm−1 (C-O deformation) (41). The FTIR spectrum of Phosal® 53MCT exhibited peaks of lecithin at 1738 cm−1 (C=O stretching), 1377 and 1243 cm−1 (C-O stretching). In the FTIR spectrum of F3, peaks corresponding to N-H stretching of ODS were shifted and broadened, which could be due to the electrostatic interaction and hydrogen bonding between PO2− of lecithin and N-H of ODS (42). No loss of other functional characteristic peaks or significant peak shift was observed, suggesting the absence of chemical interaction between ODS and lipids.
The DSC thermogram of ODS (Fig. 2d) showed a sharp endothermic peak at its melting point (184.5°C), which suggested the crystalline nature of the drug. A peak at 64.1°C was observed in the thermogram of tripalmitin, which was the fusion point of the β form of this lipid (41). The ODS endothermic peak was disappeared in the DSC thermogram of F3, likely due to the entrapment of ODS within the lipid matrix and its transformation to the amorphous state. Furthermore, the endothermic peak of tripalmitin was broadened and slightly shifted, suggesting the formation of lipid amorphous regions where ODS might be molecularly dispersed (24).
As shown in Fig. 2e, the XRD pattern of tripalmitin exhibited a crystalline diffractogram with numerous intense peaks at 4.4, 6.6, 7.4, 13.1, 17.5, and 24.1°. Pure ODS showed peaks at 12.3, 16.9, 23.1, 24.1, and 27.9° in its XRD pattern, suggesting the high crystallinity of the drug (40). In the diffractogram of F3, all the characteristic peaks of tripalmitin and ODS disappeared or reduced their intensity. These findings were consistent with the DSC results, confirming the amorphous state of ODS in the NLCs. A small proportion of tripalmitin and ODS may be still in original crystalline form as demonstrated by several low-intensity peaks.
In Vitro Drug Release
As shown in Fig. 3, F1 and F2 exhibited similar release patterns while F3 showed a remarkable improvement in drug sustained-release. It indicated that an appropriate liquid lipid level in NLCs could modify the drug release profile. A proper ratio of the two lipids could form a matrix with less crystallinity to accommodate more drugs and provide the drug sustained-release (39). The initial burst release of F3 may be due to the release of free drug, drug adsorbed on the NLCs surface and drug located underneath the NLCs stratum (36,37). The slower and sustained release phase was attributed to the diffusion of drug entrapped within the imperfect lipid matrix (10,15,24). Despite the high water solubility of ODS, the existence of lipid matrix could hinder the drug release from the NLCs, demonstrating the advantage of drug encapsulation in NLCs (37).

Drug release profiles of F1, F2, and F3 (Mean ± SD, n = 3). * Significantly different compared with F1 and F2 (p < 0.05).
Pharmacokinetic Studies
Parenteral delivery of SLNs/NLCs loaded with drugs has been studied following IV (43) or SC injection (44). To evaluate the advantages of NLCs in prolongation of the drug-release, both IV and SC administration could be used. In the case of the SC administration, the depot effect offered a new perspective to prolong the drug detainment in blood circulation (45). It could help to increase the sustained-release characteristics of the NLCs. The advantages of NLCs in increasing the circulation time could be proved through the comparison between the control solution and the optimized formulation following the same administration route (SC injection). Therefore, in this study, the pharmacokinetic evaluation was conducted following SC administration in rats. The control group received a dose of 2 mg/kg of ODS. Since the optimized NLCs formulation (F3) was expected to prolong the drug release for 2–4 days, the test group received a dose of 4 mg/kg.
The average ODS plasma concentration-time profiles in rats are presented in Fig. 4 and the pharmacokinetic parameters are summarized in Table VI. The plasma concentration of ODS in the control group was initially elevated and quickly dropped to the baseline level within 12 h with a Tmax of 0.50 h. The MRT was relatively short (3.03 h). It was due to the rapid distribution and absorption of the drug. In contrast, F3 exhibited a sustained-release characteristic with the presence of ODS in blood until 96 h. The Tmax extended to 1.80 h and the Cmax significantly decreased compared to that of the control group despite the use of a 2-time higher dose. The MRT was increased to 43.3 h by about 14.3-fold, compared with that of the control group. Furthermore, the bioavailability of F3 expressed as AUCinf/Dose increased nearly 271% in comparison with that of the control group. The pharmacokinetic results successfully demonstrated that the optimized NLCs formulation could prolong the release of ODS following SC administration in vivo. The rapid release phase of ODS within 24 h could be due to the initial burst release of the drug distributed on the NLCs surface and underneath the NLCs stratum (36,37). The prolonged release phase was probably attributed to the slow diffusion of the drug from the lipid matrix into the blood and the slow passage and distribution of NLCs through biological barriers (44). For sustained-release products, the rapid release phase could sufficiently ensure the quick achievement of the therapeutic drug concentration in the blood. In this phase, the ODS release was adequately controlled as evidenced by its concentration in the blood within the first 24 h without dramatic fluctuations. Compared to the control solution, the optimized formulation could considerably reduce the high drug level in the blood at the early state. Therefore, it could reduce side effects that usually occur in the case of conventional parenteral formulations due to the high elevation of drug concentration in blood (21). The sustained-release phase of ODS-loaded NLCs is desirable to prolong the drug action, reduce dosing frequency, and thereby enhance patient compliance. The target tissue (brain) distribution profiles of ODS were not investigated in the present study and remained one of the limitations. They would be further evaluated to provide more evidence proving the advantages of the optimized formulation.

ODS plasma concentration-time profiles obtained following SC administration of ODS solution (control, 2 mg/kg) and the optimized NLCs (F3, 4 mg/kg) to rats (Mean ± SD, n = 5). * Significantly different compared with control group (p < 0.05).
Conclusions
ODS was successfully incorporated into NLCs using solvent injection method with the enhanced drug incorporation. The pH of the aqueous phase was the most critical variable that could increase EE to >90%. ODS-loaded NLCs showed an excellent sustained-release characteristic in vitro and in vivo. The findings of this study suggest the value of the optimized formulation for clinical applications. It would be potential for improving CINV management by prolongation of drug action and reduction in dosing frequency.
Abbreviations
- CINV:
-
Chemotherapy-induced nausea and vomiting
- DL:
-
Drug loading
- DSC:
-
Differential scanning calorimetry
- EE:
-
Entrapment efficiency
- FTIR:
-
Fourier transform-infrared spectroscopy
- HPLC:
-
High performance liquid chromatography
- IS:
-
Internal standard
- LC-MS/MS:
-
Liquid chromatography – tandem mass spectroscopy
- MRM:
-
Multiple reaction monitoring
- MRT:
-
Mean residence time
- MWCO:
-
Molecular weight cut-off
- NLCs:
-
Nanostructured lipid carriers
- ODS:
-
Ondansetron hydrochloride
- SLNs:
-
Solid lipid nanoparticles
- TEM:
-
Transmission electron microscopy
- XRD:
-
X-ray powder diffraction
References
de Sousa Marcial SP, Carneiro G, Leite EA. Lipid-based nanoparticles as drug delivery system for paclitaxel in breast cancer treatment. J Nanopart Res. 2017;19(10):340.
Wissing SA, Kayser O, Müller RH. Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev. 2004;56(9):1257–72.
Tran TH, Choi JY, Ramasamy T, Truong DH, Nguyen CN, Choi H-G, et al. Hyaluronic acid-coated solid lipid nanoparticles for targeted delivery of vorinostat to CD44 overexpressing cancer cells. Carbohydr Polym. 2014;114:407–15.
Ramasamy T, Khandasami US, Ruttala H, Shanmugam S. Development of solid lipid nanoparticles enriched hydrogels for topical delivery of anti-fungal agent. Macromol Res. 2012;20(7):682–92.
Tran TH, Ramasamy T, Truong DH, Choi H-G, Yong CS, Kim JO. Preparation and characterization of Fenofibrate-loaded nanostructured lipid carriers for Oral bioavailability enhancement. AAPS PharmSciTech. 2014;15(6):1509–15.
Gupta B, Ramasamy T, Poudel BK, Pathak S, Regmi S, Choi JY, et al. Development of bioactive PEGylated nanostructured platforms for sequential delivery of doxorubicin and Imatinib to overcome drug resistance in metastatic tumors. ACS Appl Mater Interfaces. 2017;9(11):9280–90.
Ramasamy T, Ruttala HB, Gupta B, Poudel BK, Choi H-G, Yong CS, et al. Smart chemistry-based nanosized drug delivery systems for systemic applications: a comprehensive review. J Control Release. 2017;258:226–53.
Müller RH, Radtke M, Wissing SA. Nanostructured lipid matrices for improved microencapsulation of drugs. Int J Pharm. 2002;242(1):121–8.
Li Q, Cai T, Huang Y, Xia X, Cole SPC, Cai Y. A review of the structure, preparation, and application of NLCs, PNPs, and PLNs. Nanomaterials. 2017;7(6):122.
Devkar TB, Tekade AR, Khandelwal KR. Surface engineered nanostructured lipid carriers for efficient nose to brain delivery of ondansetron HCl using Delonix regia gum as a natural mucoadhesive polymer. Colloids Surf B: Biointerfaces. 2014;122(Supplement C:143–50.
Iqbal MA, Md S, Sahni JK, Baboota S, Dang S, Ali J. Nanostructured lipid carriers system: recent advances in drug delivery. J Drug Target. 2012;20(10):813–30.
Schubert MA, Müller-Goymann CC. Solvent injection as a new approach for manufacturing lipid nanoparticles – evaluation of the method and process parameters. Eur J Pharm Biopharm. 2003;55(1):125–31.
Jain S, Jain S, Khare P, Gulbake A, Bansal D, Jain SK. Design and development of solid lipid nanoparticles for topical delivery of an anti-fungal agent. Drug Deliv. 2010;17(6):443–51.
Arıca Yegin B, Benoît J-P, Lamprecht A. Paclitaxel-loaded lipid nanoparticles prepared by solvent injection or ultrasound emulsification. Drug Dev Ind Pharm. 2006;32(9):1089–94.
Pandita D, Ahuja A, Velpandian T, Lather V, Dutta T, Khar R. Characterization and in vitro assessment of paclitaxel loaded lipid nanoparticles formulated using modified solvent injection technique. Pharmazie. 2009;64(5):301–10.
Wang T, Wang N, Zhang Y, Shen W, Gao X, Li T. Solvent injection-lyophilization of tert-butyl alcohol/water cosolvent systems for the preparation of drug-loaded solid lipid nanoparticles. Colloids Surf B: Biointerfaces. 2010;79(1):254–61.
Christofaki M, Papaioannou A. Ondansetron: a review of pharmacokinetics and clinical experience in postoperative nausea and vomiting. Expert Opin Drug Metab Toxicol. 2014;10(3):437–44.
Ye JH, Ponnudurai R, Schaefer R. Ondansetron: a selective 5-HT3 receptor antagonist and its applications in CNS-related disorders. CNS Drug Rev. 2001;7(2):199–213.
Figg WD, Dukes GE, Pritchard JF, Hermann DJ, Lesesne HR, Carson SW, et al. Pharmacokinetics of ondansetron in patients with hepatic insufficiency. J Clin Pharmacol. 1996;36(3):206–15.
Roila F, Del Favero A. Ondansetron clinical pharmacokinetics. Clin Pharmacokinet. 1995;29(2):95–109.
Cho E, Gwak H, Chun I. Formulation and evaluation of ondansetron nasal delivery systems. Int J Pharm. 2008;349(1):101–7.
Mahajan HS, Gattani SG. Nasal administration of ondansetron using a novel microspheres delivery system part II: ex vivo and in vivo studies. Pharm Dev Technol. 2010;15(6):653–7.
Cho J-R, Van Duong A, Nguyen LTT, Chi S-C. Design of transdermal matrix patch containing ondansetron. J Pharm Investig. 2016;46(7):677–84.
Joshi AS, Patel HS, Belgamwar VS, Agrawal A, Tekade AR. Solid lipid nanoparticles of ondansetron HCl for intranasal delivery: development, optimization and evaluation. J Mater Sci Mater Med. 2012;23(9):2163–75.
Agrawal M, Saraf S, Saraf S, Antimisiaris SG, Chougule MB, Shoyele SA, et al. Nose-to-brain drug delivery: an update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J Control Release. 2018;281:139–77.
Becker Peres L, Becker Peres L, de Araújo PHH, Sayer C. Solid lipid nanoparticles for encapsulation of hydrophilic drugs by an organic solvent free double emulsion technique. Colloids Surf B: Biointerfaces. 2016;140:317–23.
Higuchi T, Connors K. Phase-solubility techniques. In: Reilly C, editor. Advances in analytical chemistry and instrumentation, vol. 4. NewYork: Wiley-Interscience; 1965. p. 117–212.
Liu D, Jiang S, Shen H, Qin S, Liu J, Zhang Q, et al. Diclofenac sodium-loaded solid lipid nanoparticles prepared by emulsion/solvent evaporation method. J Nanopart Res. 2011;13(6):2375–86.
Liu K, Dai X, Zhong D, Chen X. Quantitative determination of ondansetron in human plasma by enantioselective liquid chromatography-tandem mass spectrometry. J Chromatogr B. 2008;864(1):129–36.
Zhu Y, Zhang Q, Zou J, Wan M, Zhao Z, Zhu J. Pharmacokinetics and bioavailability study of two ondansetron oral soluble film formulations in fasting healthy male Chinese volunteers. Drug Des Dev Ther. 2015;9:4621.
Garcia-Fuentes M, Torres D, Alonso MJ. Design of lipid nanoparticles for the oral delivery of hydrophilic macromolecules. Colloids Surf B: Biointerfaces. 2003;27(2):159–68.
Almeida AJ, Souto E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv Drug Deliv Rev. 2007;59(6):478–90.
Song CX, Labhasetwar V, Murphy H, Qu X, Humphrey WR, Shebuski RJ, et al. Formulation and characterization of biodegradable nanoparticles for intravascular local drug delivery. J Control Release. 1997;43(2):197–212.
Govender T, Stolnik S, Garnett MC, Illum L, Davis SS. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J Control Release. 1999;57(2):171–85.
Hu FQ, Hong Y, Yuan H. Preparation and characterization of solid lipid nanoparticles containing peptide. Int J Pharm. 2004;273(1):29–35.
Vaghasiya H, Kumar A, Sawant K. Development of solid lipid nanoparticles based controlled release system for topical delivery of terbinafine hydrochloride. Eur J Pharm Sci. 2013;49(2):311–22.
Jain AK, Jain A, Garg NK, Agarwal A, Jain A, Jain SA, et al. Adapalene loaded solid lipid nanoparticles gel: an effective approach for acne treatment. Colloids Surf B: Biointerfaces. 2014;121:222–9.
Tiwari R, Pathak K. Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: comparative analysis of characteristics, pharmacokinetics and tissue uptake. Int J Pharm. 2011;415(1):232–43.
Kalam MA, Sultana Y, Ali A, Aqil M, Mishra AK, Chuttani K. Preparation, characterization, and evaluation of gatifloxacin loaded solid lipid nanoparticles as colloidal ocular drug delivery system. J Drug Target. 2010;18(3):191–204.
Park J-M, Park S-J. Preparation and characterization of water-soluble microcapsule for sustained drug release using Eudragit RS 100. Macromol Res. 2010;18(12):1191–4.
de Oliveira JL, Campos EVR, Gonçalves da Silva CM, Pasquoto T, Lima R, Fraceto LF. Solid lipid nanoparticles co-loaded with simazine and atrazine: preparation, characterization, and evaluation of herbicidal activity. J Agric Food Chem. 2015;63(2):422–32.
Patro NM, Devi K, Pai RS, Suresh S. Evaluation of bioavailability, efficacy, and safety profile of doxorubicin-loaded solid lipid nanoparticles. J Nanopart Res. 2013;15(12):2124.
Jia L, Zhang D, Li Z, Duan C, Wang Y, Feng F, et al. Nanostructured lipid carriers for parenteral delivery of silybin: biodistribution and pharmacokinetic studies. Colloids Surf B: Biointerfaces. 2010;80(2):213–8.
Han C, Qi CM, Zhao BK, Cao J, Xie SY, Wang SL, et al. Hydrogenated castor oil nanoparticles as carriers for the subcutaneous administration of tilmicosin: in vitro and in vivo studies. J Vet Pharmacol Ther. 2009;32(2):116–23.
Chen S, Tam YYC, Lin PJC, Leung AKK, Tam YK, Cullis PR. Development of lipid nanoparticle formulations of siRNA for hepatocyte gene silencing following subcutaneous administration. J Control Release. 2014;196:106–12.
Acknowledgments and disclosures
All authors declare that they have no conflict of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 29 kb)
Rights and permissions
About this article

Cite this article
Duong, VA., Nguyen, TTL., Maeng, HJ. et al. Preparation of Ondansetron Hydrochloride-Loaded Nanostructured Lipid Carriers Using Solvent Injection Method for Enhancement of Pharmacokinetic Properties. Pharm Res 36, 138 (2019). https://doi.org/10.1007/s11095-019-2672-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11095-019-2672-x