
ABSTRACT
Purpose
Isomalt is a sugar alcohol used as an excipient in commercially available solid oral dosage forms. The potential of isomalt as a novel freeze-drying excipient was studied in order to increase knowledge of the behavior of isomalt when it is freeze-dried.
Methods
Isomalt was freeze-dried in four different diastereomer compositions and its physical stability was investigated with differential scanning calorimetry, Fourier-transform infrared and Raman spectroscopy, X-ray powder diffraction, Karl-Fischer titration and thermogravimetric analysis in order to verify the solid state form of isomalt after freeze-drying and observe any changes occurring during storage in three different relative humidity conditions.
Results
Isomalt was successfully transformed into the amorphous form with freeze-drying and three diastereomer combinations remained stable as amorphous during storage; one of the diastereomer compositions showed signs of physical instability when stored in the highest relative humidity condition. The four different crystalline diastereomer mixtures showed specific identifiable solid state properties.
Conclusions
Isomalt was shown to be a suitable excipient for freeze-drying. Preferably a mixture of the diastereomers should be used, as the mixture containing only one of the isomers showed physical instability. A mixture containing a 1:1 ratio of the two diastereomers showed the best physical stability in the amorphous form.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Amorphous sugars and sugar alcohols can be used to stabilize protein drugs in freeze-dried and spray dried formulations (1,2). The amorphous form of sugars and sugar alcohols and the effect of water vapor on their crystallization has been extensively studied (3,4). This is because amorphous sugars are generally very hygroscopic by nature and can absorb large amounts of water in their structure. Water vapor acts as a plasticizer for amorphous materials lowering their glass transition temperatures (Tg). If the Tg is lowered to such an extent that it approaches the storage temperature of the product, the crystallization propensity of the amorphous material tends to increase due to greater molecular mobility (4). This will lead to loss in the protective functionality of sugars and their performance in the formulation is compromised.
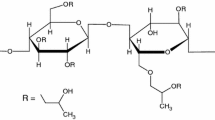
Isomalt is a sugar alcohol (i.e. a polyol) that is used in the food industry as a sugar substitute and in pharmaceutical industry as a tabletting excipient (5–8). Isomalt is produced by hydrogenating isomaltulose in a process containing two steps (5). Firstly, sucrose is transformed into isomaltulose in an enzymatic process. Secondly, the fructose molecule in isomaltulose is hydrogenated and as a result, a mixture of two diastereomers, 6-O-α-D-glucopyranosyl-D-sorbitol (1,6-GPS) and 1-O-α-D-glucopyranosyl-D-mannitol dihydrate (1,1-GPM), is obtained as an approximate equimolar mixture (9) (Fig. 1). This mixture is called isomalt. The two diastereomers, GPM and GPS, possess different physicochemical properties compared to one another. GPM crystallizes as a dihydrate and is less soluble than GPS, which crystallizes as an anhydrate. They also have differing glass transition temperatures and melting points, 66 and 168°C for GPM, and 55 and 166°C for GPS, respectively (10,11). The 3D-str uctures of the two diastereomers differ from one another, as the mannitol chain in the GPM isomer has a nearly planar zigzag conformation but the sorbitol chain in GPS exhibits a different conformation (12).

The structures of the two diastereomers, (a) GPM and (b) GPS, together comprising the sugar alcohol excipient isomalt.
In the pharmaceutical context, isomalt can be used as an excipient in tablets, capsules, sachets, effervescent tablets, chewable tablets, chewing gum, dry powders for suspension and as a sugar-free coating agent (13–15). Crystalline isomalt possesses many positive features, such as good chemical, thermal and microbiological stability, non-hygroscopicity, resistance to acidic and enzymatic hydrolysis and it does not undergo any changes in its molecular structure when melted (16). Isomalt is a non-reducing sugar, which means that it does not cause alterations in the degree of glycation of proteins in freeze-dried formulations during storage (17). Because of this fact, isomalt could be considered as a candidate excipient for freeze-dried drug formulations. Isomalt is included in the United States Pharmacopoeia National Formulary, European and British Pharmacopoeia and in Japanese Pharmaceutical Excipients (JPE) (18–20), and its use as an excipient in pharmaceutical formulations is increasing. The toxicity of orally administered isomalt has been studied in many investigations and it has been found to be a safe excipient through oral administration (21–24). The parenteral toxicity of isomalt has not been yet studied.
Sugars and sugar alcohols are commonly used excipients for protein stabilization during freeze-drying. They usually offer both efficient cryo- and lyoprotection (17,25). Because the number of protein drugs will increase in future, a broader range of excipients for formulation development of protein drugs would be desirable (26). The sugar excipients currently used for protein stabilization in freeze-dried formulations are not able to stabilize all proteins sufficiently, and the limited selection of excipients used, namely disaccharides sucrose and trehalose, is a constriction to formulation scientists. The aims of the present investigation were to thoroughly study the physicochemical properties, including stability, of amorphous isomalt, produced by freeze-drying, and the effect of variation in percentage of GPM and GPS on the freeze-drying and storage stability properties of the excipient. Analysis has involved the synergistic use of diffractometric, thermal and vibrational spectroscopic (coupled with multivariate analysis and quantum chemical calculations) methods. To the best of our knowledge, the freeze-drying properties and storage stability of various forms of isomalt after freeze-drying have not previously been studied in depth which such a range of techniques. Such in depth analysis is necessary in order to evaluate if isomalt is suitable as a novel freeze-drying excipient.
MATERIALS AND METHODS
Materials
Four different grades of isomalt were used in this study: galenIQ™ 721, galenIQ™ 720, Isomalt GM and pure GPM-diastereomer. All of the isomalt grades were kindly donated by Beneo-Palatinit GmbH (Germany). The various isomalt grades contained differing contents of GPM- and GPS-diastereomers (Table I). By using the four supplied isomalt grades, GPM-content of the freeze-dried samples could be varied from 23.5 to 99% and the GPS-content from 0.0 to 75.7%. Thus approximate GPM:GPS isomer ratios of 1:3, 1:1, 2:1 and 1:0 could be studied. The combined GPM and GPS contents of the isomalt grades were 99.2, 99.2, 99.7 and 99.0% for 1:3, 1:1, 2:1 and 1:0 GPM:GPS mixtures, respectively. Pure GPS-diastereomer was not used in the study, because it is not commercially available in sufficient quantities. Highly purified water (Milli-Q, Millipore Inc., USA) was used in all of the studies.
Preparation of the Isomalt Solutions and Their Freeze-Drying
Each of the four isomalt grades was dissolved in highly purified water to produce solutions with 100 mg/ml isomalt concentration. The prepared solutions (2 ml) were transferred to 20 ml freeze-drying vials and the solutions were freeze-dried using Lyostar II freeze-dryer (SP Industries Inc., Warminster, USA). The vials were first equilibrated at 20°C for 10 min and then at 0°C for 30 min. After that the samples were frozen at −40°C for 2 h. Primary drying was performed at −20°C and 150 mTorr for 26 h. Secondary drying was carried out in four steps: the vials were equilibrated at −10°C, 10°C, 20°C and finally at 40°C, each step with a duration of 2 h and chamber pressure of 150 mTorr. After the freeze-drying cycle was complete, the chamber temperature was lowered from 40 to 25°C and the chamber was filled with N2 gas in order to bring the pressure inside the chamber back to atmospheric pressure (1.01 × 105 Pa). After that, the vials were stoppered under N2 atmosphere inside the chamber by compression before opening the chamber door.
Samples used in XRPD tests were freeze-dried with an Epsilon 2-4 LSC laboratory scale freeze-dryer (Martin Christ GmbH, Osterode am Harz, Germany). The freeze-drying cycle used was identical to the one described in the above chapter except for the fact that, due to apparatus restrictions, the chamber could not be filled with N2 gas after the cycle was complete.
Storage Stability Testing of the Freeze-Dried Isomalt Samples
The crystallization tendency of the freeze-dried isomalt samples was studied by storing them in three different relative humidity (RH) conditions at room temperature. The samples were transferred into desiccators straight after the freeze-drying cycle was complete. The vials were stored open without stoppers in desiccators containing phosphorous pentoxide (P2O5), silica beads or saturated salt solution of lithium chloride, thus producing a relative humidity of approximately 2, 9 and 16%, respectively. The humidity conditions inside the desiccators were monitored with Tinytag data loggers (Gemini Data Loggers, Chicester, United Kingdom). The samples were stored in the desiccators for a total period of 23 days and were analyzed at regular intervals of 1, 3, 7, 10, 14, 18 and 23 days. The crystallization tendencies of amorphous isomalt lyophilizates were monitored with differential scanning calorimetry, X-ray powder diffraction, Fourier-transform infrared spectroscopy and Raman spectroscopy. The water content of the samples was studied with Karl-Fischer titration and thermogravimetric analysis during the storage testing. For each storage stability testing point, a number of sample vials needed for analysis on a given day were taken out of the desiccators and rubber stoppers were placed on the vials. All possible steps were taken to minimize ambient moisture uptake during analyses, specifically the vials were always stoppered between taking out powder samples from the vials and the analyses were conducted as rapidly as possible. Each sample vial was used for analyses only during one storage stability time point so the analyses were always carried out from untouched sample vials.
Storage stability test samples for XRPD studies were stored in desiccators containing either silica beads or saturated salt solution of lithium chloride, thus producing a relative humidity of approximately 9 and 16%, respectively. The sample vials were stored as open vials without stoppers.
The GPM anhydrate and amorphous 1:1 GPM:GPS mixtures produced by melting were prepared as a reference for storage stability studies. GPM anhydrate was produced by drying GPM dihydrate (1:0 GPM:GPS) in an oven (Memmert, Schwabach, Germany) at 105°C for 30 min and allowed to cool in a desiccator containing silica beads until room temperature. The 1:1 GPM:GPS mixture was melted rapidly with a hot plate and left to cool to room temperature in ambient conditions. After reaching room temperature, the solidified melt was ground gently with a mortar and a pestle.
Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) was used to support the freeze-drying cycle design and to monitor the crystallization of the lyophilized isomalt samples during their storage. The glass transition temperatures of the maximally freeze-concentrated aqueous solutions (Tg’) of different isomalt grades were detected from 50 mg/ml solutions with DSC823e (Mettler-Toledo International Inc., Greifensee, Switzerland). The solutions were transferred into aluminum DSC pans in a quantity of approximately 12 mg and were first cooled to −40°C at 1°C/min and held there for 10 min. After that the samples were heated from −40°C to 10°C at 5°C/min and the Tg’ of each isomalt grade was detected from the acquired thermograms. The measurements were carried out under 50 ml/min N2 purge.
The glass transition temperatures (Tg) of the freeze-dried isomalt samples were detected at regular intervals (1, 3, 7, 10, 14, 18 and 23 days) during storage by using standard and modulated DSC runs. For standard DSC runs 3–7 mg and for modulated DSC runs 9–15 mg of the sample was transferred in an aluminum DSC pan and sealed hermetically. The samples were prepared in a glove box with controlled humidity conditions. Each sample for standard DSC run was made in triplicate and the samples were taken from different vials (n = 3). The standard DSC experiments were carried out by first equilibrating the samples at 25°C for 5 min and then heating them to 150°C at 10°C/min under 50 ml/min N2 purge. The modulated runs were executed as TOPEM experiments. The samples were equilibrated at 25°C for 5 min and then heated to 110°C with a heating rate of 1°C/min. For all TOPEM experiments, a temperature amplitude of the pulses of 0.5°C was selected with a switching time range of pulse width with minimum and maximum values of 15 and 30 s, respectively. The results were analyzed with STARe Thermal Analysis Software (Mettler-Toledo International Inc., Greifensee, Switzerland).
The samples freeze-dried separately for XRPD analysis were investigated to confirm that they matched the formerly freeze-dried samples with DSC analysis. The DSC studies were carried out using a Discovery DSC (TA Instruments, Newcastle, DE, US). Samples were crimped hermetically into pans. The standard runs were recorded from 25 to 150°C using a 10°C/min heating rate. Each sample was measured in triplicate from different vials (n = 3). The modulated runs were recorded from 25 to 125°C using a 1°C/min heating rate, a modulation temperature amplitude of 1°C and a period of 60 s. Sample amounts for standard runs and modulated runs were 3–5 and 6–8 mg, respectively. Data were collected using Trios software (TA Instruments).
X-ray powder diffraction
XRPD was used to analyze the solid state properties of raw materials and isomalt lyophilizates after freeze-drying and during storage. Samples were studied in duplicate and taken from different vials (n = 2). XRPD analysis was performed using a PANalytical X’Pert PROMPD X-ray diffractometer (PANalytical, Almelo, The Netherlands) and a Ni filtered Cu Kα1 radiation (λ = 1.541 Å). Samples were measured in Bragg Brentano reflection mode in the range of 5–36°2θ using a PIXel detector (PANalytical) and a step size of 0.039°2θ. The operating current and voltage were 40 mA and 45 kV, respectively. Data were collected using X’Pert Data Collector (PANalytical). For non-isothermal (variable temperature) XRPD (VT-XRPD) measurements an Anton Paar CHC chamber (Anton Paar GmbH, Graz, Austria) was mounted on the goniometer of the diffractometer. A 0.2 mm deep sample holder was used. VT-XRPD scans were obtained from 25 to 115°C and the diffractograms were measured at 25, 45, 75, 90 and 115°C with a heating rate of 26°C/min in average between each step. The heating rate was controlled by TCU 110 temperature controller (Anton Paar GmbH) and the samples were equilibrated in each temperature step for 3 min before initiating the scan. The measurements were carried out in ambient relative humidity conditions. Calculated X-ray diffractograms obtained from Cambridge Structural Database were used as references. The reference structures were BAVCAC for the GPS diastereomer (27) and BAGZEO for the GPM diastereomer (28).
Spectroscopic Analysis
Fourier-transform infrared spectroscopy (FT-IR) and Raman spectroscopy were used to detect changes to the solid state of the isomalt samples before and after freeze-drying and during storage at regular intervals of 1, 3, 7, 10, 14, 18 and 23 days. FT-IR measurements were carried out with a Vertex 70 FT-IR spectrometer (Bruker Optics Inc., Germany) using a Miracle™ single reflection attenuated total reflectance (ATR) accessory (Pike technologies, Wisconsin, USA). All samples were analyzed in triplicate and the samples were taken from different vials (n = 3). The FT-IR spectra were recorded with OPUS 5.5 software (Bruker Optics Inc., Germany) over the spectral region 650 to 4000 cm-1 with 4 cm-1 resolution. Each spectrum was a composite of 64 co-added scans.
Raman spectroscopy was performed with a Raman Rxn1 system (Kaiser Optical Systems, Inc., MI, USA). The spectrometer was equipped with a PhAT probe consisting of an array of 50 optical fibers, a 785 nm excitation laser source and silicon charge-coupled device (CCD) detector. The spectra were recorded in triplicate from different vials (n = 3). HoloGRAMS 4.1 program (Kaiser Optical Systems, Inc., MI, USA) was used to record the spectra with an integration time of 2 s by 5 co-added scans.
Water Content Measurements
The residual water content of the freeze-dried samples was measured with Karl-Fischer titration (KF) and thermogravimetric analysis (TGA). The measurements were carried out straight after freeze-drying and during storage under different relative humidity atmospheres with V30 Volumetric KF Titrator (Mettler-Toledo International Inc., Greifensee, Switzerland). The KF measurements were conducted from the same sample vials as DSC, FT-IR and Raman experiments. The lyophilization vial was weighed, the residual sample amount of approximately 80 mg (n = 3) was poured in the KF titration vessel and the vial was weighed yet again to calculate the sample mass. The results are presented as an average of three sample vials.
TGA analysis was conducted for some of the samples to verify the results of the KF measurements. The studies were carried out using a Discovery TGA (TA Instruments, Newcastle, DE, US). Samples of approximately 7 mg were weighted into open platinum pans. The runs were recorded from 25 to 150°C using a 10°C/min heating rate. Data were collected using Trios software (TA Instruments, Newcastle, DE, US).
The theoretical water contents of the raw materials were calculated based on the mass percentage of GPM and GPS in each raw material using the molecular masses of GPM dihydrate (380.32 g/mol), GPS/GPM anhydrate (344.32 g/mol) and calculating the theoretical water content resulting from dihydrate water of GPM in each GPM:GPS mixing ratio.
Computational Quantum Mechanical Modelling
Quantum chemical calculations of fundamental molecular vibrational modes were performed using the Gaussian09 package (29). Geometry optimization and harmonic vibrational frequency calculations were performed in vacuo using density functional theory (DFT), employing the B3LYP functional. Calculated vibrational spectra were generated using Gaussum v2.2.5 software, and scaled to give the lowest value for the mean absolute deviation for band position from experimental data, with scale factors typically around 0.975. Vibrational modes were illustrated using Molden (30).
Multivariate Data Analysis
In order to better understand the differences between the raw materials and stability of processed samples over time, principal component analysis (PCA) of the FT-IR and Raman spectral data were performed with Simca software (version 13.5 Unicode, Umetrics AB, Umeå, Sweden). For the most effective comparison, the most relevant regions of the spectra were chosen with regions containing little to no useful vibrational information excluded. The data was pre-processed with standard normal variant (SNV) transformation and mean-centering before PCA to remove intensity variation of the spectra unrelated to sample composition. The FT-IR spectra were pre-processed and analyzed over the spectral regions 820 to 1500 and 2750 to 3600 cm-1. The Raman spectra were pre-processed and analyzed over the spectral region 300 to 1500 cm-1. PCA was carried out with 7 groups for cross validation.
RESULTS
Characterization of Raw Materials and Freshly Freeze-Dried Isomalt
Diffractograms of the raw materials of different GPM:GPS mixing ratios are presented in Fig. 2. Based on the measured diffractograms the raw materials are clearly crystalline and the differences in diastereomer contents are also reflected in the diffractograms as they are slightly different from one another. The raw materials containing both isomers, 1:3, 1:1 and 2:1 GPM:GPS, share peaks at the same positions, but in many cases their intensities increase or decrease as the GPS percentage in the raw material increases. Peaks at positions 15.6, 17.3, 22.0, 22.7/22.8, 24.1/24.2 and 25.4/25.5 °2θ increase in intensity as the GPS content increases. The peaks in some cases also shift by 0.1 °2θ to the right with increasing GPM concentration. The X-ray diffractogram of the raw material containing only GPM diastereomer, 1:0 GPM:GPS, deviates clearly from the other raw material diffractograms. It is affected by preferred orientation, because the crystal habit of the 1:0 GPM:GPS raw material was short needle-like crystals and they were not ground for the measurement. The diffractogram of 1:0 GPM:GPS raw material shares the same peaks as the literature reference for GPM dihydrate from Cambridge Structural Database (CSD) (28). Some of the common peaks are highlighted in Fig. 2. It can be seen from the literature reference diffractogram of GPS (BAVCAC) that the peaks at 15.6, 17.3 and 22.0 °2θ clearly originate from the GPS isomer for they are absent from the GPM isomer diffractogram (27). The peaks at the aforementioned positions in the measured raw material diffractograms show a clear trend of growing intensity as their GPS percentage increases. In contrast, the peaks at 18.8 and 21.3 °2θ originate from GPM isomer, as they are present only in the GPM dihydrate reference diffractogram and in the measured raw material diffractograms, which show a trend of the peak at 21.3 °2θ increasing in intensity as the GPM concentration of the raw materials increases (Fig. 2). As isomalt mixtures are composed of two diastereoisomers, the molecular structure of isomalt could potentially either be a cocrystal, eutectic or physical mixture. Based on the measured raw material X-ray diffraction data, the GPM:GPS mixtures form eutectics because the mixtures have similar unit cell structures to those of the components they are composed of. If they would form cocrystals, the X-ray crystal structure of the mixtures would be different from those of the individual diastereomers, GPM and GPS (31). After the freeze-drying process was completed, the diffractograms of the freshly freeze-dried samples were measured and they showed clear amorphous halos suggesting that all of the different GPM:GPS mixing ratios were successfully transformed into the amorphous form during the process (Fig. 2).

The X-ray powder diffractograms of raw materials and freeze-dried GPM:GPS mixtures and calculated diffractograms BAGZEO and BAVCAC from CSD shown as reference.
The different GPM:GPS mixing ratios were analyzed using complementary vibrational spectroscopic methods IR and Raman spectroscopy. The FT-IR spectra of all of the raw materials with varying diastereomer contents were very complex, as it is typical for sugar alcohols and sugars (32). FT-IR and Raman spectra of the raw materials revealed that different mixing ratios of the diastereomers could be identified based on differences in the spectra (Figs. 3a and 4a). In the IR and Raman spectra there was a trend in the observed band positions and intensities when moving from high GPM content to high GPS content. Despite the GPS not coming in the 100% form, the spectral features associated with this isomer were deduced by spectral subtraction from the high GPS content sample.

FT-IR spectroscopic analysis of the raw and freeze-dried materials. (a) FT-IR spectra of the various GPM:GPS ratio materials before and after freeze-drying. (b) PCA scores plot of the FT-IR spectra for the raw and freeze-dried materials and the effect of different storage conditions over time. The first two PCs describe 67% of the total spectral variance with the loadings for PC1 (c), and PC2 (d) also given.

Raman spectroscopic analysis of the raw and freeze-dried materials. (a) Raman spectra of the various GPM:GPS ratio materials before and after freeze-drying. (b) PCA scores plot of the Raman spectra for the raw and freeze-dried materials and the effect of different storage conditions over time. The first two PCs describe 74% of the total spectral variance with the loadings for PC1 (c), and PC2 (d) also given.
The vibrational frequencies for the Raman and IR spectra for the separate isomers GPM and GPS were calculated using DFT to assign the vibrational transitions observed in the experimental spectra. The vibrations tend to be centered on the chain or the ring. An illustration of chain-centered, ring-centered and non-centered vibrations are illustrated in Figure S1 in the supplementary material. These vibrational locations have been used to describe the vibrational assignments for GPM and GPS. The calculated assignments are given in Table SI. These assignments are consistent with what has been reported previously in the literature (32).
The IR spectra show many noticeable changes to peak position and intensity across the vibrational spectra in the 650 to 1500 and 2800 to 3600 cm−1 spectral regions. Some notable changes include the shifting of the chain centered mixed vibration at 1005 cm−1 in GPM to 991 cm−1 in GPS, the presence of the mixed (non-centered) transition at 1038 cm−1 in GPS, which is barely observable as a shoulder in GPM. Features at 1057 (mixed non-centered), 1086 (mixed chain centered) and 1107 cm−1 (mixed chain centered) in GPM shift to 1038, 1079 and 1115 cm−1 in GPS, respectively. There are also numerous changes in the 830–950 cm−1 region associated with mixed (ring, chain and non-centered) transitions and the 1150 to 1500 cm−1 region, associated with numerous C-C-H wagging and mixed transitions. The O-H stretching region also contains significant differences, particularly the presence of the band at 3362 cm−1 in GPM which is absent in GPS and the band at 3522 cm−1 present in GPS which is absent in GPM.
The FT-IR spectra of the different mixing ratios measured after freeze-drying were nearly identical as observed in Fig. 3a. When multivariate analysis methods were applied, these spectra formed a general trend in PC space instead of distinct groups further exemplifying the similarity in these freeze-dried spectra. Generally the multiple peaks in the region of 1200–950 cm−1 in the raw material spectra have merged together forming broad features around 1150 and 1017 cm−1 and a shoulder ~1074 cm−1 in the spectra measured after freeze-drying. This type of broadening and shifting of the bands are commonly seen in the formation of the amorphous form. This is consistent with the XRPD results which also suggests the formation of the amorphous form upon freeze-drying.
Like IR, the Raman spectra of GPM and GPS have observable spectral differences. Whilst there are many subtle shifts in band position and intensity, some notable differences include a shift in the feature at 532 cm−1 (O-H wag, chain centered) in GPM to 546 cm−1 (mixed non-centered) in GPS, and the presence of a mixed chain centered transition at 665 cm−1 and a mixed ring centered transition at 834 cm−1 in GPS which is absent in GPM. GPM contains a C-C ring centered transition at 855 cm−1 which is absent in GPS. The intensity of the mixed ring centered transition at 875 cm−1 is more intense in GPM, with GPS having a second peak at 886 cm−1 assigned as a mixed ring centered transition.
Like what was observed with the IR data, the Raman spectra of all the freeze-dried GPM:GPS mixtures were nearly identical as seen in Fig. 4a. After freeze-drying the spectrum is clearly different to the raw material spectra with broadening and merging of peaks in the regions 800–950 and 1000–1200 cm−1. This broadening and shifting of the bands after freeze-drying is consistent with changes expected to be observed when going from a crystalline to amorphous state, in which the increased disorder in the samples leads to unique stretching environments of slightly different energies (band broadening).
DSC measurements of the raw materials showed differences in thermal behavior between different isomer mixing ratios (Table II). The 1:3, 1:1 and 2:1 GPM:GPS mixtures exhibited a dehydration peak at 99-105°C, after which the 1:1 and 2:1 GPM:GPS mixtures showed a dissolution peak of anhydrous GPM and GPS at 128°C and 135°C, respectively. The 1:3 GPM:GPS mixture showed a melting peak of anhydrous GPM and GPS at 151°C. The 1:0 GPM:GPS mixture exhibited three peaks in its thermograms: first a melting peak of GPM dihydrate at 112°C, then an exothermic crystallization peak to anhydrous GPM at 121°C and finally a dissolution/melting peak of GPM anhydrate at 127°C. The glass transition temperatures of maximally freeze-concentrated isomalt solutions (Tg’) were measured for each diastereomer grade before freeze-drying using DSC and values of –35, –34, –34 and –32°C were acquired for 1:3, 1:1, 2:1 and 1:0 GPM:GPS mixtures, respectively.
The isomalt samples exhibited good cake structures after freeze-drying and no collapse was observed (Fig. 5). They all had glass transition temperatures below 65°C (heating rate of 1°C/min), the Tg rising as the residual water content of the different mixtures decreased (Table II). Table II shows the residual water contents of the freeze-dried samples varied between 0.5 and 3.1%. The residual water contents also showed a trend as water content rose as the GPS content increased. The measured water contents of the raw materials matched well with their theoretical values and the water content of the different GPM:GPS mixes rose as the GPM concentration increased (Table II).

The cake appearance of freshly freeze-dried isomalt samples.
Storage Stability Testing
All of the four studied isomalt grades had a good cake structure after freeze-drying and they retained this structure well throughout the storage stability studies in all of the three relative humidity conditions. No cake collapse was detected in samples stored even in the highest relative humidity condition (16%). The diffractograms measured with variable temperature XRPD during storage for up to 23 days revealed no signs of crystallization. All GPM:GPS mixing ratios consistently showed amorphous halos even during storage in the highest humidity condition, 16% RH. During storage stability testing, as the samples were heated up to 115°C with VT-XRPD, all of the samples showed amorphous diffractograms all the way during heating at 25, 45, 75, 90 and 115°C (data not shown).
DSC results showed some clear differences between the different isomer mixtures during storage in different relative humidity conditions. The mixture containing the most GPS isomer, 1:3 GPM:GPS, was stable as amorphous when stored in 2 and 9% RH desiccators (Table IIIa). When these samples were stored at 16% RH, crystallization exotherms were observed during heating after 1 and 23 days of storage. The freeze-dried 1:1 GPM:GPS isomer mixture was physically the most stable out of the four mixtures based on DSC results, because it did not crystallize during heating after storage at any of the relative humidity conditions (Table IIIb). As the GPM content increased in the mixtures, more events started to occur in the thermograms measured during storage. The 2:1 GPM:GPS isomer mixture showed no crystallization peaks in any of the humidity storage conditions but the thermograms had an endotherm around 60–80°C, slightly varying depending on the humidity at which the samples were stored. This endotherm could be already seen when the samples were measured straight after freeze-drying and it remained in the thermograms throughout storage at all of the relative humidities (Table IIIc). The 1:0 GPM:GPS samples containing only GPM isomer showed multiple events in the thermograms. Straight after freeze-drying and during storage in 2 and 9% RH, the thermograms showed two endothermic events in the region of approximately 70–100°C (Table IIId). When stored in 16% RH, in addition to the two endotherms between approx. 60 to 85°C, there was also a crystallization peak around 100°C which was seen immediately after 1 day of storage and throughout the storage time of 23 days. The residual water measurements showed that as the humidity percent during storage was higher, the samples also contained higher water amounts, as expected (Table III). Freeze-dried isomalt samples were also stored in 23% RH and in these conditions some of the diastereomer mixtures (1:3 and 1:1 GPM:GPS) collapsed and the Tg of the samples was not visible anymore so they did not remain stable in this higher RH condition (results not shown). It must be however noticed that all of the vials were stored open without stoppers, so 23% RH is a high humidity condition for unprotected freeze-dried cakes.
PCA of the IR spectra collected from the raw materials and freeze-dried samples stored under different conditions over time was used to inspect the spectral differences observed in the samples and monitor changes to the freeze-dried samples over time. The scores plot for the first two principal components is given in Fig. 3b where it can be seen that PC1 separates crystalline GPM signal (positive PC1) from the amorphous freeze-dried spectral signals (negative PC1). The second PC then separate the GPS signal (positive PC2) from the GPM signal (negative PC2). This separation is corroborated with the associated loading plots (Fig. 3c and d) which contains spectral features consistent with GPM in positive PC1 loadings and with amorphous in negative PC1 loadings and positive PC2 loadings with GPS like spectral features and negative with GPM like spectral features.
The raw materials distinctly separated based on the GPM:GPS ratios, however the freeze-dried samples only gave a trend based on the GPM:GPS content. This is likely due to the amorphous form having less distinct spectral features in which to separate these samples. All freeze-dried samples which were stored under different conditions did not appear to move significantly in PC space indicating that these samples were stable and did not recrystallize in the timeframe studied.
GPM anhydrate was produced and its spectra were included in the PCA plots in order to detect differences between GPM dihydrate and anhydrate, and also see if the freeze-dried samples showed similarities with it. Both PCA plots separate GPM dihydrate from the anhydrate form (Figs. 3b and 4b) showing differences in their FT-IR and Raman spectral band positions. The freeze-dried samples do not show similarities with GPM anhydrate spectra, for they are separated along PC1 which differentiates amorphous and crystalline (Figs. 3b and 4b). Amorphous 1:1 GPM:GPS produced by melting was also added to the PCA plots to compare if the amorphous forms of isomalt differ when produced in different methods. Molten amorphous 1:1 GPM:GPS shows clear similarities to the freeze-dried amorphous 1:1 GPM:GPS, although it is slightly separated from the freeze-dried samples along PC2 (Fig. 3b) and PC1 (Fig. 4b). Isomalt has very good thermal stability and does not undergo changes in its molecular structure when melted, so the negligible changes between molten and freeze-dried amorphous isomalt are not related to thermal degradation. The cause of this is different conformations of isomalt in solutions and in a melt (33).
PCA analysis was also performed on the Raman spectra. The resulting scores plot (Fig. 4b) shows clear separation of the freeze-dried samples from the raw materials along PC1 which is associated with 59% of the overall variance between the spectra. Inspection of the associated loadings plot (Fig. 4c) shows spectral features consistent with crystalline material in positive PC1 and features consistent with the amorphous/freeze-dried material in negative PC1 space.
The second PC separates based on the differences between the GPM and GPS spectra, with the highest proportion of GPM (and GPM anhydrate) signal separating into positive PC2 space and highest levels of signal associated with GPS in negative PC2 space. The associated loadings plot (Fig. 4d) shows spectral signals consistent with this separation. The raw materials separate more strongly than their freeze-dried counterparts. However a trend along PC2 based on the ratio of GPM:GPS is still observed for the freeze-dried samples.
The Raman based PCA suggests that after 23 days storage at 2, 9 or 16% relative humidity, the freeze dried material remains amorphous for all GPM:GPS ratios in the time period studied. In general, both the FT-IR and Raman score scatter plots had no clear differences between the positions of samples stored in different storage conditions (Figs. 3b and 4b).
Crystallization Tendency of the 1:0 GPM:GPS Isomer Mixture During Heating
When the isomalt grade containing only the GPM diastereomer was stored in 16% RH, crystallization peaks could be seen in the modulated DSC runs around 100°C. The FT-IR spectrum of the sample remaining in the hermetically sealed DSC pan after heating to 110°C at 1°C/min was measured to verify its crystallinity. The resulting FT-IR spectrum showed clearly that the sample was crystalline after the slow heating with spectral features consistent with the anhydrate rather than the dihydrate (starting material) form (Fig. 6). To verify whether the 1:0 GPM:GPS stored in 16% RH crystallizes when it is heated at a faster rate than 1°C/min, a small amount of the sample was heated on a hot plate set to 110°C and heated as fast as possible (rate not controlled) with the FT-IR spectrum collected after cooling. The comparison of FT-IR spectra of these two samples heated at a different rates clearly showed that 1:0 GPM:GPS remained amorphous when heated with a fast heating rate (Fig. 6). The same studies, where the FT-IR spectrum was measured after heating, were also performed for the other mixtures containing 1:3, 1:1 or 2:1 GPM:GPS stored in 16% RH, but only amorphous spectra were observed (data not shown), resulting in the conclusion that the other mixtures remained amorphous during the slow heating rate up to 110°C.

FT-IR spectra of the 1:0 GPM:GPS mixture after 7 days of storage in 16% RH as is (dark green dashed) and after heating to 110°C with slow (grey) and fast (black) heating rates in comparison to the anhydrate (pale green) and dehydrate form (solid dark green).
Variable temperature XRPD measurements conducted in non-hermetic conditions, where the 1:0 GPM:GPS samples stored in 16% RH were heated to 115°C at 1°C/min and the diffractogram was measured during heating at 5 temperature steps, were used to see if any crystalline peaks could be seen. The slow heating rate was used to simulate the modulated DSC runs, that produced the crystallized 1:0 GPM:GPS samples. Despite the slow heating rate, the X-ray powder diffractograms showed only amorphous halos and no peaks could be observed even when measured at 115°C (data not shown).
DISCUSSION
The raw materials of different GPM:GPS isomer mixtures were studied with various complimentary analytical techniques and based on the results it can be concluded that the crystalline raw materials could be identified from each other even though they consist of simply two diastereomers mixed in different ratios. The diastereomers of isomalt form eutectic mixtures based on X-ray diffraction and DSC results obtained from the crystalline raw materials and based on former knowledge of isomalt (34,35). Unfortunately, as the pure GPS diastereomer was not able to be included in the study, a full series of the mixing ratios was not possible. Therefore the results of the samples with a high GPS content cannot be fully compared to the solid state properties of pure GPS isomer.
The measured melting points of the raw materials, between 127 and 151°C, are lower than those reported for pure GPM and GPS isomers, 168 and 166°C, respectively (11). In addition to these high melting point values, a melting point of 107.9°C has been reported for the GPM isomer, describing that GPM dihydrate melts at this temperature, after which the crystal water begins to evaporate and after that peaks caused by evaporation of residual crystal water and melting of GPM monohydrate at 120–150°C are observed, and finally the melting point of GPM anhydrate takes place at approx. 170°C (36). Because the existence of GPM monohydrate has not been confirmed by X-ray diffraction, this conclusion partly lacks evidence and thus in this current study our speculation is that first a melting peak of GPM dihydrate at 112°C is observed, then a crystallization peak to anhydrous GPM at 121°C and finally a dissolution/melting peak of GPM anhydrate at 127°C, for some proof is found in the literature for this conclusion (35). Our results and the literature studies suggest that the thermal behavior of the GPM diastereomer is quite complex. This is also reflected in the thermal behavior of the different GPM:GPS mixes. Mixtures containing the two isomers are reported to have a clearly lower melting point compared to their constituents alone (10,34). The two diastereomers form a eutectic mixture between the isomers, and this mixture has a significantly lower melting temperature compared to the pure isomers, with the melting peak position depending on the ratio of the isomers (34). Eutectics traditionally exhibit this behavior (37). It has been reported that isomalt mixtures containing various amounts of the two diastereomers, with composition of the GPS isomer ranging from 45 to 100%, were observed to have a broad melting peak in the range between 130 and 154°C (34). In this current study a melting peak at 151°C was observed for the 1:3 GPM:GPS mixture, complying well with these previous measurements. The 1:1 and 2:1 GPM:GPS mixtures showed an endothermic peak at 128 and 135°C, respectively, and these peaks were interpreted as dissolution peaks of anhydrous GPM and GPS based on their shapes, which indicate a dissolution process starting at the end of the dehydration process and proceeding with increasing temperature. The possible difference in the sealing method of DSC pans (hermetic/non-hermetic) between this current study and the literature references can affect the melting behavior of different isomalt grades and the melting peak positions in their thermograms. Also the fact that the raw materials were not ground for the DSC measurements of this current study might have a lowering effect on the melting peak positions of isomalt (38) and it may also have an effect on the water content of the crystalline raw materials thus also affecting their melting points.
The vibrational spectral data clearly shows differences between the crystalline raw materials and freeze-dried samples. As seen in Fig. 3, the peaks in FT-IR spectra of the freshly freeze-dried samples are broader and less sharp compared to the raw materials, which is typical for amorphous materials in general. The assigned bands of the measured FT-IR spectrum of 1:0 GPM:GPS raw material match very well with results previously published regarding the FT-IR spectrum of GPM dihydrate as are the assignments of the vibrational transitions based on our DFT calculations (Supplementary information, Table SI) (32).
Based on the diffractograms, thermograms and Raman and FT-IR spectra measured straight after freeze-drying, all of the isomalt grades with different GPM:GPS mixing ratios were successfully transformed into amorphous during the freeze-drying process and they did not crystallize during the process. The different isomer mixing ratios all exhibited only one Tg, demonstrating that they are single phase systems. For pure GPM and GPS isomers, Tgs of 65.8 and 55.0°C have been previously measured with heating rate of 10°C/min (11). Because of the differences in heating rates and in residual water contents, the results are not comparable to those of the current study. When the samples of this study were measured with standard DSC runs with the heating rate of 10°C/min, the glass transition temperatures measured straight after freeze-drying were more in line with those reported previously (10). Despite this, the results from the modulated runs with heating rate of 1°C/min showed the thermal events much more clearly and precisely, and were thus presented in Tables II and III. The Tg values measured with samples stored in 2% RH having a low residual moisture contents were very close to the previously reported literature values for pure GPM isomer and for 1:1 mixture of GPM and GPS (10,11). A Gordon-Taylor model has been previously plotted for amorphous isomalt and experimental measurements with isomalt containing up to approximately 22% (w/w) water complied well with the theoretical Gordon-Taylor equation values (35).
As seen in Table III, the 1:1 GPM:GPS mixture showed no other events than a glass transition in any of the storage humidity conditions tested, which demonstrates its excellent stability in the amorphous form. The 1:1 mixture behaves like a single phase system since only one Tg and no other events are detected in DSC analysis. It can be concluded to be the most stable mix in the amorphous form out of the four isomer compositions studied. The 1:1 ratio has been previously found to be the most stable one, when blends of two excipients other than isomalt where studied for their physical stability in the amorphous form (39). The favorable stability of the 1:1 mixture was supposed to result from stabilization of molecular interactions, which can be the case also with isomalt. The 1:3 GPM:GPS mixture also showed very good stability, although when stored in 16% RH, exothermic events, interpreted as crystallization, were observed on two stability testing time points. Because crystallization is a stochastic phenomenon, it is reasonable that crystallization was not observed in all samples. In the thermal measurements of the 2:1 GPM:GPS mixture, an endothermic event was observed already when measured after freeze-drying and throughout the storage stability testing in all humidity conditions. The position of the endotherm varied slightly between the humidity conditions but was consistently in the same area during the 1–23 days of stability testing of a given humidity condition. Given the area in which the endotherm was observed, between 55 and 85°C, and based on their appearance in the modulated DSC measurements, the endotherms, showing as non-reversing events, were concluded to be caused by evaporation of water. Thermogravimetric analysis of the samples showed loss of weight during heating starting approximately at 50°C, but the difference in the heating rates and pan types (open in TGA vs hermetically sealed in modulated DSC) of the TGA and modulated DSC runs results in difference as to where the water evaporation begins to occur. The 1:0 GPM:GPS mixture showed the most complex thermal behavior. All of the samples showed a glass transition temperature, indicating that the samples remained amorphous in all of the humidity conditions, 2, 9 and 16% RH. Two endothermic events, in some cases partly overlapping, were observed after freeze-drying and throughout the storage stability studies. The endothermic events were in the region between 60 and 100°C and given the region, as with the 2:1 GPM:GPS mixture, the non-reversing endothermic events were resulted from water removal. When the mixture containing only GPM was stored in the highest humidity condition, 16% RH, a clear and sharp crystallization peak was seen around 100°C, which was not present in the thermograms of the samples stored in 2 or 9% RH. 1:0 GPM:GPS mixture adsorbed more water at higher relative humidity and it crystallized readily during the slow heating of the modulated DSC run. Based on literature, amorphous isomalt, the diastereomer content of which was not specified, has been observed to remain partly amorphous for up to 25 days in very high relative humidity conditions (80% RH), also suggesting that isomalt is relatively stable as amorphous (40).
The water content measurements straight after freeze-drying revealed that the different mixtures 1:3, 1:1, 2:1 and 1:0 GPM:GPS contained 3.1, 2.8, 0.8 and 0.5% of water, respectively. The TGA measurements (data not shown) showed that after freeze-drying the GPS containing mixtures had a loss of weight profile occurring in two steps under 150°C, while the 1:0 GPM:GPS mixture showed only one step. Weight loss at the given area is caused by water loss and the TGA results indicate that the GPS containing mixtures contain water in two phases, one from which water is easily removed after Tg, and one from which water requires more energy to be removed, caused possibly by being part of structures with some degree of order in the amorphous phase. Because of this, the GPM:GPS mixtures containing GPS dry inferiorly during freeze-drying compared to pure GPM isomer. During storage, the mixtures with the highest GPS content, 1:3 and 1:1 GPM:GPS, became drier when stored in 2 and 9% RH, and retained approximately the same level of water content when stored in 16% RH, when compared to the results measured straight after freeze-drying. The mixtures with high GPM content, 2:1 and 1:0 GPM:GPS, had a very low water content after freeze-drying and it remained at the same level when they were stored in 2% RH, but increased as the humidity of the storage condition was higher, growing up to 2.3 and 2.5%, respectively, during storage in 16% RH. The pure isomers, GPS and GPM, in the amorphous form are hygroscopic and adsorb large amounts of water, especially when stored in high relative humidity conditions (41). The hygroscopic nature of amorphous isomalt after freeze-drying is evident in the water content measurements of the 2:1 and 1:0 GPM:GPS mixtures, which adsorbed water even when stored in as low a humidity as 9% RH. Even though the studied samples did not crystallize in room temperature during the study period, the adsorbed water acts as a plasticizer which promotes crystallization.
The PCA analysis of the spectral data during the storage stability testing confirmed that no crystallization of the amorphous samples happened in any of the humidity conditions. No trends of crystallization in the amorphous samples could be seen in the PCA scatter plots of FT-IR or Raman spectra. In both of the PCA models, there were trends in one or both PC’s that separated crystalline and amorphous samples, it would be along this axis that changes in crystallinity would be expected to be observed. The amorphous GPM:GPS mixtures containing different ratios of the two diastereomers formed a trend with increasing GPM content, even though overlapping partly in the PCA plots. Samples stored in different relative humidity conditions among all of the amorphous samples of a given mixture were randomly situated compared to one another and no trends of increasing humidity in the storage conditions could not be noticed (Figs. 3b and 4b). It would be expected that if crystallization were occurring at the surface of the freeze dried particles the ATR-IR would detect this over the Raman measurements as with the instruments utilized in this study the Raman measures the bulk material and the ATR-IR measures the particle surface in contact with the ATR-IR crystal. Previously, crystallization of approx. 1:1 GPM:GPS mixture of amorphous isomalt (produced by melting) was tried to inhibit by adding oligomeric or polymeric additives to isomalt, but it was noticed that pure isomalt samples had the least crystal growth compared to samples with additives (42). This supports the good physical stability of amorphous isomalt, especially the 1:1 GPM:GPS mixture, proven in this current study.
The 1:0 GPM:GPS mixture remained amorphous and no crystallization peaks were observed in thermograms when stored in 2 and 9% RH. When stored in 16% relative humidity, the mixture adsorbed higher amounts of water and was more susceptible for crystallization during slow heating in DSC, even though it still remained at least partly amorphous in room temperature up to 23 days. According to Dr. Karin Gehrich from BeneoPalatinit GmbH, moisture has an effect on the crystallization tendency of isomalt and it can even undergo cold crystallization in the presence of moisture (Karin Gehrich, oral communication, 18th November, 2015). When the FT-IR spectrum of the sample containing only GPM isomer, stored in 16% RH, was measured after heating it in a hermetically sealed DSC pan at 1°C/min to 110°C, a crystalline spectrum was observed. The slow heating rate made it possible for the 1:0 GPM:GPS amorphous mixture to crystallize at about 100°C. No crystallization happened when the same sample was heated rapidly on a hot plate, nor did the diffractograms, obtained from standard DSC runs with a heating rate of 10°C/min, show any crystallization exotherms, possibly due to the fact that the heating rate was too fast for nucleation and subsequent crystallization to occur. It is noted that no signs of the incipient instability of the 1:0 GPM:GPS amorphous mixture stored in 16% RH, were seen in either FT-IR or Raman PCA analysis. The X-ray powder diffractograms measured from the 1:0 GPM:GPS samples stored in 16% RH with heating at 1°C/min to 115°C showed only amorphous halos throughout the measurements up to 115°C, which suggests that either the crystals were too small for the XRPD to detect or that the crystallization event is sensitive to ambient moisture. The XRPD measurement was an open system, where the sample was inevitably exposed to moisture. The obtained crystalline FT-IR spectrum from the slowly heated 1:0 GPM:GPS sample was consistent with anhydrate forms of GPM isomer (Fig. 6). It could be concluded that the 1:0 GPM:GPS sample, stored in 16% RH, remained predominantly amorphous during storage, however slow heating in a hermetically sealed DSC pan resulted in crystallization into the GPM anhydrate. This was not observed for any of the other isomer combinations, further demonstrating the stabilizing effect of the combination of the two diastereomers.
Sucrose and trehalose are the most frequently used cryo- and lyoprotecting sugar excipients in freeze-dried protein formulations. Like isomalt, sucrose and trehalose can be transformed from crystalline to amorphous with freeze-drying and another important common feature which they share is that they are all non-reducing sugars. Almost all disaccharides and polyols used as protein stabilizing excipients in freeze-dried formulations are non-reducing by nature because reducing sugars can cause changes in the degree of glycation of proteins, which can be harmful to the proteins and affect their biological response. The physical stability of amorphous sucrose and trehalose have been extensively studied and they have been found to have good physical stability in the amorphous form (43,44). Amorphous trehalose has been found to be more robust and have a lower crystallization tendency than amorphous sucrose (3). At 23°C and 33% RH amorphous sucrose has been observed to crystallize after 10 days of storage. Although isomalt was not studied in humidity conditions as high as these, in 16% RH three out of the four studied isomalt grades showed good physical stability in the amorphous form during the whole study period of 23 days. Dry sucrose and trehalose have Tgs of 70 and 121°C, respectively (45,46). The Tgs of the four studied isomalt grades were lower than those of sucrose and trehalose, although the Tg of GPM isomer (1:0 GPM:GPS) stored in 2% RH, with a water content of 0.5% (w/w), had a Tg of approximately 64°C, very close to that of dry sucrose. Unlike sucrose and trehalose, which are comprised of only one type on molecules, isomalt consists of a mixture of two diastereomers. The advantage of isomalt is that its properties can be altered by adjusting the diastereomer ratio of GPS and GPM in the mixture, which is not possible with sucrose or trehalose. This feature is already exploited with crystalline isomalt when different isomalt grades are produced for specific application needs. The same could apply to freeze-dried amorphous isomalt, because as seen by the results of this investigation, the ratio of the two diastereomers has an effect on their Tg, residual water content and physical stability during storage.
CONCLUSION
This study represents a thorough analysis of the solid state structure and behavior of crystalline and amorphous GPM:GPS isomer mixtures using a range of complimentary analytical techniques. Crystalline raw materials of the different GPM:GPS isomer mixtures could be differentiated from each other when studied with various analytical techniques. All of the studied isomalt grades were successfully transformed from crystalline to amorphous with freeze-drying. The different GPM:GPS diastereomer mixtures behaved slightly differently during storage compared to one another. The amorphous mixtures containing 1:3, 1:1 and 2:1 GPM:GPS were stable during storage, as no obvious signs of crystallinity were observed. The amorphous isomalt grade containing only the GPM isomer was stable when stored up to 23 days in the lower humidity conditions, but during storage at 16% RH, the excipient started to exhibit instability. Crystallization occurred when the sample was heated slowly during thermal analysis in hermetic conditions. Amorphous GPM transformed in situ to anhydrate when using sealed pans in thermal analysis. Based on all of the findings of this study, isomalt could be a suitable excipient for freeze-dried formulations and it is all in all stable in the amorphous form when stored in low relative humidity conditions. Based on the DSC and FT-IR results obtained, preferably a mixture containing both the GPM and GPS isomers should be selected for freeze-drying based on its better stability. Out of the four studied mixing ratios, the 1:1 GPM:GPS mixture, which behaved like a single phase system, appeared to be the best one based on its physical stability in the amorphous form. A drug formulation containing amorphous isomalt with only GPM diastereomer would be risky because of its crystallization tendency during storage if the residual water content of the sample would be high. Isomalt could be a suitable excipient for freeze-dried formulations with small molecule active pharmaceutical ingredients and based on its physical properties, isomalt has potential to be further studied as a novel excipient for lyophilized protein formulations. For this purpose though the cryo- and lyoprotecting properties of isomalt should be thoroughly investigated with different proteins in further studies. The intravenous toxicity of isomalt must also be studied before it could be used in freeze-dried formulations intended for parenteral use.
Abbreviations
- DSC:
-
Differential scanning calorimetry
- FT-IR:
-
Fourier-transform infrared spectroscopy
- GPM:
-
1-O-α-D-glucopyranosyl-D-mannitol dihydrate
- GPS:
-
6-O-α-D-glucopyranosyl-D-sorbitol
- HPLC:
-
High performance liquid chromatography
- KF:
-
Karl-Fischer titration
- TGA:
-
Thermogravimetric analysis
- VT-XRPD:
-
Variable temperature X-ray powder diffraction
- XRPD:
-
X-ray powder diffraction
REFERENCES
Shire SJ. Formulation and manufacturability of biologics. Curr Opin Biotechnol. 2009;20:708–14.
Heljo V, Jouppila K, Hatanpää T, Juppo A. The use of disaccharides in inhibiting enzymatic activity loss and secondary structure changes in freeze-dried β-galactosidase during storage. Pharm Res. 2011;28:540–52.
Heljo V, Nordberg A, Tenho M, Virtanen T, Jouppila K, Salonen J, et al. The effect of water plasticization on the molecular mobility and crystallization tendency of amorphous disaccharides. Pharm Res. 2012;29:2684–97.
Hunter NE, Frampton CS, Craig DQM, Belton PS. The use of dynamic vapour sorption methods for the characterisation of water uptake in amorphous trehalose. Carbohydr Res. 2010;345:1938–44.
Sentko A, Willibald-Ettle I. Isomalt. In: O’Donnell K, Kearsley MW, editors. Sweeteners and sugar alternatives in food technology. 2nd ed. Chichester: Wiley-Blackwell; 2012. p. 243–74.
Bolhuis GK, Engelhart JJP, Eissens AC. Compaction properties of isomalt. Eur J Pharm Biopharm. 2009;72:621–5.
Ndindayino F, Henrist D, Kiekens F, Van den Mooter G, Vervaet C, Remon JP. Direct compression properties of melt-extruded isomalt. Int J Pharm. 2002;235:149–57.
Sáska Z, Dredán J, Balogh E, Luhn O, Shafir G, Antal I. Effect of isomalt as novel binding agent on compressibility of poorly compactable paracetamol evaluated by factorial design. Powder Technol. 2010;201:123–9.
Gau W, Kurz J, Müller L, Fischer E, Steinle G, Grupp U, et al. Analytische charakterisierung von palatinit. Z Lebensm Unters Forsch. 1979;168:125–30.
Cammenga HK, Figura LO, Zielasko B. Thermal behaviour of some sugar alcohols. J Therm Anal. 1996;47:427–34.
Cammenga HK, Zielasko B. Thermal behaviour of isomalt. Thermochim Acta. 1996;271:149–53.
Lichtenthaler FW, Immel S, Kreis U. Evolution of the structural representation of sucrose [1]. Starch-Starke. 1991;43:121–32.
Kállai N, Luhn O, Dredán J, Kovács K, Lengyel M, Antal I. Evaluation of drug release from coated pellets based on isomalt, sugar, and microcrystalline cellulose inert cores. AAPS PharmSciTech. 2010;11:383–91.
Sentko A, Bernard J. Isomalt. In: O’Brien-Nabors L, editor. Alternative sweeteners. 4th ed. Boca Raton: CRC Press; 2011. p. 275–96.
Bolhuis GK, Rexwinkel EG, Zuurman K. Polyols as filler-binders for disintegrating tablets prepared by direct compaction. Drug Dev Ind Pharm. 2009;35:671–7.
Ndindayino F, Henrist D, Kiekens F, Vervaet C, Remon JP. Characterization and evaluation of isomalt performance in direct compression. Int J Pharm. 1999;189:113–24.
Carpenter J, Pikal M, Chang B, Randolph T. Rational design of stable lyophilized protein formulations: some practical advice. Pharm Res. 1997;14:969–75.
European Pharmacopoeia Online 8th Edition 2014 (8.5). 2015 May 8; Available at: http://online6.edqm.eu/ep805/.
British Pharmacopoeia Commission. British pharmacopoeia 2015. London: TSO; 2015.
United States Pharmacopeial Convention. United States Pharmacopeia and National Formulary (USP 38-NF 33). Slp ed. (May 2015). Rockville: United States Pharmacopeial; 2014.
Smits-Van Prooije AE, De Groot AP, Dreef-Van Der Meulen HC, Sinkeldam EJ. Chronic toxicity and carcinogenicity study of isomalt in rats and mice. Food Chem Toxicol. 1990;28:243–51.
Waalkens-Berendsen DH, Koëter HBWM, van Marwijk MW. Embryotoxicity/teratogenicity of isomalt in rats and rabbits. Food Chem Toxicol. 1990;28:1–9.
Waalkens-Berendsen DH, Koëter HBWM, Sinkeldam EJ. Multigeneration reproduction study of isomalt in rats. Food Chem Toxicol. 1990;28:11–9.
Storey DM, Lee A, Zumbé A. The comparative gastrointestinal response of young children to the ingestion of 25 g sweets containing sucrose or isomalt. Br J Nutr. 2002;87:291–7.
Wang W. Lyophilization and development of solid protein pharmaceuticals. Int J Pharm. 2000;203:1–60.
Jorgensen L, Hostrup S, Moeller EH, Grohganz H. Recent trends in stabilising peptides and proteins in pharmaceutical formulation - considerations in the choice of excipients. Expert Opin Drug Deliv. 2009;6:1219–30.
Lichtenthaler FW, Lindner HJ. The preferred conformations of glycosylalditols. Liebigs Ann Chem. 1981;2372–2383.
Lindner HJ, Lichtenthaler FW. Extended zigzag conformation of 1-O-α-D- glucopyranosyl-D-mannitol. Carbohydr Res. 1981;93:135–40.
Frisch M, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 09, Revision A. 02. Wallingford: Gaussian Inc.; 2009.
Schaftenaar G, Noordik JH. Molden: a pre- and post-processing program for molecular and electronic structures*. J Comput Aided Mol Des. 2000;14:123–34.
Cherukuvada S, Nangia A. Eutectics as improved pharmaceutical materials: design, properties and characterization. Chem Commun. 2014;50:906–23.
Perkkalainen P, Pitkänen I, Huuskonen J. Crystal structure and IR spectrum of 1-O-α-d-glucopyranosyl-d-mannitol–ethanol (2/1). J Mol Struct. 1999;510:179–89.
Karin Gehrich. Phasenverhalten einiger Zucker und Zuckeraustauschstoffe. Technische Universität Braunschweig. 2002.
Borde B, Cesàro A. Thermal properties of isomalt: a diastereomer mixture. J Therm Anal Calorim. 2002;69:267–80.
Zielasko B. Ermittlung physikalisch-chemischer Daten von Isomalt und seinen Komponenten. Clausthal-Zellerfeld. Technical University Braunschweig Carolo-Wilhelmina. 1997.
Perkkalainen P, Halttunen H, Pitkänen I. Thermal behaviour of anhydrous, dihydrate and (2/1) ethanol forms of 1-O-α-d-glucopyranosyl-d-mannitol. Thermochim Acta. 1998;320:215–21.
Cherukuvada S, Guru Row TN. Comprehending the formation of eutectics and cocrystals in terms of design and their structural interrelationships. Cryst Growth Des. 2014;14:4187–98.
Borde B, Cesaro A. A DSC study of hydrated sugar alcohols: isomalt. J Therm Anal Calorim. 2001;66:179–95.
Hoppu P, Jouppila K, Rantanen J, Schantz S, Juppo AM. Characterisation of blends of paracetamol and citric acid. J Pharm Pharmacol. 2007;59:373–81.
McFetridge J, Rades T, Lim M. Influence of hydrogenated starch hydrolysates on the glass transition and crystallisation of sugar alcohols. Food Res Int. 2004;37:409–15.
Cammenga HK, Zielasko B. Glasses of sugars and sugar substitutes. Ber Bunsenges Phys Chem. 1996;100:1607–9.
Raudonus J, Bernard J, Janßen H, Kowalczyk J, Carle R. Effect of oligomeric or polymeric additives on glass transition, viscosity and crystallization of amorphous isomalt. Food Res Int. 2000;33:41–51.
Carstensen JT, Van Scoik K. Amorphous-to-crystalline transformation of sucrose. Pharm Res. 1990;7:1278–81.
Crowe LM, Reid DS, Crowe JH. Is trehalose special for preserving dry biomaterials? Biophys J. 1996;71:2087–93.
Orford PD, Parker R, Ring SG. Aspects of the glass transition behaviour of mixtures of carbohydrates of low molecular weight. Carbohydr Res. 1990;196:11–8.
Hinrichs WLJ, Sanders NN, De Smedt SC, Demeester J, Frijlink HW. Inulin is a promising cryo- and lyoprotectant for PEGylated lipoplexes. J Control Release. 2005;103:465–79.
ACKNOWLEDGMENTS AND DISCLOSURES
The authors wish to acknowledge Karin Gehrich, Oliver Luhn and Beneo-Palatinit GmbH for kindly donating the isomalt grades used in the study and assisting in DSC data interpretation. Dr. Jacco van de Streek from the Division of Pharmaceutical Technology and Engineering, University of Copenhagen, is acknowledged for the helpful conversations regarding interpretation of the results. The support of the Dodd-Walls Centre is gratefully acknowledged (KCG and JEB).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Koskinen, AK., Fraser-Miller, S.J., Bøtker, J.P. et al. Physical Stability of Freeze-Dried Isomalt Diastereomer Mixtures. Pharm Res 33, 1752–1768 (2016). https://doi.org/10.1007/s11095-016-1915-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-016-1915-3