
Abstract
Purpose
siRNA may be delivered as electrostatic complexes with cationic lipids (lipoplexes) or polycations (polyplexes). The purpose of this project was to determine the effect of cellular internalization mechanism(s) on siRNA-mediated gene silencing efficiency.
Methods
Lipoplexes were formed comprising siRNA and N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP), cholesterol and dioleoyl phosphatidylethanolamine (DOPE), and polyplexes comprised siRNA with polyethylenimine (PEI). During transfections, specific uptake mechanisms were inhibited by pharmacological agents and RNAi-mediated knockdown of proteins involved in various endocytosis pathways. Confocal fluorescence microscopy further elucidated the predominant endocytic pathways of siRNA delivery via colocalization of vectors with endocytic vesicle markers.
Results
Inhibition of macropinocytosis (MP), caveolin-mediated endocytosis (CvME), flotillin-mediated endocytosis (FME) and knockdown of ARF6 significantly decreased PEI/siRNA-mediated gene silencing. Inhibition of endocytosis pathways, however, had negligible effect on lipoplex uptake and gene silencing mediated by lipoplexes. Rather, internalization of lipoplexes and subsequent siRNA-mediated gene silencing occurred via an energy-independent process.
Conclusions
MP, CvME and FME, but not the acidified clathrin-mediated pathway, lead to effective gene silencing by PEI/siRNA polyplexes. Lipoplexes, in contrast, deliver siRNA primarily by direct fusion of the liposomal and cellular membranes. These results provide a new understanding of the mechanisms of siRNA delivery materials in HeLa cells and may aid in design of more effective RNAi strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
RNA interference (RNAi) is a ubiquitous post-transcriptional gene silencing mechanism (1,2) capable of inhibiting expression of virtually any gene and has been recognized to be of immense therapeutic potential (3,4). RNAi can be initiated by cytosolic delivery of 19–21 nucleotide duplexes, small interfering RNA (siRNA) (4), that guide cleavage of complementary mRNA by the RNA-induced silencing complex (RISC) (5). siRNA is a relatively large, polyanionic and fragile molecule that cannot readily cross cellular membranes. In addition, RNases present in serum degrade siRNAs in a matter of minutes (6–8). As a result, RNAi requires delivery agents to protect the siRNA, facilitate cellular internalization, and deliver the nucleic acid cargo to the cytosol (9–11).
Nanoparticles including siRNA delivery vectors commonly enter cells via endocytosis, which involves de novo production of internal vesicles from the plasma membrane lipid bilayer and consists of several different mechanisms (12,13). These mechanisms include macropinocytosis and clathrin-, caveolin-, flotillin-, and ARF6-mediated endocytosis. The fates of the resulting vesicles are different depending on the proteins promoting their formation and on their cargo.
Macropinocytosis (MP) is among the earliest discovered pathways (14) and is characterized by the formation of large vesicles (0.2–10 μm in diameter) (15) called macropinosomes. This pathway does not discriminate against cargo; any molecules or particles attached to the cell membrane or in the extracellular fluid near the cell surface may be internalized by MP. Macropinosomes are more “leaky” compared to other types of endocytic vesicles (16,17). Clathrin-mediated endocytosis (CME) is a heavily investigated pathway and accounts for nearly half of all cellular internalization (12). CME is characterized by the formation of a clathrin cage around invaginations at the plasma membrane. The resulting clathrin-coated vesicles fuse with early endosomes from which cargo can be recycled to the cell surface or trafficked to late endosomes (pH 5–6) and lysosomes (pH 4–5). Caveolin-mediated endocytosis (CvME) originates from 50- to 100-nm flask-like structures containing caveolin-1 on the plasma membrane and is associated with lipid rafts (12). Caveolin-1 proteins assemble on the plasma membrane by binding to cholesterol. The resulting vesicles, termed caveosomes, are in some cases not acidified and travel to Golgi and/or endoplasmic reticulum, in contrast to endosomes associated with CME (18,19). Flotillin-mediated endocytosis (FME) is a recently discovered pathway that has yet to be explored in depth. Flotillin-1 is homologous to caveolin-1 (12) and is necessary for dynamin-dependent but caveolin- and clathrin-independent uptake of negatively charged plasma membrane proteoglycans that are transported to late endosomes in HeLa cells (20,21). The FME pathway may be responsible for fluid-phase uptake in HeLa cells, since many flotillin-positive structures were found to be co-localized with dextran (a fluid-phase marker) (13,21). ARF6-dependent endocytosis (ADE) is associated with vesicles that contain ADP ribosylation factor 6 (ARF6) (12), one of the many members of the ADP ribosylation factor family of GTP-binding proteins that influence membrane trafficking, actin cytoskeleton structure, and membrane lipid modification (22). The trafficking of vesicles formed from ADE is not well-understood, but some evidence suggests that they fuse with early endosomes and traffic to lysosomes or recycle to the plasma membrane (22,23).
Internalization of DNA delivery vectors by some endocytosis pathways results in greater transfection than other endocytosis pathways (24–34). For example, CvME is more effective than CME for polyethylenimine (PEI)- and poly(amidoamine) dendrimer-mediated delivery of plasmid DNA (24,25). The most efficient uptake pathway varies among cell lines and may be different for various vectors in a given cell line. Furthermore, because the ultimate destinations of DNA and siRNA are different (nucleus and cytosol, respectively), one may expect that efficient siRNA delivery may occur through a different pathway than DNA delivery. The rational design of siRNA delivery vectors would, therefore, benefit from understanding of the predominant uptake pathways that can lead to efficient RNAi.
Chemical inhibitors can be used to inhibit the various uptake and intracellular trafficking mechanisms. Chlorpromazine is commonly used as a CME inhibitor, preventing the dissociation of the clathrin lattice (17) by relocating the clathrin and protein adaptor complex AP2 from the cell surface to intracellular vesicles (35). Amantadine was found to stabilize clathrin-coated vesicles and block their budding from the plasma membrane (36), thus inhibiting CME. Genistein is a tyrosine kinase inhibitor that causes local disruption of the actin network and can inhibit the recruitment of dynamin to plasma membranes, both of which are critical participants in CvME (17,37). Methyl-β-cyclodextrin (mβCD) is routinely used to deplete plasma membranes of cholesterol (17,38) thereby inhibiting CvME. Amiloride is a specific inhibitor of MP (16) regulating the Na+/H+ exchange process by lowering submembrane pH and preventing the activation of Rac1 and Cdc42 proteins (39).
We have characterized the mechanisms of uptake and RNAi of two different model vectors in HeLa cells. Polyethylenimine (PEI) was used as a typical cationic polymer that binds siRNA to form polyplexes. Lipoplexes were formed by complexation of siRNA with liposomes comprising a cationic lipid, N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP), and two “helper” lipids, cholesterol and dioleoyl phosphatidylethanolamine (DOPE). The presence of cholesterol in the liposomes greatly improves the cell binding of lipoplexes (40) and has been widely used in vivo (41–43), while DOPE facilitates the release of the encapsulated cargo in response to pH changes (42,44). Our findings indicate that PEI/siRNA polyplexes internalize through multiple pathways but CvME and MP allow for more effective RNAi than CME. On the other hand, internalization of and siRNA-mediated gene silencing by lipoplexes is highly dependent on cholesterol content in cells and largely proceeds through direct fusion of lipoplexes with the plasma membrane.
Materials and Methods
Materials
HeLa-Luc (luciferase-expressing cervical cancer) cells were a kind gift from Prof. Mark Davis at the California Institute of Technology. The cells were cultured according to ATCC protocols in DMEM with 10% (v/v) FBS (Gemini Bio-Products, West Sacramento, CA) and incubated at 37°C and 5% CO2. The anti-luciferase siRNA duplex (siGL3) and human Lamin A/C siRNA duplex (siLam) were purchased from Dharmacon (Lafayette, CO). The siRNA sequences are presented in Supplementary Table I. siRNA tagged with AlexaFluor 647 (AF647-siRNA) and the siRNAs that target clathrin heavy chain, caveolin-1, flotillin-1 and ARF6 proteins were purchased from Qiagen (Germantown, MD). Lipids were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Unless otherwise noted, all other reagents were purchased from Sigma-Aldrich (St. Louis, MO). Branched, 25-kDa PEI was dissolved in DI water at a concentration of 1 mg/ml. The stock was kept at room temperature prior to being used.
Formation of siRNA Polyplexes and Lipoplexes
To form liposomes, DOTAP, DOPE and cholesterol were separately dissolved in chloroform at a concentration of 10 mg/ml and mixed in glass tubes in amounts to give the desired DOTAP:DOPE:cholesterol ratios. The mixtures were dried under vacuum overnight leading to the formation of a thin lipid film which was resuspended in PIPES buffer (20 mM PIPES, 150 mM NaCl) at 0.2 mg/ml using a water-bath sonicator. The lipid suspensions were extruded through a 100-nm polycarbonate membrane 11 times using a Mini-Extruder (Avanti Polar Lipids). The lipid suspensions were kept at 4°C until use. To prepare polyplexes and lipoplexes, an appropriate amount of the 20 μM stock siRNA solution in RNase-free water was first added to PIPES buffer. Liposomes or PEI (at concentrations required to give the final required vehicle:siRNA ratio) were then added to the siRNA solution. The final suspension containing the complexes was incubated at room temperature for 30 min prior to transfections.
siRNA Transfection
HeLa-Luc cells were seeded in 12-well plates at 1 × 105 cells/well 24 h prior to transfections. The culture media was replaced by 1 ml of serum-free media, and 50 μl of the siRNA-vector suspension was added to each well to achieve final concentration of 5 nM siRNA. After 4 h, the transfection media was replaced by culture media. After 20 h, the cells were washed once with PBS and 100 μl of cell culture lysis reagent (CCLR) were added to each well. Twenty microliters of each sample lysate were used to evaluate luciferase expression using the Promega luciferase assay (Promega, Madison, WI) and Lumat LB 9507 luminometer (Berthold, Oak Ridge, TN) according to the manufacturer’s protocol. The total protein content was evaluated for each lysate sample using the Pierce BCA assay (Peirce, Rockford, IL) and used to normalize luciferase content of each sample to total cellular protein expression. Knockdown is calculated as 1 – (RLU/mg)/(RLU/mg)control, where (RLU/mg)control is the luciferase activity after transfection with the control siLamin siRNA.
Cytotoxicity
Cytotoxicity was determined using the CellTiter-Blue cell viability assay (Promega, Madison, WI) according to the manufacturer’s instructions. Briefly, HeLa-Luc cells were seeded at 1 × 104 cells/well in 96-well plates. After 24 h, the media was aspirated and 100 μl of serum-free media was added. In addition, inhibitors at various concentrations were added to the cells and allowed to incubate for 4 h. The drug-containing media was then replaced with culture media, and the cells were placed in the incubator for 20 h. The CellTiter-Blue reagent was then added (20 μl/well), and the cells were incubated for 4 h. The absorbance at 570 nm and 600 nm was read on a SpectraMAX 340 microplate reader (Molecular Devices, Sunnyvale, CA). To obtain viability, the absorbance values at 600 nm were subtracted from the values at 570 nm and normalized to the untreated cells.
Cellular Internalization
HeLa-Luc cells were seeded 24 h before transfection on 12-well plates at 1 × 105 cells/well. The vectors were formed as described earlier using AF647-siGL3. The cells were transfected in serum-free media for 2 h and washed once with 1 ml of 0.001% SDS in PBS and once with 1 ml of PBS to remove fluorescently labeled vectors on the surface of the cells. Following the washing, 200 μl of 0.25% trypsin in PBS solution were added to each well, and the plates were allowed to incubate for 5 min. The cell suspensions were mixed by pipetting and added to 300 μl of FBS. The cells were stored on ice prior to being analyzed. The cells were analyzed on BD FACSCanto flow cytometer (BD Biosciences, San Jose, CA), and the data were analyzed using FCS Express v.3.0 software (De No Software, Los Angeles, CA). The gate was set based on the forward and side scattering parameters of living cells, and at least 2000 cells were measured in each group.
Transfections in the Presence of Chemical Inhibitors
The stock solutions of chlorpromazine (5 mg/mL), amiloride (5 mg/mL), amantadine (50 mg/mL) and methyl-β-cyclodextrin (250 mg/mL) (Roquette, Gurnee, IL) were prepared in water. The stock of genistein was prepared at 50 mg/ml in DMSO. Prior to transfections as described above, the stock solutions were mixed with culture media to achieve the desired final concentration. All of the drugs were added to HeLa-Luc cells at least 30 min before addition of siRNA vectors as described above.
RNAi Knockdown of Endocytosis Pathways
To knockdown proteins associated with the various endocytosis pathways, 2 × 105 HeLa-Luc cells/well were seeded in 6-well plates. After 24 h, the cells were transfected in a serum-free media using the Lipofectamine RNAiMAX reagent (Life Technologies, Carlsbad, CA) with final concentration of 20 nM siRNA according to the manufacturer’s protocol. Briefly, for each experimental group, the required amount of siRNA and 3.5 μl RNAiMAX were each diluted in 150 μl of OptiMEM (Invitrogen), and the RNAiMAX solutions were added dropwise to the siRNA solutions. The complexes were left at room temperature for 15 min and mixed with DMEM just before transfection. Four hours post-transfection, the media was replaced with the serum-containing media and the cells were placed in the incubator for 24 h. Control groups included cells to which no siRNA was added and cells transfected with 20 nM siLam. The cells were washed with PBS, trypsinized with 320 μl of trypsin (Cellgro, Manassas, VA) and equally partitioned into 6 wells of a 12-well plate. The plates were placed in incubator and cultured for 20 h. Subsequently, the cells were either used in further transfections or validated for a successful knockdown of the targeted proteins by Western blots (Supplementary Figure 2).
Confocal Laser Scanning Microscopy
HeLa-Luc cells were seeded on microscope cover slips that were placed in 6-well plates at 1 × 105 cells/well and allowed to incubate for 48 h. Polyplexes and lipoplexes were formed with fluorescently labeled siGL3 as described earlier. The cells were transfected with polyplexes and lipoplexes at final siRNA concentration of 5 nM in serum-free media for 2 h after which the media was aspirated and the cells were rinsed three times with PBS prior to being fixed with 3.7% formaldehyde in PBS for 10 min. The cells were permeabilized with 0.25% Triton X-100 in PBS solution for 10 min, and incubated at room temperature in 10% FBS in PBS solution to prevent non-specific staining. The cells were then incubated in the primary antibody solution (Supplementary Table II) for 1 h, washed three times with PBS and incubated in 5 μg/ml of secondary AlexaFluor 488-labeled antibody (Invitrogen). Finally, the cells were washed thrice with PBS, the cover slips were removed from the wells and mounted on standard microscopy slides containing a drop of Prolong Gold (Invitrogen) reagent. The slides were left in the dark overnight and cover-slips were sealed the following day by nail polish. AlexaFluor 568-labeled 10 kDa dextran (Invitrogen) was used as a marker for fluid phase uptake. As described above, the transfection of fluorescently labeled siGL3 was performed on cells cultured on the cover slips, but 20 min before the end of transfection, dextran was added to wells to yield a final concentration of 1.5 mg/ml. The media was then aspirated, the wells were washed three times with PBS, and the cells were fixed with a 3.7% solution of formaldehyde. The cover slips were extracted from the wells and mounted as described above. Micrographs were obtained using LSM 700 confocal fluorescence microscope (Zeiss, Oberkochen, Germany). The images were processed using deconvolution plugin for ImageJ (45).
Energy Dependency of Uptake and RNAi
To determine if uptake and transfection were due to passive membrane fusion, the seeding, transfections, luciferase expression and vector uptake measurements were carried out the same way as described above. However, during the incubation post-transfection, one plate was incubated at 4°C while and other at 37°C (2 h for uptake and 4 h for transfection). After the incubation period, cells for the uptake study were prepared for flow cytometry as described above. For transfections, the cells were washed with PBS, the media was replaced with serum-containing media and the plates from both groups were returned to the 37°C incubator.
Vector Sizing
All sizing was done using dynamic light scattering (DLS). Vectors were prepared in PIPES buffer as described earlier with the final siRNA concentration three times higher than in the solution used for transfection at the desired vector/siRNA ratios. The solutions were placed in transparent cuvettes, allowed to incubate at room temperature for 15 min and measured 10 times each using Brookhaven Instruments Corporation 90 Plus Particle Size Analyzer (Holtsville, NY).
Statistics
If not otherwise stated, the experiments were performed in triplicates. The error bars on plots represent standard deviation. Statistical significance was computed using one-way ANOVA with Tukey means comparison test in conjunction with equivalent surrogate data technique (46) using OriginPro8.6 (Northampton, MA). The significance level (α) was set to 0.05. An asterisk above the presented data (*) indicate statistical significance compared to the control group (p < 0.05).
Results
Selection of Vector Composition
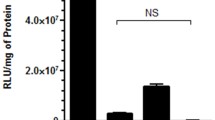
HeLa-Luc cells were transfected using lipoplexes of various compositions in order to determine the optimal lipid composition and lipid/siRNA ratio. Liposomes containing ≥50 wt% DOTAP promoted higher knockdown than vectors with higher content of the helper lipids (cholesterol and DOPE) (Fig. 1). The knockdown was in the range of 7–25% for 1:1 (w:w) and 30–70% for 10:1 (w:w) of lipid:siRNA. Liposomes comprising DOTAP:DOPE:cholesterol 11:2:7 (w:w:w) at 10:1 lipid:siRNA (w:w) (N/P 2.6), providing ~75% knockdown, were used for the remainder of this study. Based on previous work, the composition of polyplexes was fixed at 10:1 PEI:siRNA (w:w) (N/P 76) (47).

Knockdown of luciferase expression in HeLa-Luc cells via siRNA lipoplexes of varied compositions (DOTAP:DOPE:cholesterol, w:w:w) at (a) 1:1 and (b) 10:1 lipid:siRNA (w:w) (n = 3, error bars represent standard deviation).
Energy Dependence of Vector Internalization and RNAi
To investigate if internalization of the vectors was energy dependent, transfections were performed at 4 and 37°C (Table I). Uptake of lipoplexes was unaffected by temperature while polyplex uptake was reduced 10-fold at 4°C compared to 37°C. In addition, at the lower temperature, knockdown was reduced for both vectors, with the effect being dramatic in the case of the polyplexes (~6-fold reduction). These results suggest that PEI/siRNA polyplexes are internalized primarily by energy-dependent endocytic mechanisms in HeLa cells. Internalization of the lipoplexes, in contrast, can occur through energy-independent mechanisms such as direct fusion with the plasma membrane. The decrease in lipoplex-mediated knockdown at 4°C, however, suggests that such an energy-independent mechanism may be less effective for siRNA delivery.
Effect of Endocytosis-Inhibiting Drugs on Vector Internalization
Chemical inhibitors of endocytosis were used to investigate the uptake pathways of the siRNA vectors at concentrations previously found to provide inhibition of uptake with low cytotoxicity (Supplementary Figure 1) (24,25). Uptake of PEI/siRNA polyplexes was reduced by ~60% in the presence of ≥3 μg/ml chlorpromazine and by ~40% in the presence of ≥150 μg/ml amantadine, suggesting CME as a significant internalization pathway (Fig. 2). Similarly, the uptake of polyplexes decreased by ~40–70% in the presence of genistein (Fig. 3) and by ~40% in the presence of amiloride (Fig. 4), suggesting a role of CvME and MP in their cellular internalization. While the presence of mβCD did not alter polyplex uptake (Fig. 3), the uptake of lipoplexes was reduced by ~25–60%, underscoring the importance of plasma membrane cholesterol in lipoplex uptake (Fig. 3). The presence of amantadine (Fig. 2) reduced the uptake of lipoplexes by ~25% while the presence of chlorpromazine (Fig. 2) or amiloride (Fig. 4) did not affect lipoplex internalization. Most significantly, in the presence of genistein, an ~20% reduction in uptake of lipoplexes occurred only at 70 μg/ml (Fig. 3). Hence, it is likely that both CME and CvME play minor roles in the uptake of lipoplexes.

Uptake of and knockdown of luciferase expression via polyplexes and lipoplexes in HeLa-Luc cells in the presence of chemical inhibitors of CME (chlorpromazine and amantadine). For each vector, uptake in the presence of drugs was normalized to uptake in the drug-free control. (n = 3, error bars represent standard deviation; *, p < 0.05 relative to untreated control group).

Uptake of and knockdown of luciferase expression via polyplexes and lipoplexes in HeLa-Luc cells in the presence of chemical inhibitors of CvME (genistein and mβCD). For each vector, uptake in the presence of drugs was normalized to uptake in the drug-free control. (n = 3, error bars represent standard deviation; *, p < 0.05 relative to untreated control group).

Uptake of and knockdown of luciferase expression via polyplexes and lipoplexes in HeLa-Luc cells in the presence of chemical inhibitor of MP (amiloride). For each vector, uptake in the presence of drugs was normalized to uptake in the drug-free control. (n = 3, error bars represent standard deviation; *, p < 0.05 relative to untreated control group).
Effect of Endocytosis-Inhibiting Drugs on siRNA Delivery
The effect of endocytosis-inhibiting drugs on the efficacy of siRNA-mediated gene silencing was investigated in a similar manner. Knockdown via PEI/siRNA polyplexes was significantly reduced to ~8% in the presence of genistein (Fig. 3) and to ~10% in the presence of amiloride (Fig. 4), while the presence of amantadine, mβCD and chlorpromazine had little effect (Figs. 2 and 3). This suggests that even though the siRNA polyplexes entered the cells through multiple pathways, only those that entered through CvME and MP led to significant knockdown. The presence of genistein and mβCD (Fig. 3) reduced the knockdown mediated by lipoplexes to ~25% and <5%, respectively, while chlorpromazine and amantadine had little effect (Fig. 2). Thus, lipoplex uptake via CvME led to significant knockdown, and cholesterol was essential to the process. The presence of amiloride inhibited lipoplex-mediated knockdown at 27 and 40 μg/mL, but not at 50 μg/mL, possibly because more efficient pathways became increasingly accessible as MP was inhibited (Fig. 4).
Effect of Knockdown of Proteins Associated with Endocytosis on Vector Uptake and RNAi
To confirm the effects of drug-mediated inhibition of CME, CvME and MP, and to investigate the roles of pathways for which small-molecule inhibitors are not available (FME and ADE), expression of proteins known to be essential in these endocytosis pathways was knocked down prior to polyplex- or lipoplex-mediated transfection. The knockdown of these proteins was confirmed by Western blot (Supplementary Figure 2). The absence of caveolin, flotillin and ARF6 proteins significantly reduced the uptake of PEI/siRNA polyplexes by 50–60%, while knockdown of clathrin had no effect (Fig. 5a). However, polyplex-mediated knockdown was significantly reduced (to <2%) in cells lacking ARF6 (Fig. 5b). In the absence of caveolin, lipoplex uptake increased by over 20% (Fig. 5a). This could be due to structural changes in the plasma membranes or perhaps that pathways other than CvME facilitated compensatory uptake. Knockdown of other proteins had no effect on lipoplex uptake. Most importantly, the absence of clathrin, caveolin, ARF6 or flotillin did not significantly affect lipoplex-mediated knockdown (Fig. 5b).

(a) Normalized uptake and (b) knockdown of luciferase expression in HeLa-Luc cells by siRNA polyplexes and lipoplexes upon siRNA-mediated knockdown of several membrane trafficking proteins – clathrin (siCLT), caveolin (siCav), flotillin (siFLT) and ARF6 (siARF). For each vector, uptake up membrane trafficking protein knockdown was normalized to uptake in the untreated control. (n = 3, error bars represent standard deviation; *, p < 0.05 relative to untreated control group).
Colocalization of siRNA with Endocytic Vesicle Marker Proteins
To visualize colocalization of polyplexes and lipoplexes with specific endocytic vesicle markers, HeLa-Luc cells were transfected with vectors containing AF647-siRNA, and proteins associated with specific endocytosis pathways were subsequently immunofluorescently stained (Fig. 6, Supplementary Figures 3–12). When delivered with PEI, the siRNA colocalized with caveolin and flotillin, providing evidence of the significance of CvME and FME in intracellular processing of polyplexes. Minor colocalization of polyplex-delivered siRNA with ARF6 was also observed. None of the proteins showed strong co-localization with siRNA in the lipoplexes. Fluorescent dextran was also co-delivered with siRNA vectors to determine if the latter enter the cells through MP. Strong colocalization between dextran and polyplexes was observed (Supplementary Figure 7). No such co-localization was observed with lipoplexes, suggesting that they do not utilize MP as an uptake mechanism (Supplementary Figure 12).

Representative confocal micrographs of fluorescently labeled siRNA (red) with various proteins associated with endocytosis – clathrin, caveolin, flotillin and ARF6 – immunologically stained (green). Fluorescent dextran (green) served as a MP marker. All panels are 50.81 × 50.81 μm2. Colocalization is indicated by white arrowheads.
Vector Size
The size of the vectors can influence endocytosis and their subsequent processing within the cells (48). The sizes of the lipid vesicles alone were significantly reduced upon extrusion to <500 nm (Table II). However, upon mixing with siRNA to form vectors, the sizes increased significantly to 468 nm. The sizes of the siRNA polyplexes (~200 nm) were smaller than siRNA lipoplexes.
Discussion
Multiple intracellular barriers have been investigated with regard to their impact on the optimal design of gene delivery vectors. From extracellular clearance to intracellular degradation, gene delivery vectors should be designed to optimize their interactions with their environment at every stage of their activity from administration to therapeutic expression. Intracellular trafficking within the endolysosomal pathways remains the most studied parameter affecting the functions of these vectors.
Because naked siRNA is highly susceptible to intracellular degradation (49), designing the appropriate supramolecular structure that can protect them is important. siRNA delivery requires only efficient cellular internalization and entry to the cytosol. Hence, release from endocytic vesicles that allow for their uptake and subsequent transport is important. In fact, recent research has shown that recycling of vesicles containing siRNA cargo, leading to cargo transport out of the cell rather than entry into the cytosol, is a major barrier in siRNA delivery (50). To improve siRNA vector release from vesicles, several approaches have been utilized. For example, polymers and dendrimers have been tagged with histidine-containing peptides (51,52), to increase their proton buffering capacity, or with melittin (53), which destabilizes membranes resulting in efficient release of the vectors into the cytosol without negatively impairing their gene transfection efficiency. Specific uptake mechanisms have also been targeted to increase delivery efficiency such as by conjugation of transferrin or cholera toxin B, which are known to be internalized by CME and CvME, respectively. Further, targeting MP via the use of TAT peptide within the vector design element dramatically improves gene delivery efficiency (17,54), as macropinosomes are inherently leaky. Ultimately, the aim is to improve the chemistry and supramolecular form of the vector to match its biological environment so as to maximize its working potential. Accordingly, both chemical and biological methods for selective inhibition of endocytosis pathways were used in this study to determine the uptake mechanisms that play a significant role in efficient siRNA delivery by PEI and liposomes composed of DOTAP, DOPE and cholesterol to investigate how vector macromolecular chemistry may be exploited for efficient siRNA delivery.
Performing transfections at a reduced temperature is widely used to study the dependence of cellular uptake and activity of vectors on energy-dependent mechanisms (55,56). Reduced temperature inhibits active processes including endocytosis, while passive entry mechanisms such as membrane fusion are still functional. The decrease in PEI/siRNA polyplex uptake and the resulting knockdown at 4°C (Table I) is consistent with previously published work that reported that the uptake of PEI-based siRNA polyplexes occurs through endocytosis (4,57). In contrast, the uptake of lipoplexes was unaffected by low temperatures while RNAi was slightly decreased (not statistically significant, Table I). We cannot rule out the possibility that some polyplexes and lipoplexes remained attached to the cells surface upon increasing the temperature and were subsequently internalized during the following 20 h. This may account for the transgene expression observed after transfection with polyplexes at 4°C. The much higher transgene expression upon lipoplex-mediated transfection at 4°C, however, suggests that significantly more lipoplexes were internalized at this reduced temperature. Thus, internalization of lipoplexes and the majority of the subsequent RNAi likely occur through passive mechanisms. This result is consistent will previous observations of fusion of lipoplexes with the plasma membrane (58,59).
Chemical inhibitors are commonly employed by many researchers and, thus, are important for comparison with previous studies. Cytotoxicity and possible non-specificity should be considered in interpretation of the results, however (60). The slight increase in siRNA-mediated gene silencing by polyplexes in the presence of chlorpromazine (Fig. 2, not statistically significant) is likely due to increased internalization of polyplexes via clathrin-independent pathways that are upregulated and more effective for siRNA delivery. The effect of chlorpromazine became apparent, however, only at concentrations at or above 3 μg/mL, where significant cytotoxicity was observed (Supplementary Figure 1). Further, knockdown by polyplexes was not significantly affected by amantadine (Fig. 2) or by knockdown of clathrin-1 expression (Fig. 5). The differing effects of the chemical inhibitors and knockdown of clathrin expression on PEI/siRNA uptake is most likely due cytotoxicity and/or non-specific effects of the inhibitors. The lack of change in uptake upon knockdown of clathrin expression is consistent with the absence of colocalization of the polyplexes with clathrin in confocal micrographs (Fig. 6). Taken together, the decrease in uptake siRNA-mediated gene silencing in the presence of genistein (Fig. 3) and amiloride (Fig. 4) and upon knockdown of caveolin-1 expression (Fig. 5), and the strong colocalization of polyplexes with caveolin and dextran (Fig. 6), suggest that effective PEI/siRNA-mediated gene silencing occurs primarily through CvME and MP, but not CME.
Knockdown of ARF6 resulted in reduced uptake of polyplexes and also dramatically reduced RNAi (Fig. 5). Since ARF6 protein has been implicated in multiple pathways, it is difficult to determine which pathway was inhibited (22), more so since ARF6 also did not colocalize with siRNA (Fig. 6). ARF6 is associated with vesicle fusion events involving exchange of intraluminal contents and membranes, and budding of vesicles such as recycling endosomes (61,62), which have recently been implicated as a possible limitation to siRNA delivery (50). The likelihood of escape of vectors from early endocytic vesicles likely increases during these budding and fusion events, during which vesicle membranes are deformed. Thus, knockdown of ARF6 may reduce the number of opportunities for vectors to escape into the cytosol. Moreover, since uptake was measured 2 h post transfection, it is also possible that the reduction in the recycling mechanism did not dramatically influence the uptake, but significantly affected eventual RNAi that was evaluated 24 h post-transfection.
Neither the endocytosis inhibitors nor knockdown of membrane trafficking proteins significantly affected uptake of lipoplexes or lipoplex-mediated knockdown. No co-localization between any of the endocytic vesicle marker proteins and lipid-delivered siRNAs was observed. In addition, fewer punctate aggregates of siRNA were detected in lipoplexes than in PEI/siRNA polyplexes, even though higher knockdown was observed with lipoplexes compared to polyplexes. The larger size of lipoplexes may also prevent internalization by some endocytic pathways. These data confirm the energy independence of lipoplex internalization and together suggest that lipoplexes enter cells and deliver siRNA primarily by fusion of liposomes with the cell membrane.
Cholesterol can play a major role in multiple endocytosis pathways, and altering the distribution and organization of cholesterol in the plasma membrane may also affect fusion with lipoplexes. In addition, the depletion of cholesterol from the plasma membrane might reduce the amount of cholesterol inside the cells, thereby affecting the fusion events that could occur within endocytic vesicles. The consistent inverse correlation between the concentration of mβCD and uptake of and knockdown by lipoplexes is most likely due to a reduction of fusion events between the lipoplexes and plasma membranes. It is interesting that mβCD was the only drug that affected the lipoplexes to a higher degree than PEI/siRNA polyplexes. This observation is consistent with previously published data showing mβCD had a larger effect on uptake of lipoplexes than on polyplexes (63).
Previous studies have discussed the possibilities of endocytosis pathways that can mediate siRNA delivery that were not active in pDNA delivery. Lu and co-workers used a commercially available lipid-based siRNA transfection reagent (DharmaFECT1) in various cell lines and determined that although 95% of particles enter cells through endocytosis, the inhibition of CME, CvME, MP and lipid-raft mediated endocytosis did not lead to reduction in RNAi. The depletion of cholesterol from the plasma membrane also did not alter the uptake of the lipoplexes, but the siRNA-mediated gene silencing was reduced. Low temperatures during transfections also inhibited knockdown thus indicating that direct fusion between the lipoplexes and plasma membranes is likely to be responsible for siRNA delivery by this vector (55). Another study investigated intracellular trafficking of antisense oligonucleotides that only expressed when delivered to nucleus (splice shifting oligonucleotide) using two commercially available delivery reagents: jetPEI (derivative of linear polyethylenimine) and Lipofectamine 2000. They concluded that effective delivery using the lipid-based system occurred through direct fusion with plasma membrane, and PEI-based delivery occurred through a dynamin- and caveolin-independent pathway that is also not CME (63).
The possibility that other endocytic mechanisms are active in transfections cannot be excluded. The vesicles that are formed in these different endocytosis processes have different surfaces, lumina and physico-chemical properties that allow them to process cargo in various ways. It is evident that the vesicles preserve the “memory” of their origin using protein or lipid markers (12,64). Other mechanisms such as the CLIC/GEEC pathway characterize clathrin- and dynamin-independent endosomal compartments enriched with GPI-AP proteins that do not have cytosolic extensions. The vesicles that are formed in this pathway are tubular in shape, form special early endosomal compartments and do not travel to Golgi (13).
Conclusion
In HeLa cells, PEI/siRNA polyplexes use energy-dependent endocytosis mechanisms predominantly relying on MP and CvME for efficient siRNA delivery while ARF6 is crucial for efficient intracellular processing of polyplexes. In contrast, lipoplexes enter cells and deliver siRNA primarily by cholesterol-dependent fusion with the cell membrane. The results of this study suggest that for design of polyplexes for siRNA delivery to these cells, attention should be given to release of the particles from endosomes, but not based on the acidification of CME-originated vesicles. Rather, the disruption of endosomal membranes by other means may be considered since pathways that do not lead to acidic endosomes are highly significant for PEI-mediated siRNA delivery. Lipoplexes remain promising vectors, since they allow for release of siRNA directly into the cytosol and evade limitations of intracellular trafficking and endocytic release. In conclusion, siRNA-mediated gene silencing efficiency depends critically on the mechanism of cellular uptake, and the mechanisms should be taken into account in design of siRNA vectors.
Abbreviations
- ADE:
-
ARF6-dependent endocytosis
- ARF6:
-
ADP ribosylation factor 6
- CME:
-
Clathrin-mediated endocytosis
- CvME:
-
Caveolin-mediated endocytosis
- DOPE:
-
Dioleoyl phosphatidylethanolamine
- DOTAP:
-
N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate
- FME:
-
Flotillin-mediated endocytosis
- MP:
-
Macropinocytosis
- mβCD:
-
Methyl-β-cyclodextrin
- PEI:
-
Polyethylenimine
- RISC:
-
RNA-induced silencing complex
- RNAi:
-
RNA interference
- siRNA:
-
Small interfering RNA
References
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–8.
Fire A, Albertson D, Harrison SW, Moerman DG. Production of antisense RNA leads to effective and specific-inhibition of gene-expression in c-elegans muscle. Development. 1991;113(2):503–14.
Davidson BL, McCray PB. Current prospects for RNA interference-based therapies. Nat Rev Genet. 2011;12(5):329–40.
Malefyt AP, Angart PA, Chan C, Walton SP. SiRNA therapeutic design: tools and challenges. In: Mallick B, Ghosh Z, editors. Regulatory RNAs: basics, methods and applications. Berlin: Springer; 2012. p. 475–503.
Rivas FV, Tolia NH, Song JJ, Aragon JP, Liu J, Hannon GJ, et al. Purified argonaute2 and an siRNA form recombinant human RISC. Nat Struct Mol Biol. 2005;12(4):340–9.
van de Water FM, Boerman OC, Wouterse AC, Peters JGP, Russel FGM, Masereeuw R. Intravenously administered short interfering RNA accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab Dispos. 2006;34(8):1393–7.
Layzer JM, McCaffrey AP, Tanner AK, Huang Z, Kay MA, Sullenger BA. In vivo activity of nuclease-resistant siRNAs. RNA. 2004;10(5):766–71.
Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432(7014):173–8.
Gary DJ, Puri N, Won YY. Polymer-based siRNA delivery: perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. J Control Release. 2007;121(1–2):64–73.
Gao K, Huang L. Nonviral methods for siRNA delivery. Mol Pharm. 2009;6(3):651–8.
Frohlich T, Wagner E. Peptide- and polymer-based delivery of therapeutic RNA. Soft Matter. 2010;6(2):226–34.
Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902.
Kumari S, Swetha MG, Mayor S. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 2010;20(3):256–75.
Swanson JA, Watts C. Macropinocytosis. Trends Cell Biol. 1995;5(11):424–8.
Swanson JA. Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol. 2008;9(8):639–49.
Kerr MC, Teasdale RD. Defining macropinocytosis. Traffic. 2009;10(4):364–71.
Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58(1):32–45.
Xiang SN, Tong HJ, Shi Q, Fernandes JC, Jin T, Dai KR, et al. Uptake mechanisms of non-viral gene delivery. J Control Release. 2012;158(3):371–8.
Reilly MJ, Larsen JD, Sullivan MO. Polyplexes traffic through caveolae to the golgi and endoplasmic reticulum en route to the nucleus. Mol Pharm. 2012;9(5):1280–90.
Payne CK, Jones SA, Chen C, Zhuang XW. Internalization and trafficking of cell surface proteoglycans and proteoglycan-binding ligands. Traffic. 2007;8(4):389–401.
Glebov OO, Bright NA, Nichols BJ. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat Cell Biol. 2006;8(1):46–U16.
Donaldson JG. Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J Biol Chem. 2003;278(43):41573–6.
Kalia M, Kumari S, Chadda R, Hill MM, Parton RG, Mayor S. Arf6-independent GPI-anchored protein-enriched early endosomal compartments fuse with sorting endosomes via a Rab5/phosphatidylinositol-3′-kinase-dependent machinery. Mol Biol Cell. 2006;17(8):3689–704.
Gabrielson NP, Pack DW. Efficient polyethylenimine-mediated gene delivery proceeds via a caveolar pathway in HeLa cells. J Control Release. 2009;136(1):54–61.
Hwang ME, Keswani RK, Pack DW. Dependence of PEI and PAMAM gene delivery on clathrin- and caveolin-dependent trafficking pathways. Pharm Res. 2015;32(6):2051–9.
Rejman J, Bragonzi A, Conese M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol Ther. 2005;12(3):468.
Goncalves C, Mennesson E, Fuchs R, Gorvel JP, Midoux P, Pichon C. Macropinocytosis of polyplexes and recycling of plasmid via the clathrin-dependent pathway impair the transfection efficiency of human hepatocarcinoma cells. Mol Ther. 2004;10(2):373–85.
von Gersdorff K, Sanders NN, Vandenbroucke R, De Smedt SC, Wagner E, Ogris M. The internalization route resulting in successful gene expression depends on both cell line and polyethylenimine polyplex type. Mol Ther. 2006;14(5):745–53.
Manunta M. Gene delivery by dendrimers operates via different pathways in different cells, but is enhanced by the presence of caveolin. J Immunol Methods. 2006;314(1–2):134–46.
van der Aa MAEM, Huth WS, Hafele SY, Schubert R, Oosting RS, Mastrobattista E, et al. Cellular uptake of cationic polymer-DNA complexes via caveolae plays a pivotal role in gene transfection in Cos-7 cells. Pharm Res. 2007;24(8):1590–8.
Douglas KL. Toward development of artificial viruses for gene therapy: a comparative evaluation of viral and non-viral transfection. Biotechnol Prog. 2008;24(4):871–83.
Diaz-Moscoso A, Balbuena P, Gomez-Garcia M, Mellet CO, Benito JM, Le Gourrierec L, et al. Rational design of cationic cyclooligosaccharides as efficient gene delivery systems. Chem Commun. 2008;17:2001–3.
McLendon PM, Fichter KM, Reineke TM. Poly(glycoamidoamine) vehicles promote pDNA uptake through multiple routes and efficient gene expression via caveolae-mediated endocytosis. Mol Pharm. 2010;7(3):738–50.
Vercauteren D, Piest M, van der Aa LJ, Al Soraj M, Jones AT, Engbersen JFJ, et al. Flotillin-dependent endocytosis and a phagocytosis-like mechanism for cellular internalization of disulfide-based poly(amido amine)/DNA polyplexes. Biomaterials. 2011;32(11):3072–84.
Wang LJ, Rothberg KF, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123:1107–17.
Phonphok Y, Rosenthal KS. Stabilization of clathrin coated vesicles by amantadine, tromantadine and other hydrophobic amines. FEBS Lett. 1991;281(1–2):188–90.
Vercauteren D, Vandenbroucke R, Jones A, Rejman J, Demeester J, De Smedt S. The use of inhibitors to study endocytic pathways of gene carriers: optimization and pitfalls. Mol Ther. 2010;18(3):561–9.
Nabi IR, Le PU. Caveolae/raft-dependent endocytosis. J Cell Biol. 2003;161(4):673–7.
Koivusalo M, Welch C, Hayashi H, Scott C, Kim M, Alexander T. Amiloride inhibits macropinocytosis by lowering submembranous ph and preventing rac1 and cdc42 signaling. J Cell Biol. 2010;188(4):547–63.
Crook K, Stevenson BJ, Dubouchet M, Porteous DJ. Inclusion of cholesterol in DOTAP transfection complexes increases the delivery of DNA to cells in vitro in the presence of serum. Gene Ther. 1998;5(1):137–43.
Liu Y, Mounkes LC, Liggitt HD, Brown CS, Solodin I, Heath TD, et al. Factors influencing the efficiency of cationic liposome-mediated intravenous gene delivery. Nat Biotechnol. 1997;15:167–73.
Pedroso de Lima MC, Simoes S, Pires P, Faneca H, Duzgunes N. Cationic lipid-DNA complexes in gene delivery: from biophysics to biological applications. Adv Drug Deliv Rev. 2001;47(2–3):277–94.
Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol. 2007;25(10):1149–57.
Schroeder A, Levins CG, Cortez C, Langer R, Anderson DG. Lipid-based nanotherapeutics for siRNA delivery. J Intern Med. 2010;267(1):9–21.
Vonesch C, Unser M. A fast thresholded landweber algorithm for wavelet-regularized multidimensional deconvolution. IEEE Trans Image Process. 2008;17(4):539–49.
Larson DA. Analysis of variance with just summary statistics as input. Am Stat. 1992;46(2):151–4.
Wong LS. Elucidation of design criteria for siRNA delivery in mammalian cells using polyethylenimine. Ph.D. Thesis. University of Illinois, Urbana-Champaign; 2009.
Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377:159–69.
Li CX, Parker A, Menocal E, Xiang S, Borodyansky L, Fruehauf JH. Delivery of RNA interference. Cell Cycle. 2006;5(18):2103–9.
Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol. 2013;31(7):653–U119.
Chang K-L, Higuchi Y, Kawakami S, Yamashita F, Hashida M. Development of lysine–histidine dendron modified chitosan for improving transfection efficiency in HEK293 cells. J Control Release. 2011;156(2):195–202.
Chang K-L, Higuchi Y, Kawakami S, Yamashita F, Hashida M. Efficient gene transfection by histidine-modified chitosan through enhancement of endosomal escape. Bioconjug Chem. 2010;21(6):1087–95.
Pichon C, Billiet L, Midoux P. Chemical vectors for gene delivery: uptake and intracellular trafficking. Curr Opin Biotechnol. 2010;21(5):640–5.
Kaplan IM, Wadia JS, Dowdy SF. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Release. 2005;102(1):247–53.
Lu JJ, Langer R, Chen JZ. A novel mechanism is involved in cationic lipid-mediated functional siRNA delivery. Mol Pharm. 2009;6(3):763–71.
Perez AP, Romero EL, Morilla MJ. Ethylendiamine core PAMAM dendrimers/siRNA complexes as in vitro silencing agents. Int J Pharm. 2009;380(1–2):189–200.
Juliano RL, Ming X, Nakagawa O. Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides. Bioconjug Chem. 2012;23(2):147–57.
Pires P, Simoes S, Nir S, Gaspar R, Duzgunes N, de Lima MCP. Interaction of cationic liposomes and their DNA complexes with monocytic leukemia cells. Biochim Biophys Acta-Biomembr. 1999;1418(1):71–84.
Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, et al. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci U S A. 1987;84:7413.
Ivanov AI. Pharmacological inhibition of endocytic pathways: is it specific enough to be useful? Methods Mol Biol. 2008;440:15–33.
Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10(9):597–608.
Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30(17):3481–500.
Ming X, Sato K, Juliano RL. Unconventional internalization mechanisms underlying functional delivery of antisense oligonucleotides via cationic lipoplexes and polyplexes. J Control Release. 2011;153(1):83–92.
Angers CG, Merz AJ. New links between vesicle coats and Rab-mediated vesicle targeting. Semin Cell Dev Biol. 2011;22(1):18–26.
ACKNOWLEDGMENTS AND DISCLOSURES
We thank Sandy Mattick at the Cell Culture Media Facility at the University of Illinois for help with preparation of cell media. We also thank Maureen K. Thompson for assistance with cell culturing and flow cytometry experiments, Dr. B. Pilas for help with flow cytometry and Dr. M. Sivaguru for the help with microscopy.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 8362 kb)
Rights and permissions
About this article

Cite this article
Lazebnik, M., Keswani, R.K. & Pack, D.W. Endocytic Transport of Polyplex and Lipoplex siRNA Vectors in HeLa Cells. Pharm Res 33, 2999–3011 (2016). https://doi.org/10.1007/s11095-016-2022-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-016-2022-1