
ABSTRACT
An increasing number of newly discovered drugs are poorly water-soluble and the use of natural and synthetic lipids to improve the oral bioavailability of these drugs by utilizing the digestion pathway in-vivo has proved an effective formulation strategy. The mechanisms responsible for lipid digestion and drug solubilisation during gastrointestinal transit have been explored in detail, but the implications of drug precipitation beyond the potential adverse effect on bioavailability have received attention only in recent years. Specifically, these implications are that different solid forms of drug on precipitation may affect the total amount of drug absorbed in-vivo through their different physico-chemical properties, and the possibility that the dynamic environment of the small intestine may afford re-dissolution of precipitated drug if present in a high-energy form. This review describes the events that lead to drug precipitation during the dispersion and digestion of lipid based formulations, common methods used to inhibit precipitation, as well as conventional and newly emerging characterization techniques for studying the solid state form of the precipitated drug. Moreover, selected case studies are discussed where drug precipitation has ensued from the digestion of lipid based formulations, as well as the apparent link between drug ionisability and altered solid forms on precipitation, culminating in a discussion about the importance of the solid form on precipitation with relevance to the total drug absorbed.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
The aim of using lipid based drug delivery systems (LBDDS) is to enable the effective oral delivery of lipophilic drugs. Issues of formulation dispersion, digestion and post-absorptive fate of both drug and lipid have all been the subject of increasingly intense research in the field. A limited number of products, including Sandimmune®, Neoral® (1) and Aptivus® have achieved some success on the market as LBDDS (2), however their widespread commercialization has been hampered by a lack of understanding on several important fronts. Perhaps one of the most critical of these is the need for a thorough understanding of drug precipitation during the digestion of LBDDS with regard to the solid state form of the drug, and subsequent effects on re-dissolution in the presence of an absorptive sink (e.g., in-vivo). In this review the current level of understanding around factors that govern drug precipitation during the dispersion and digestion of LBDDS are examined, in addition to describing the common methods used to inhibit drug precipitation, and perspectives on the emerging realization in the field that the paradigm of preventing drug precipitation during lipolysis may not be necessary are offered that may provide a way forward in understanding how to optimally design LBDDS.
Poorly Water Soluble Drugs and Lipid Based Drug Delivery
The discovery of drugs with poor aqueous solubility is becoming increasingly common because of bias towards such molecules in combinatorial chemistry and high throughput screening. Both of these drug discovery strategies aim to maximize drug–receptor complementarity, stressing hydrophobic drug moieties that improve ligand–receptor affinity through key interactions (3,4). Improved ligand–receptor affinity, however, generally remains juxtaposed with decreasing aqueous solubility. Current predictions suggest that over 70% of newly-discovered drugs are poorly water-soluble, and lipophilic (5), or class 2 compounds as termed by the Biopharmaceutics Classification System (BCS) (6). These class 2 compounds are highly permeable across the intestinal membrane, but have limited solubility by gastrointestinal fluids and hence may be slow to dissolve in the gastrointestinal tract, thereby limiting their absorption (7–9).
Advances in oral drug delivery may present formulation options that overcome the solubility limitations of such compounds, thus enabling administration of drug in a form that addresses the issues with poor aqueous solubility through formulation, rather than at the drug molecular structure level.
Poorly water-soluble drugs often exhibit slow dissolution in the gastrointestinal tract, ultimately limiting their absorption (10). Slow dissolution can lead, potentially, to incomplete dissolution of drug on passage through the gastrointestinal tract and excretion of undissolved material. Formulation approaches to improve the bioavailability of such drugs are therefore focused on modifying drug dissolution and solubility characteristics, or avoiding the need for dissolution through the use of a solution dosage form. The Noyes-Whitney dissolution equation dictates that the solubility in the surrounding medium relative to the concentration of dissolved drug, the drug diffusion coefficient and surface area of the solute determine the dissolution rate of a solid material (11). As a consequence, dissolution is affected by the disposition of gastrointestinal fluids - their pH and solubilizing components, agitation, degree of saturation, and the presence of molecular species that may modify drug dissolution characteristics by adsorption to the solute particles (12). There have been many different formulation approaches to address the slow dissolution of drug by impacting on these variables at both the solid state and solution levels (13).
For BCS Class 2 compounds in particular, the lipophilicity of the drug leads intuitively to the use of lipids to provide a dosage form in which the drug is dissolved prior to administration, and during the gastrointestinal processing of the formulation (dispersion and digestion), to make the drug available in a highly solubilized state for absorption. The use of lipids to improve bioavailability could range from simple fatty meals to highly engineered LBDDS as unit dosage forms. With the exception of LBDDS containing a high fraction of hydrophilic components, such as Type 3B and Type 4 formulations, which will be discussed in a later section, a common aspect is that LBDDS generally require digestion to provide the optimal pre-absorptive environment to maximize bioavailability (9,14–20).
The digestion of lipids is a complex combination of biochemical and physicochemical processes. Many reviews exist that describe these processes in detail (9,14–20). Figure 1 provides an overview of the digestion process and the potential fate of both lipids and lipophilic drugs.

An overview of the digestion of lipids in the stomach and small intestine, and the potential fate of lipids and drug. Adapted from Porter et al. (18).
Briefly, with relevance to LBDDS administered in a capsule, the digestion of lipids starts in the stomach, where enzyme-driven hydrolysis renders triglycerides to 1,2(2,3)-diglycerides and free fatty acid molecules. Up to 30% of total triglyceride is initially broken down by gastric lipase, aided by agitation from the stomach to form a crude emulsion (21). Transit to the small intestine then triggers the major aspect of lipid digestion, where the pancreatic lipase/colipase complex breaks down any diglycerides and remaining triglycerides at the oil–water interface, producing an sn2-monoglyceride and two free fatty acid molecules per molecule of triglyceride (10). Biliary secretions released from the gallbladder, containing other amphiphilic endogenous molecules, such as bile salts, phospholipids and cholesterol, then aid in emulsifying and solubilizing the lipid digestion products by forming different colloidal structures, principally vesicles and mixed micelles (22–26). The commonly proposed mechanism of transport of lipids towards the epithelial surface occurs via mass transport, in which the final colloidal phases (typically mixed bile salt micelles) carry the digestion products across the unstirred water layer separating the absorptive cells from bulk intestinal fluid (18).
The premise of digestion-enabled drug delivery is therefore to provide a solubilizing medium (e.g., the mixed micelles) for poorly water-soluble, lipophilic, compounds. This is most simply achieved by administering drug immediately after consuming a high fat meal (10). However, inter-patient differences in the fat content and temporal aspects of taking the medicine often lead to variable pharmacokinetics and consequently variable therapeutic outcomes (27). LBDDS often achieve more consistent absorption profiles, at least in part by providing drug in a pre-dissolved state, thus avoiding the rate-limiting dissolution step (9,17,19). There is clear evidence in the literature that when compared to an aqueous suspension, drug administered in solution in a lipid vehicle almost always provides improved absorption (28–31). It is less clear whether lipid solutions are beneficial over lipid suspensions from a bioavailability perspective (32). Other drawbacks for lipid suspensions, such as Ostwald ripening and dose uniformity, mean that they have not been as extensively studied as lipid solutions.
There are different types of LBDDS composed of varying amounts of lipids, surfactants (either hydrophobic or hydrophilic) and co-solvents. The lipid formulation classification system (LFCS) groups LBDDS based on their composition and physical properties (9). Briefly, Type 1 formulations comprise oil excipients only, such as triglycerides and other glycerides, requiring digestion for dispersion to occur. Type 2 formulations are self-emulsifying, meaning that upon dilution and provided gentle agitation, a dispersed system forms with a droplet size in the range of 0.25–2 μm. Oils and non-ionic surfactants generally make up Type 2 formulations. Type 3 formulations produce very fine dispersions with droplet sizes < 100 nm, due to the inclusion of water-soluble components, they are also optically clear. Subtypes A and B exist for Type 3 formulations, where the discriminating factor is the fraction of total formulation consisting of water-soluble parts and co-solvents. On the extreme end of the LFCS are Type 4 formulations, which do not contain any lipid excipients, and are purely composed of surfactants to form fine micellar solutions.
There is growing evidence to suggest that LBDDS improve the extent of absorption of lipophilic, poorly water-soluble drugs. Current approaches to the formulation of LBDDS, however, continue to be largely empirical, based on drug solubility in the formulation, and dispersibility of the formulation during in-vitro dispersion testing. The complexity of the digestive environment and the dynamic compositional changes over time are currently not well understood, but are critical in dictating drug disposition. To date there has been a focus on drug solubilization, but the solid state aspects of drug disposition are emerging as an important aspect of overall drug behavior during dispersion, digestion and absorption from LBDDS. Solid state aspects that could influence drug disposition are those of the formulation in the case of a suspension formulation, and those of solid material resulting from precipitation during dispersion and digestion. The latter is the focus of this review article.
Supersaturation and Drug Precipitation
Precipitation of drug during the dispersion and digestion of LBDDS may reduce the total amount of drug absorbed, and therefore decrease oral bioavailability (7,14,33–36) on the basis that re-dissolution of the drug does not take place. Precipitation is the nucleation and phase separation of solid drug particles from a supersaturated liquid system. A supersaturated state is a prerequisite for precipitation (37), and there are different mechanisms by which supersaturation of drug can occur during digestion. The pH shift mechanism is apparent during the gastric emptying of basic drugs, where supersaturation occurs due to the movement of drug from the highly solubilising, low pH environment of the stomach (~pH = 2), to the higher pH of the small intestine. Consequently, the solubility of the basic drug is reduced resulting in a transiently supersaturated state (7,36,38–41). Another mechanism by which a supersaturated state forms, relevant to LBDDS, is where dispersion and digestion in the small intestine leads to a decrease in solubilisation capacity of the formulation for the drug (20,34,36,40,42).
Under conditions of supersaturation, nucleation and crystal growth are inevitable. Solute molecules initially gather, either on the surface of an impurity or in three dimensional clusters, to form stable nuclei that are of a sufficient size for subsequent growth to take place (36,37,43). Nucleation is aided by the presence of impurities, as they decrease the energy barrier towards successful cluster formation. An increased degree of supersaturation and sufficient level of impurities lessen the time taken for precipitation to occur. Moreover, recent findings suggest the existence of precursor complexes that lead to nucleation and potentially influence precipitation, and therefore the solid material produced. A detailed discussion on the function of these precursors can be found elsewhere (44).
Crystal growth begins once stable nuclei are formed, but growth and nucleation continue concurrently from this point. Dominance of one mechanism over the other largely determines crystal size, for example a few stable nuclei would give rise to a few large crystals, whereas continuous nucleation would form many small crystals. Growth of crystals is driven by the attachment of solute molecules to energetically-favoured growth sites, and the macroscale form of a crystal, or habit, is highly variable, depending on factors such as rate of growth, nature of solvent, degree of supersaturation and agitation, as well as the presence of impurities or excipient material (37,44,45). In a system reverting to thermodynamic stability, precipitation proceeds only to the point where there is no longer a supersaturated state.
Three theories exist on the progression of crystallisation, namely the surface energy theory, the diffusion theory and the adsorption layer theory (36). The surface energy theory states that a drop of fluid is most stable when its area/surface energy is at a minimum, and in special cases this minimum is achieved through crystal growth. However, this theory is not well supported due to its inability to explain supersaturation and varying growth rates (36). Diffusion theory describes the integration of units to a lattice structure, where controlled diffusion of the components occurs and a stagnant film can be found on the surface of the crystal. Modifications to diffusion theory were necessary to account for the effects of agitation, which would decrease the thickness of the stagnant film and theoretically lead to rapid crystal growth, but this is never observed in real systems. Therefore, a two-step process has been proposed where, first, diffusion carries solute molecules to the crystal surface, and then rearrangement takes place prior to integration (36). Lastly, the adsorption layer theory is based on the thermodynamics of adsorbed layers at a crystal face. Where adsorbed solute molecules do not immediately integrate on crystal faces, but are rather adsorbed and free to diffuse around the surface, thus creating an equilibrium between adsorbed solute molecules constituting the layer and the bulk solution containing solute molecules. Attractive areas on the crystal face yield growth sites or kinks for crystal step growth (36,46).
In-Vitro Lipolysis and Maximum Supersaturation Ratio (SRM)
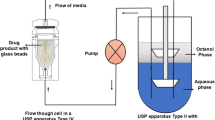
In-vitro lipolysis models are often used to study the solubilisation and precipitation of drug during dispersion and digestion of LBDDS (18). Briefly, in-vitro digestion/lipolysis experiments attempt to simulate the processes occurring on oral administration of lipid formulations by mimicking the in-vivo environment of the gastrointestinal tract, most often conditions reflecting the upper small intestine. Hydrolysis of lipids occurs upon the addition of lipases to simulated intestinal fluid containing the dispersed lipid formulation in a thermostatted (37°C) glass vessel (17). The simulated intestinal fluid is composed of bile salt and phospholipids in buffer. An automated titration unit maintains pH, generally at 6.5, which is a compromise between the pH of the upper small intestine (47) and the optimal pH for the lipolytic activity of pancreatin enzyme (48). Sodium hydroxide solution is used as the titrant to neutralize free fatty acids liberated by lipase-mediated degradation of triglycerides (49). Information with regard to the extent of digestion of the formulation and the distribution of drug in different phases is commonly acquired from lipolysis experiments.
After a designated time period the in-vitro lipolysis can be arrested by addition of a lipase inhibitor to the glass vessel, after which the digested contents are centrifuged to produce the relevant post-digestion phases, namely undigested oil (if present), aqueous colloidal phase and a semi-solid or solid pellet (50). Distribution of lipids and drug between the different phases can then be assessed analytically, where precipitated drug in the pellet and solubilized drug in the colloidal phase indicate the likelihood of drug precipitation in-vivo.
However, the prediction of drug precipitation in-vivo based on the results from in-vitro lipolysis experiments may be overestimated (34,51–53). The absence of an absorptive sink in-vitro is considered a major drawback of lipolysis experiments, and the lack of a product-removal mechanism implies exaggerated results with regard to drug precipitation. Furthermore, the variables such as type and concentration of bile, pH and type and activity of the enzyme have made it difficult to compare results across groups, or to draw definitive conclusions on the biorelevance of the lipolysis methods, leading to recent efforts to standardise in-vitro digestion protocols for LBDDS (54–57).
As mentioned earlier, solubilisation of drug in the aqueous colloidal phase may be an indicator of likely in-vivo performance. A high amount of solubilized drug in the aqueous colloidal phase may increase the thermodynamic driving force for absorption, leading to a recent focus on supersaturation of drug in the colloidal phase during and after digestion as a more appropriate indication of the potential for absorption from a LBDDS. The degree to which digestion produces a supersaturated drug-containing medium has been termed the maximum supersaturation ratio (SRM) (56).
The SRM describes the maximum amount of supersaturation obtained before drug precipitation is observed on digestion of lipid based formulations. The SRM is a useful tool with respect to quantifying the influence of formulation components on the kinetic aspects of supersaturation. The absolute value between drugs, however, does not take account of several issues including drug participation in self-assembly processes (58), impact of drug on kinetic aspects of digestion (59), or importantly, differences in solid state between drug in solubility measurements and precipitated drug in dynamic lipolysis experiments. One particular area of conjecture remains the nature of the centrifuged pellet phase with regard to its molecular composition, and effect on drug precipitation. It has been proposed that during in-vitro lipolysis the liberation of fatty acids from the breakdown of glycerides yields calcium-fatty acid complexes, which form insoluble soaps residing in the pellet of a centrifuged digestion sample (51). Insoluble soaps are an undesired non-biorelevant byproduct from sequestering liberated fatty acids with calcium in an effort to maximize lipase action. This fatty acid-calcium matrix could, potentially, affect the drug precipitation process. Therefore, if the pellet itself is influencing the solid state form of precipitated drug in the in-vitro model, this would not be the case in-vivo, as the absorption of fatty acids would relieve inhibition of lipase.
Conventional Characterization Techniques for Analyzing Precipitated Drug
There are several commonly used characterization methods for determining the solid state of precipitated drug during digestion-related experiments. These include X-ray diffraction (XRD), Cross Polarized Light Microscopy (CPLM), dissolution testing, Differential Scanning Calorimetry (DSC) and Fourier Transform Infrared Spectroscopy (FTIR).
XRD is typically performed on the pellet phase of a digested formulation, after isolation of the precipitated material. The basis of how XRD works to obtain the structural information of solids is well documented and a detailed review may be consulted for a basic understanding (60). Briefly, X-rays pass through a sample and diffract at angles unique to the crystal structure, effectively providing a fingerprint for the molecular arrangement characteristic of the solid state of the material. Therefore, polymorphic forms of a drug may be identified due to the unique molecular arrangements of the polymorphic crystals. Amorphous drug forms, lacking long range orientational and positional order are also identifiable by XRD, albeit indirectly, by an absence of diffraction peaks and the presence of a largely undefined halo region in the resultant intensity vs angle diffractograms. XRD thus provides an easy means to differentiate between amorphous and crystalline drug forms; however, analyzing precipitated drug in the pellet phase from digested lipid formulations is difficult, due to sample preparation and analysis time requirements, and the resulting diffractograms need to be interpreted with caution. Typically, isolation of the pellet phase requires a period of centrifugation ranging from 30 to 90 min, which is followed by removal of supernatant and sometimes drying before performing the XRD measurement which requires an additional 30 min per sample. Whether the final and observed solid state form of the drug after performing XRD truly reflects the precipitated drug during the digestive process remains unclear (i.e., amorphous to crystalline conversion of precipitated drug), but can potentially be confirmed with the development of in-situ characterization methods, which will be discussed in a later section.
CPLM is an imaging technique that can identify drug crystals in the pellet phase, with birefringence indicating crystalline material. Calcium-fatty acid soaps, however, may also be present in the isolated pellet and show birefringence on CPLM due to their anisotropic lamellar liquid crystalline structure, which makes amorphous drug difficult to detect. Similar to XRD, invasive sample preparation and the potential for temporal changes in structure during sampling and experiment apply for CPLM. In any case, CPLM remains a sound visualization technique that provides a link between crystal morphology and XRD data. A thorough understanding behind the principles of CPLM may be obtained elsewhere (61).
Dissolution testing of precipitated drug in bio-relevant media can inform the likelihood of re-dissolution of drug during the digestion of lipid-based formulations in the presence of an absorptive sink (e.g., in-vivo). Dissolution tests can be performed on isolated and extracted pellet phases in a standard USP-2 paddle apparatus (62,63). The solid state form of the pelleted drug precipitated during the digestion of lipid formulations can be compared to the reference crystalline form by spiking crystalline drug into the isolated pellets of corresponding blank (drug-free) lipid formulations (40,62,64). Solid state transformations upon contact with solvent should also be considered, where amorphous drug can convert to a crystalline or polymorphic form. Recently, an in-situ approach that combines dissolution testing with Raman spectroscopy was able to monitor the transition of amorphous indomethacin to its crystalline forms (65).
DSC can be used identify key thermal events intrinsic to the precipitated drug found in the pellet phase after digestion of lipid formulations. A glass transition temperature (Tg) or recrystallization event would suggest amorphous drug has precipitated during lipolysis. In contrast, exothermic melting should be observed for crystalline drug. However, as discussed above the pellet is not limited in composition to precipitated drug, with calcium-fatty acid complexes also being present, and thermal events due to these components need to be distinguished from those of the drug. A detailed review on the applications of DSC can be found elsewhere (66,67).
Delaying the Onset of Drug Precipitation with Polymeric Precipitation Inhibitors (PPIs)
Polymeric materials, such as polyvinylpyrrolidone (PVP), or hydroxypropylmethyl cellulose (HPMC), when administered with poorly water-soluble drugs can act to inhibit precipitation of drug for a given time period. Delayed onset of drug precipitation can be a result of these polymers working to inhibit both the nucleation and crystal growth mechanisms, and have not been shown to operate exclusively on one or other of these processes (68,69). Kinetic stability of supersaturated states is thought to be achieved through polymer-drug interactions, such as hydrogen bonding, hydrophobic interactions or steric disruption of the crystallization phase (70,71). By doing so, PPIs effectively maintain a supersaturated environment that allows for rapid drug absorption to occur (34). Typically, PPIs do not produce an effect on equilibrium solubility of the drug. PPIs slow the onset of drug precipitation; however, they do not completely stop precipitation, as precipitation is thermodynamically favored. The “spring and parachute” concept has been used in the literature when describing the action of PPIs (72), where drug is maintained in a supersaturated state and the PPIs slow down the otherwise rapid decrease in solubilized drug, eventually relaxing to a thermodynamically-favored precipitated state (36,72). In addition, once the drug starts to precipitate and undissolved particles are present, the PPIs may continue to exert their effect by retarding the crystal growth mechanism and slow down further precipitation.
The reported mechanisms and sites of action for PPIs include changing surface tension at the bulk-solution interface (36,73), changing the adsorption layer at the crystal-solution interface (36,74), adsorbing to the crystal-surface interface (36,70), adsorption to growth terraces and blocking access of solute to these growth sites (36,75), adsorption to surface crevices creating a smooth surface free of growth areas and altering the surface energy of crystal faces and changing the level of solvation (36). Factors that influence polymer-drug binding have been cited previously and include temperature, molecular weight, viscosity, dielectric constant and hydrogen bonding (36). High temperatures lead to greater drug solubility, which weakens intermolecular bonding. Increasing the molecular weight of the polymer generally strengthens polymer-drug interactions through an increased viscosity and number of functional groups for bonding. The rate of diffusion of drug is inversely proportional to the viscosity of the aqueous medium. For poorly water-soluble drugs, lowering the dielectric constant is expected to increase drug solubility, thus negatively affecting drug-polymer binding. Finally, hydrogen bonding between polymer and drug is greater where there are more hydrogen bonding sites on the polymer (36). These act as merely generalized observations and are not necessarily always the case for a given drug-polymer combination.
Link Between Drug Ionisability and Solid State form on Precipitation
Until recently the pervasive view was that drug precipitation is an undesirable event during the dispersion and digestion of lipid based formulations, as the precipitated drug may not be absorbed and therefore could result in decreased bioavailability. Drug precipitation can lead to changes in the rate of drug absorption regardless of where precipitation takes place along the gastrointestinal tract and this will likely have some effect on therapeutic outcomes. However, this view of drug precipitation inherently leading to negative therapeutic outcomes is more applicable where the gastric precipitation of drug upon dispersion of the formulation in the stomach is expected, which in turn is more likely to occur when relatively hydrophilic formulations are administered, for instance Type 3B and Type 4 formulations. A loss in solubilisation capacity for the drug upon dispersion is most often seen with these hydrophilic formulations and generally is not considered a major hurdle for more lipophilic Type 1, Type 2 and Type 3A formulations. The absence of self-assembled lipid digestion products and the low pH environment of the stomach means the potential for re-dissolution of the precipitated drug would be different to that of the small intestine. Conversely, the likelihood of drug precipitation following the administration of more lipophilic formulations (Type 1, Type 2 and Type 3A) is greatest in the small intestine upon digestion of the lipid components in the formulation. However, drug precipitation does not necessarily preclude re-dissolution in the dynamic environment of the small intestine, where sink conditions may be re-instated, through transient changes in lipid composition, changes in colloidal structures, or absorption of lipid or drug. The solubility of drug in the changing medium will also depend on the solid state form of precipitated drug, hence it is possible that differences in solid state of the precipitated material during administration may account for varying extents of total drug absorption, and bioavailability. Despite the likely importance of the solid state of precipitated drug in understanding the performance of LBDDS, the issue has received only recent attention in the lipid-based formulation field. There are currently a handful of studies in the literature where the solid state form of precipitated drug has been investigated after the digestion of lipid and non-lipid formulations, the findings are summarized in Table I. These studies point to a strong correlation between the ionisability of the drugs tested and an altered solid state form on precipitation, particularly for lipid formulations tested with in-vitro lipolysis experiments.
Cinnarizine is a selective calcium antagonist that prevents contraction of arterial smooth muscle, is poorly water-soluble, and is often used as a model drug in LBDDS experiments (41,42,62,81–84). A recent study by Sassene et al. showed that cinnarizine precipitated in a non-crystalline form during in-vitro lipolysis of a self-micro-emulsifying drug delivery system (SMEDDS) (62). Crossed polarised light microscopy (CPLM) and X-ray diffraction (XRD) suggested that the precipitated drug was in an amorphous form. Dissolution studies performed on solid material collected post-digestion, the results of which are shown in Fig. 2, revealed a ten-fold higher dissolution rate for precipitated cinnarizine in the pellet, compared to neat crystalline drug powder spiked in a blank pellet.

Dissolution rate of pellet with cinnarizine from endpoint in vitro lipolysis (circles) and blank pellet spiked with crystalline cinnarizine (squares). Reproduced with permission from Wiley (62).
XRD patterns were compared for raw crystalline cinnarizine, precipitated cinnarizine in the pellet from the digestion sample, a blank pellet from the digestion of drug-free formulation and a blank pellet spiked with crystalline cinnarizine. These diffraction patterns are presented in Fig. 3.

XRD pattern of (a) starting crystalline cinnarizine used in SMEDDS, (b) pellet from the lipolysis of SMEDDS with cinnarizine; CIN pellet, (c) pellet from the lipolysis of SMEDDS without cinnarizine; blank pellet, (d) blank pellet spiked with cinnarizine; blank pellet + CIN. Numbers over the peaks in (c) indicate d-spacings and are also applicable for the peaks of (b) Reproduced with permission from Wiley (62).
From Fig. 3, raw cinnarizine spiked in a blank pellet from a drug-free formulation (d) showed diffraction peaks that correlated to reference XRD data for cinnarizine (a). Conversely, precipitated drug from the pellet phase post-digestion did not show these peaks (b), suggesting a lack of crystalline cinnarizine in the sample. The conclusion was that cinnarizine, post-digestion, presented itself in a non-crystalline, amorphous form, which may have favourable physicochemical properties for drug absorption. This paper was the first to acknowledge that the solid state form of drug upon precipitation during digestion may play a role in determining the total amount of drug absorbed.
The same research group also examined the precipitation of the neutral, poorly water-soluble compound, danazol (79) from the same SMEDDS formulation as for cinnarizine in the aforementioned study. In contrast to the cinnarizine study, danazol precipitated in crystalline form according to XRD data. The authors attributed this finding to the different physicochemical properties of cinnarazine and danazol, and acknowledged that these properties may be important in determining the solid state formed upon precipitation.
In analogous studies, the in-vitro precipitation behavior and solid state characteristics of the poorly water-soluble, weakly basic drugs, loratadine and carvedilol, were studied on in-vitro digestion of lipid-based formulations (40). Formulations containing these drugs were prepared with drug dissolved at up to 80% total drug saturation. Three different formulations were tested with the two compounds, resembling medium-chain Type 3A, medium-chain Type 3B and Type 4 LBDDS. In this study the effects of both formulation dispersion and digestion on precipitated drug were investigated, as opposed to digestion alone. Although the dispersion tests were carried out at a pH of 7.5, the precipitation of drug upon the dispersion of LBDDS can indicate the likelihood of gastric precipitation, as these formulations are first diluted in the stomach before passing to the small intestine, which is the main site for lipid digestion. The solubility of the weakly-basic drugs decreased in dispersed systems compared to undiluted formulations, due to a loss of solubilising capacity upon dispersion of the formulation. As in the case of Sassene et al. above, XRD was used to determine the crystallinity of precipitated drug in the pellet. After a dispersion period of 1 h, both drugs precipitated from the Type 3B formulation and the resultant pellet was analysed with XRD, which showed that both drugs had precipitated in their thermodynamically stable crystalline form. After a digestion period of 30 min, however, the drugs precipitated from all three formulations tested, where loratadine was crystalline after digestion in all cases, but the digestion of carvedilol produced an amorphous precipitate, analogous to the cinnarizine study already discussed (62). An explanation for this discrepancy in the solid state form of precipitated drug on digestion between these two basic compounds is discussed in a later section.
The neutral drug fenofibrate and the weak acid tolfenamic acid, both poorly water-soluble highly lipophilic compounds, were also tested for their solid state form upon precipitation during the in-vitro digestion of lipid formulations (56). The presence of fenofibrate crystals after digestion (60 min) was detected by CPLM, and XRD data supported this observation as characteristic peaks corresponding to the thermodynamically stable crystalline form were present. Tolfenamic acid also precipitated during digestion, and crystals were also observed using CPLM, but the crystal form was dependent on the type of lipid formulation. Whilst Type 3B lipid formulations produced crystalline tolfenamic acid in the same form as the reference drug, Type 4 lipid formulations produced a different (“yellow”) polymorph upon digestion, as is shown via XRD and CPLM in Fig. 4. This latter finding suggests that the composition of the formulation and physicochemical properties of the precipitated drug products are interrelated.

XRD patterns for precipitated tolfenamic acid (TA) after in-vitro lipolysis of Type 3B and Type 4 lipid based formulations. Digestion of Type 4 formulations at 80 and 100% drug saturation levels produced the ‘yellow polymorph’ Reproduced with permission from Springer (56).
The link between drug precipitation in-vitro and formulation performance in-vivo has also been examined for fenofibrate using a self-nano emulsifying drug delivery systems (SNEDDS), at different drug saturation levels (53). Fenofibrate was shown to precipitate in its thermodynamically stable crystalline form during in-vitro lipolysis, however, this did not correlate well with in-vivo data, leading to the idea that the high lipophilicity and permeability of fenofibrate may dictate that a supersaturated state is not maintained for sufficient time in-vivo for precipitation to occur (7).
A separate study by the same researchers examined the in-vitro and in-vivo performance in beagle dogs of SNEDDS and super-SNEDDS formulations containing the poorly water-soluble, weakly basic, drug halofantrine (64). The in-vitro digestion of these SNEDDS formulations rapidly induced the precipitation of halofantrine. XRD was performed on the pellet phases after digestion (60 min), where halofantrine was detected in a non-crystalline form. Dissolution of the non-crystalline precipitate was performed in lipolysis media, and the dissolution was shown to be enhanced in comparison to the starting material, similar to the previous observation with cinnarizine discussed above. This enhanced dissolution of the precipitated halofantrine correlated with improved bioavailability in-vivo, as two SNEDDS capsules were dosed in order to reach a similar AUC and Cmax as a single super-SNEDDS capsule. Findings from this paper are therefore also in direct contrast to the logic that drug precipitation during in-vitro digestion is inherently linked with a decreased amount of total absorbed drug.
The general trend with the studies discussed thus far, and summarized in Table I, is that poorly water-soluble, weakly-basic drugs tend to precipitate in a non-crystalline form during in-vitro digestion (cinnarizine, halofantrine and carvedilol), whilst neutral and acidic drugs precipitate in a crystalline form (fenofibrate, danazol and tolfenamic acid, although polymorphism was observed with the latter). Simvastatin, however, which is a neutral compound, went against this trend in a study that compared the performance of super-SNEDDS formulations, in that case at 200% drug loading (80). No sign of crystalline simvastatin within the digested pellet of the 200% super-SNEDDS formulation was seen in the XRD data, which suggests that it is not an absolute rule that only basic drugs precipitate during in-vitro digestion in a non-crystalline or amorphous form.
Solid State Precipitation Behavior in the Absence of Lipid Formulation
The general trend of basic drugs forming non-crystalline precipitates upon digestion is not as apparent in the absence of lipids. For example, precipitation of mebendazole from dimethylacetamide solutions on dilution with aqueous media resulted in crystalline drug in-vitro and in-vivo (77). In-vivo experiments were performed on dogs with intestinal stomas for sampling, and precipitation of drug in a crystalline form did not appear to negatively impact bioavailability. The results from this study suggest that the components present during and after lipid digestion may play an integral role in dictating the solid state form of precipitated drug during the digestion of lipid formulations.
Studies on aspirated intestinal samples from humans have shown that the poorly water-soluble, weak base, ketoconazole appears to precipitate in an amorphous form in-vivo (76), but the drug precipitates in a crystalline form during in-vitro experiments that were designed to simulate similar conditions (78). This discrepancy in the solid state form of the precipitated drug between these two studies could be related to the more complex and dynamic environment of humans in-vivo. As lipids were not used as part of the formulation in either study, however, the amorphous precipitate observed from in-vivo samples could not be attributed to the presence of lipid digestion products, but could be formed via another unknown mechanism. This observed amorphous precipitate from human intestinal samples implies that the precipitation of weakly basic compounds in the small intestine, in some cases even in the absence of lipids, may not lead to a decrease in the total amount of drug absorbed; as precipitation of a poorly water-soluble drug in an amorphous form may promote re-dissolution and subsequent absorption. In-vitro experiments were also performed on the weakly basic compound, AZD0865, which was shown to precipitate in a different polymorphic form to the reference drug material (78).
The way certain drugs form supersaturated systems has also been considered a possible determinant of the formation of amorphous or crystalline precipitates. Recently, the solid state properties of 10 different weakly-basic compounds were characterized upon precipitation induced via a pH shift in the dissolution medium (85). Drugs were dissolved at low pH before a base was added as titrant to promote precipitation, subsequently small amounts of base or acid were added to form sub- and supersaturated states in solution, and the extent and duration of supersaturation was determined. Two trends were observed with regard to supersaturation behavior, which were reflected in the solid state properties of the precipitated drug. Type 1 basic compounds, as described by the authors, exhibited a short-lived, but relatively much longer supersaturation period prior to precipitation compared to their Type 2 counterparts, which appeared to phase separate immediately above the equilibrium solubility of the crystalline form. Consequently, crystalline drug precipitated in experiments containing Type 1 compounds, where molecules supposedly had sufficient opportunity to rearrange themselves during supersaturation to later form a solid crystal, whereas the rapid onset of precipitation of Type 2 compounds gave rise to amorphous, randomly-oriented solid forms. This study provides a mechanistic explanation for the solid state properties observed with precipitated weak bases by linking the amorphous or crystalline nature of precipitated drug to the intrinsic supersaturation phenomena of the compounds, reflected by one of the two observed trends (i.e., Type 1 or 2 compounds). However, whether this supersaturation behavior is affected by the complex and dynamic environment of the digestive tract remains unclear.
A follow up study by the above researchers examined the pH-induced precipitation of ionisable drugs, this time in the presence of polymers (86). These polymers were proposed to alter the supersaturation behavior intrinsic to the drugs tested, and this change in supersaturation behavior was achieved primarily via the polymer-driven inhibition of nucleation, as discussed above. Drugs were selected based on their previously determined tendency to rapidly crystallize with the pH shift method. It was shown that whilst some drug-polymer combinations led to stable amorphous precipitates, others did not. Molecular complexity of the drug and the ability to form tautomers were cited as reasons why different polymers were able to maintain the amorphous forms of drugs such as glyburide and warfarin, whereas dipyridamole predominantly precipitated in crystalline form.
The case studies above indicate that the solid state of some poorly water-soluble compounds is affected by the presence of digested lipids, however, there is a lack of understanding as to why these different solid forms are generated, and exactly what their implication is on drug absorption. A shortcoming of the solid state analysis methods used thus far to characterize digested drug pellets is that no real information can be deduced with regard to the mechanistic aspects of the solid structures formed, instead CPLM and XRD data alone has pushed forward the notion that amorphous precipitates or polymorphs are formed exclusively. However, solid state properties are complex and the dynamic compositional changes occurring during digestion provide for a number of other scenarios that could take place upon precipitation, which ultimately affects the solid state and re-dissolution potential of these drugs, as well as total amount absorbed.
Amorphous Drug Precipitation or Other Solid State Forms?
The amorphous and crystalline forms of a drug exhibit different dissolution properties, and these differences may have an impact on bioavailability. An amorphous solid consists of randomly oriented molecules and lacks the long-range order of thermodynamically favoured crystal forms (65,87). As a result, amorphous forms have high free energy, display faster dissolution rates and have lower melting temperatures than crystalline forms (88), which are generally all beneficial properties for oral drug delivery. A significant focus of industry has been to manufacture amorphous forms of drugs with an enhanced aqueous solubility and dissolution rate for solid oral dose forms; hence it makes sense that in-situ generation of similar material should be seen as beneficial over precipitation in crystalline form. The studies presented in Table I that indicate precipitation of amorphous weak bases during the digestion of lipid formulations, in some cases suggest an implication of other components in the digested matrix. The precipitated material may or may not be amorphous drug per se, but could be in alternative amorphous solid state forms, such as amorphous salts or co-amorphous systems (87,89). Moreover, precipitated drug also has the potential to undergo solution-mediated transformations, as well as taking part in excipient-drug interactions.
Whilst significant effort is geared towards stabilizing the supersaturated state (7,20,33,34,36,42,69,72), it appears inevitable that precipitation is likely to occur at least to some degree in-vivo with poorly water-soluble drugs in digestible formulations, especially at high drug loadings often seen with LBDDS. The precipitation of drug is highly dependent on the drug loading relative to the solubility of the drug in the different lipid excipients used in the formulation, and also the Type of formulation. For instance preparing a Type 2 formulation with drug loaded at 80% of its solubility in the formulation may be less likely to produce a precipitate on digestion, whereas a saturation level of 80% in a Type 3B formulation would be more likely to produce a precipitate. Hence, a challenge for formulation science is to control the solid state form of drug on precipitation during digestion and prevent rapid conversion to the crystalline form.
In consideration of the many ionisable species present during lipid digestion, the possibility of ion-pairing between precipitating drug and liberated fatty acids, bile salts or other excipients in the formulation has received no direct attention in the literature. The literature evidence to date indicates that, generally, basic drugs appear to precipitate from digesting lipid-based formulations in an amorphous form, while non-ionisable compounds appear to precipitate in a crystalline form, suggesting a possible link between ionisability and solid state form on precipitation, as has been suggested by Stillhart et al.(40) previously with the evidence presented in Table I largely supporting this logic. Therefore, it is reasonable to assume that weakly-basic drugs are precipitating during the digestion of lipid formulations as amorphous salts with fatty acid as the counter-ion. Moreover, when the pKa values of these weakly basic drugs, which appear to be precipitating in an amorphous form, are compared to the pH level of the digestion medium in their respective experiments, the link between ionisability and amorphous precipitation becomes more apparent. For example, cinnarizine has a pKa value of 7.4 and the in-vitro lipolysis experiment performed by Sassene et al. (62), mentioned earlier, was set to pH 6.5. At this pH level cinnarizine would be predominantly ionized and available for pairing with oppositely-charged species that are present during digestion, most likely with fatty acids and bile salts, which will also be at least partly ionized at this pH. In contrast, the basic drug loratadine has a pKa value of 5.0 and showed quite different behavior in the study by Stillhart et al. (40), where in this case the pH of the digestion medium was set to 7.5 and the drug precipitated in crystalline form after digestion, presumably due to the unionized state of the drug at this pH and therefore an inability to form the amorphous salt on precipitation. Whilst the characterization techniques mentioned thus far in the case studies presented above are able to discriminate between amorphous and crystalline drug, they are unable to elicit ionic-interactions, hence other characterization techniques are needed for this purpose. If this observation is founded in the chemistry as a general effect, it could potentially be utilized directly as a tool in formulation to induce precipitation of drug in an amorphous form.
Re-dissolution Potential and Dynamic Processing During Digestion
Controlling drug precipitation with regard to the solid state form can only be a driver of enhanced drug absorption if re-dissolution takes place in-vivo. In-vitro case studies have shown that drugs can precipitate in non-crystalline form, however, in closed in-vitro models re-dissolution is not possible. Dynamic changes in composition and drug absorption in-vivo, however, are anticipated to favor re-dissolution of precipitated drug, especially for high-energy solid forms, such as amorphous salts of drug-fatty acids or drug-bile salt compositions discussed above. The proposed scenario would involve generation of a supersaturated state by the earlier mentioned mechanisms during dispersion and digestion of the formulation, inducing drug precipitation and simultaneous absorption of drug and digestion products. As this dual mechanism proceeds, the level of saturation will decrease and promote dissolution of the high-energy solid/precipitated drug (e.g., amorphous salt). Meanwhile, digestion will continue to produce a high concentration of colloidal phases (mixed micelles and vesicles), which act to further facilitate the re-dissolution of drug. The role of colloidal structures formed during digestion and changes to their self-assembly and drug carrying capacity during the digestion process is of vital importance in generating the re-dissolution sink and mechanism of transport across the unstirred water layer. In order to consolidate the behavior in these systems beyond the observations to date, there remain two major challenges, namely to determine the kinetics of drug precipitation during digestion using real-time methods, and to determine the composition of the precipitated material, for example whether drug is actually ion-pairing with fatty acids and/or bile salts.
Newly Emerging In-Situ Characterization Techniques for Analyzing Precipitated Drug
The need for in-situ methods that enable the detection and structural interpretation of precipitated drug during digestion is increasingly recognized. Recently, the kinetics of drug precipitation were studied using an in-situ digestion model with in-line Raman spectroscopy (90). A SMEDDS formulation was loaded with the neutral drug fenofibrate at different concentrations, and in-vitro digestion with simultaneous Raman was performed. Conventionally, Raman has been used to detect high concentrations of crystallized material in a constant medium, although in this case, a low drug concentration (precipitated drug) needed to be detected and differentiated from the hydrolysis products in a dynamic environment. The kinetics of fenofibrate precipitation was non-linear with respect to lipolysis of the formulation. This finding suggests a supersaturated state formed for several minutes prior to fenofibrate precipitation, and is in contrast to the linear relationship reported between lipolysis progression and the extent of precipitation by Sassene et al. with the weakly basic drug cinnarazine (62).
In addition to kinetics, in-situ structural elucidation may also provide key insights into the solid state behavior of precipitated drug. The relevance of the solid material obtained from the pellet phase after digestion, subsequent to isolation and extraction, has been questioned in the past, due to sample handling and its potential to accelerate the precipitation of drug. To date, structural characterization using in-situ lipolysis and SAXS has been limited to that of the colloidal phases produced on lipid digestion in a number of studies (23,91–93). Recently, however, in-situ lipolysis and SAXS have also been used to follow the structure of precipitated fenofibrate during the in-vitro digestion of a highly saturated medium chain lipid formulation, or super-SNEDDS (94), and will further be used to probe the precipitation behavior of ionisable compounds during the digestion of lipid formulations with regard to solid state.
Similar time-resolved scattering approaches may involve performing experiments comparable to those reported by Hsieh et al. (86), where lipolysis was carried out and samples were taken at different time-points, rapidly centrifuged and placed in the SAXS beamline for analysis. This type of experiment may reduce the influence of sample preparation on drug precipitation, and may be described as ex-situ. Overall, there is much scope to improve and develop new in-situ techniques for following drug precipitation during digestion of lipid formulations, as studies look to such techniques for an understanding of what takes place in real-time (35,95).
As outlined above, experiments that may lead to a mechanistic understanding as to why amorphous and other solid forms of drugs are precipitating during the digestion of lipid formulations is key to designing optimal lipid based formulations for poorly water-soluble compounds. From the trends observed for weakly-basic drugs in Table I, potential interactions between drugs and formulation excipients, as well as drugs and endogenous species in the digestive environment may affect the final solid state, especially for ionisable compounds. Spectroscopic techniques, such as FTIR and NMR, can potentially reveal these key interactions. For example, if drug-fatty acid binding upon precipitation during digestion is expected to form an amorphous salt, the resultant FTIR, or NMR spectra would elucidate this change via changes to the absorption bands of the functional groups involved in the ionic interaction.
CONCLUSION
Lipid based drug delivery has proven to be an effective means to deliver poorly water soluble compounds via the oral route. A major drawback of the formulation, however, remains the loss of solubilisation capacity upon dilution and dispersion in the gastrointestinal tract, which can lead to supersaturation and precipitation of drug. Traditionally, drug precipitation during gastrointestinal transit was believed to be detrimental to bioavailability, and conventional formulation approaches used to inhibit precipitation have been discussed here, nevertheless, recent studies have demonstrated that precipitation of drug is not always confined to poorly water-soluble crystal forms. Consequently, solid state variability upon precipitation can lead to different bioavailibilities in some instances, especially where amorphous precipitation is observed. The potential effect of the digestion environment and formulation excipients on the final solid state of precipitated drug have been explored, as well as the possibility of re-dissolution for precipitated high energy solid forms during digestion. Commonly used characterization methods for analyzing the solid state of precipitated drug have been mentioned, moreover, the emergence of highly relevant in-situ characterization techniques have been touched upon. It is becoming increasingly apparent that the solid state of precipitated drug from lipid based formulations can act as a driver for the total amount of drug absorbed. A better understanding behind the mechanisms of drug precipitation during digestion, and why the solid state of drug is often altered, are required. Thereby, controlling the solid state form of precipitated drug during digestion (e.g., amorphous vs crystalline) may prove a valuable formulation strategy to increase the total amount of absorbed drug.
Abbreviations
- LBDDS:
-
Lipid based drug delivery systems
- BCS:
-
Biopharmaceutics classification system
- LFCS:
-
Lipid formulation classification system
- SRM :
-
Maximum supersaturation ratio
- XRD:
-
X-ray diffraction
- CPLM:
-
Crossed polarized light microscopy
- DSC:
-
Differential scanning calorimetry
- FTIR:
-
Fourier transform infrared spectroscopy
- USP:
-
United States pharmacopeia
- PPI:
-
Polymeric precipitation inhibitor
- PVP:
-
Polyvinylpyrrolidone
- HPMC:
-
Hydroxypropylmethyl cellulose
- MC:
-
Medium chain
- LC:
-
Long chain
- SNEDDS:
-
Self nano-emulsifying drug delivery system
- SMEDDS:
-
Self micro-emulsifying drug delivery system
- SAXS:
-
Small-angle x-ray scattering
- NMR:
-
Nuclear magnetic resonance
REFERENCES
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58(3):173–82.
Chen S, Dudhedia MS, Wang Z, Darrington RT, Tamblyn T, Smoliga JA, et al. Drug-excipient complexation in lipid based delivery systems: an investigation of the Tipranavir-1,3-dioctanolyglycerol complex. J Pharm Sci. 2009;98(5):1732–43.
Ku MS. Use of the biopharmaceutical classification system in early drug development. AAPS J. 2008;10(1):208–12.
Lindenberg M, Kopp S, Dressman JB. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system. Eur J Pharm Biopharm. 2004;58(2):265–78.
Gupta S, Kesarla R, Omri A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharm. 2013;2013:848043.
Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20.
Bevernage J, Brouwers J, Annaert P, Augustijns P. Drug precipitation-permeation interplay: supersaturation in an absorptive environment. Eur J Pharm Biopharm. 2012;82(2):424–8.
Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44(1):235–49.
Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3–4):278–87.
MacGregor KJ, Embleton JK, Lacy JE, Perry EA, Solomon LJ, Seager H, et al. Influence of lipolysis on drug absorption from the gastro-intestinal tract. Adv Drug Deliv Rev. 1997;25(1):33–46.
Dokoumetzidis A, Macheras P. A century of dissolution research: from Noyes and Whitney to the biopharmaceutics classification system. Int J Pharm. 2006;321(1–2):1–11.
Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm. 2011;420(1):1–10.
Grohganz H, Priemel PA, Lobmann K, Nielsen LH, Laitinen R, Mullertz A, et al. Refining stability and dissolution rate of amorphous drug formulations. Expert Opin Drug Deliv. 2014;11(6):977–89.
Mohsin K, Long MA, Pouton CW. Design of lipid-based formulations for oral administration of poorly water-soluble drugs: precipitation of drug after dispersion of formulations in aqueous solution. J Pharm Sci. 2009;98(10):3582–95.
Mu H, Holm R, Mullertz A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int J Pharm. 2013;453(1):215–24.
O’Driscoll CM, Griffin BT. Biopharmaceutical challenges associated with drugs with low aqueous solubility--the potential impact of lipid-based formulations. Adv Drug Deliv Rev. 2008;60(6):617–24.
Porter CJ, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–91.
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–48.
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11 Suppl 2:S93–8.
Williams HD, Trevaskis NL, Yeap YY, Anby MU, Pouton CW, Porter CJ. Lipid-based formulations and drug supersaturation: harnessing the unique benefits of the lipid digestion/absorption pathway. Pharm Res. 2013;30(12):2976–92.
Liao TH, Hamosh P, Hamosh M. Fat digestion by lingual lipase: mechanism of lipolysis in the stomach and upper small intestine. Pediatr Res. 1984;18(5):402–9.
Holm R, Mullertz A, Mu H. Bile salts and their importance for drug absorption. Int J Pharm. 2013;453(1):44–55.
Phan S, Hawley A, Mulet X, Waddington L, Prestidge CA, Boyd BJ. Structural aspects of digestion of medium chain triglycerides studied in real time using sSAXS and Cryo-TEM. Pharm Res. 2013;30(12):3088–100.
Trevaskis NL, Porter CJ, Charman WN. Bile increases intestinal lymphatic drug transport in the fasted rat. Pharm Res. 2005;22(11):1863–70.
Yeap YY, Trevaskis NL, Quach T, Tso P, Charman WN, Porter CJ. Intestinal bile secretion promotes drug absorption from lipid colloidal phases via induction of supersaturation. Mol Pharm. 2013;10(5):1874–89.
Kossena GA, Charman WN, Boyd BJ, Porter CJ. Influence of the intermediate digestion phases of common formulation lipids on the absorption of a poorly water-soluble drug. J Pharm Sci. 2005;94(3):481–92.
van Mourik ID, Thomson M, Kelly DA. Comparison of pharmacokinetics of Neoral and Sandimmune in stable pediatric liver transplant recipients. Liver Transpl Surg. 1999;5(2):107–11.
Han SF, Yao TT, Zhang XX, Gan L, Zhu C, Yu HZ, et al. Lipid-based formulations to enhance oral bioavailability of the poorly water-soluble drug anethol trithione: effects of lipid composition and formulation. Int J Pharm. 2009;379(1):18–24.
Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997;25(1):103–28.
Chakrabarti S, Belpaire FM. Biovailability of phenytoin in lipid containing dosage forms in rats. J Pharm Pharmacol. 1978;30(5):330–1.
Carrigan PJ, Bates TR. Biopharmaceutics of drugs administered in lipid-containing dosage forms. I. GI absorption of griseofulvin from an oil-in-water emulsion in the rat. J Pharm Sci. 1973;62(9):1476–9.
Porter CJ, Kaukonen AM, Taillardat-Bertschinger A, Boyd BJ, O’Connor JM, Edwards GA, et al. Use of in vitro lipid digestion data to explain the in vivo performance of triglyceride-based oral lipid formulations of poorly water-soluble drugs: studies with halofantrine. J Pharm Sci. 2004;93(5):1110–21.
Brouwers J, Brewster ME, Augustijns P. Supersaturating drug delivery systems: the answer to solubility-limited oral bioavailability? J Pharm Sci. 2009;98(8):2549–72.
Anby MU, Williams HD, McIntosh M, Benameur H, Edwards GA, Pouton CW, et al. Lipid digestion as a trigger for supersaturation: evaluation of the impact of supersaturation stabilization on the in vitro and in vivo performance of self-emulsifying drug delivery systems. Mol Pharm. 2012;9(7):2063–79.
Arnold YE, Imanidis G, Kuentz MT. Advancing in-vitro drug precipitation testing: new process monitoring tools and a kinetic nucleation and growth model. J Pharm Pharmacol. 2011;63(3):333–41.
Warren DB, Benameur H, Porter CJ, Pouton CW. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: a mechanistic basis for utility. J Drug Target. 2010;18(10):704–31.
Lindfors L, Forssen S, Westergren J, Olsson U. Nucleation and crystal growth in supersaturated solutions of a model drug. J Colloid Interface Sci. 2008;325(2):404–13.
Bevernage J, Brouwers J, Brewster ME, Augustijns P. Evaluation of gastrointestinal drug supersaturation and precipitation: strategies and issues. Int J Pharm. 2013;453(1):25–35.
Kostewicz ES, Wunderlich M, Brauns U, Becker R, Bock T, Dressman JB. Predicting the precipitation of poorly soluble weak bases upon entry in the small intestine. J Pharm Pharmacol. 2004;56(1):43–51.
Stillhart C, Durr D, Kuentz M. Toward an improved understanding of the precipitation behavior of weakly basic drugs from oral lipid-based formulations. J Pharm Sci. 2014.
Yeap YY, Trevaskis NL, Porter CJ. The potential for drug supersaturation during intestinal processing of lipid-based formulations may be enhanced for basic drugs. Mol Pharm. 2013;10(7):2601–15.
Yeap YY, Trevaskis NL, Porter CJ. Lipid absorption triggers drug supersaturation at the intestinal unstirred water layer and promotes drug absorption from mixed micelles. Pharm Res. 2013;30(12):3045–58.
Kashchiev D. Forms and applications of the nucleation theorem. J Chem Phys. 2006;125(1):014502.
Horn D, Rieger J. Organic nanoparticles in the aqueous phase-theory, experiment, and use. Angew Chem Int Ed Engl. 2001;40(23):4330–61.
Rodriguez-Hornedo N, Murphy D. Significance of controlling crystallization mechanisms and kinetics in pharmaceutical systems. J Pharm Sci. 1999;88(7):651–60.
JW, M, Crystallisation. Oxford: Butterworth-Heinemann; 2001.
Dressman JB, Berardi RR, Dermentzoglou LC, Russell TL, Schmaltz SP, Barnett JL, et al. Upper gastrointestinal (GI) pH in young, healthy men and women. Pharm Res. 1990;7(7):756–61.
Gargouri Y, Moreau H, Verger R. Gastric lipases: biochemical and physiological studies. Biochim Biophys Acta. 1989;1006(3):255–71.
Zangenberg NH, Mullertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model. I. Controlling the rate of lipolysis by continuous addition of calcium. Eur J Pharm Sci. 2001;14(2):115–22.
Sek L, Porter CJ, Kaukonen AM, Charman WN. Evaluation of the in-vitro digestion profiles of long and medium chain glycerides and the phase behaviour of their lipolytic products. J Pharm Pharmacol. 2002;54(1):29–41.
Devraj R, Williams HD, Warren DB, Mullertz A, Porter CJ, Pouton CW. In vitro digestion testing of lipid-based delivery systems: calcium ions combine with fatty acids liberated from triglyceride rich lipid solutions to form soaps and reduce the solubilization capacity of colloidal digestion products. Int J Pharm. 2013;441(1–2):323–33.
Shono Y, Jantratid E, Dressman JB. Precipitation in the small intestine may play a more important role in the in vivo performance of poorly soluble weak bases in the fasted state: case example nelfinavir. Eur J Pharm Biopharm. 2011;79(2):349–56.
Thomas N, Richter K, Pedersen TB, Holm R, Mullertz A, Rades T. In Vitro lipolysis data does not adequately predict the in vivo performance of lipid-based drug delivery systems containing fenofibrate. AAPS J. 2014.
Williams HD, Sassene P, Kleberg K, Bakala-N’Goma JC, Calderone M, Jannin V, et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 1: method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. J Pharm Sci. 2012;101(9):3360–80.
Williams HD, Anby MU, Sassene P, Kleberg K, Bakala-N’Goma JC, Calderone M, et al. Toward the establishment of standardized in vitro tests for lipid-based formulations. 2. The effect of bile salt concentration and drug loading on the performance of type I, II, IIIA, IIIB, and IV formulations during in vitro digestion. Mol Pharm. 2012;9(11):3286–300.
Williams HD, Sassene P, Kleberg K, Calderone M, Igonin A, Jule E, et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 3: understanding supersaturation versus precipitation potential during the in vitro digestion of type I, II, IIIA, IIIB and IV lipid-based formulations. Pharm Res. 2013;30(12):3059–76.
Thomas N, Holm R, Rades T, Mullertz A. Characterising lipid lipolysis and its implication in lipid-based formulation development. AAPS J. 2012;14(4):860–71.
Salentinig S, Salentinig S, Tangso KJ, Hawley A, Boyd BJ. pH-driven colloidal transformations based on the vasoactive drug nicergoline. Langmuir. 2014;30(49):14776–81.
Arnold YE, Imanidis G, Kuentz M. Study of drug concentration effects on in vitro lipolysis kinetics in medium-chain triglycerides by considering oil viscosity and surface tension. Eur J Pharm Sci. 2011;44(3):351–8.
Harris KD. Powder diffraction crystallography of molecular solids. Top Curr Chem. 2012;315:133–77.
RA, C. Chapter 2: polarized light microscopy, in pharmaceutical microscopy. 2011. p. 321p. 139 illus, 102 illus. in color.
Sassene PJ, Knopp MM, Hesselkilde JZ, Koradia V, Larsen A, Rades T, et al. Precipitation of a poorly soluble model drug during in vitro lipolysis: characterization and dissolution of the precipitate. J Pharm Sci. 2010;99(12):4982–91.
Carstensen JT, Lai TY, Prasad VK. USP dissolution IV: comparison of methods. J Pharm Sci. 1978;67(9):1303–7.
Thomas N, Holm R, Mullertz A, Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J Control Release. 2012;160(1):25–32.
Savolainen M, Kogermann K, Heinz A, Aaltonen J, Peltonen L, Strachan C, et al. Better understanding of dissolution behaviour of amorphous drugs by in situ solid-state analysis using Raman spectroscopy. Eur J Pharm Biopharm. 2009;71(1):71–9.
Verdonck E, Schaap K, Thomas LC. A discussion of the principles and applications of Modulated Temperature DSC (MTDSC). Int J Pharm. 1999;192(1):3–20.
Gill P, Moghadam TT, Ranjbar B. Differential scanning calorimetry techniques: applications in biology and nanoscience. J Biomol Tech. 2010;21(4):167–93.
Patel DD, Anderson BD. Effect of precipitation inhibitors on indomethacin supersaturation maintenance: mechanisms and modeling. Mol Pharm. 2014;11(5):1489–99.
Warren DB, Bergstrom CA, Benameur H, Porter CJ, Pouton CW. Evaluation of the structural determinants of polymeric precipitation inhibitors using solvent shift methods and principle component analysis. Mol Pharm. 2013;10(8):2823–48.
DiNunzio JC, Miller DA, Yang W, McGinity JW, Williams 3rd RO. Amorphous compositions using concentration enhancing polymers for improved bioavailability of itraconazole. Mol Pharm. 2008;5(6):968–80.
Rupprecht H, Ziller KH. Characterization of the crystallization behavior of poorly soluble drugs in suspensions. Pharmazie. 1981;36(4):298.
Augustijns P, Brewster ME. Supersaturating drug delivery systems: fast is not necessarily good enough. J Pharm Sci. 2012;101(1):7–9.
Machefer S, Huddar MM, Schnitzlein K. Effect of polymer admixtures on the growth habit of ionic crystals. Study on crystal growth kinetics of potassium dihydrogen phosphate in water/polyol mixtures. J Cryst Growth. 2008;310(24):5347–56.
Raghavan SL, Trividic A, Davis AF, Hadgraft J. Crystallization of hydrocortisone acetate: influence of polymers. Int J Pharm. 2001;212(2):213–21.
Gao P, Akrami A, Alvarez F, Hu J, Li L, Ma C, et al. Characterization and optimization of AMG 517 Supersaturatable Self-Emulsifying Drug Delivery System (S-SEDDS) for improved oral absorption. J Pharm Sci. 2009;98(2):516–28.
Psachoulias D, Vertzoni M, Goumas K, Kalioras V, Beato S, Butler J, et al. Precipitation in and supersaturation of contents of the upper small intestine after administration of two weak bases to fasted adults. Pharm Res. 2011;28(12):3145–58.
Carlert S, Akesson P, Jerndal G, Lindfors L, Lennernas H. Abrahamsson, B, In vivo dog intestinal precipitation of mebendazole: a basic BCS class II drug. Mol Pharm. 2012;9(10):2903–11.
Psachoulias D, Vertzoni M, Butler J, Busby D, Symillides M, Dressman J, et al. An in vitro methodology for forecasting luminal concentrations and precipitation of highly permeable lipophilic weak bases in the fasted upper small intestine. Pharm Res. 2012;29(12):3486–98.
Larsen AT, Sassene P, Mullertz A. In vitro lipolysis models as a tool for the characterization of oral lipid and surfactant based drug delivery systems. Int J Pharm. 2011;417(1–2):245–55.
Thomas N, Holm R, Garmer M, Karlsson JJ, Mullertz A, Rades T. Supersaturated self-nanoemulsifying drug delivery systems (Super-SNEDDS) enhance the bioavailability of the poorly water-soluble drug simvastatin in dogs. AAPS J. 2013;15(1):219–27.
Boyd BJ, Khoo SM, Whittaker DV, Davey G, Porter CJ. A lipid-based liquid crystalline matrix that provides sustained release and enhanced oral bioavailability for a model poorly water soluble drug in rats. Int J Pharm. 2007;340(1–2):52–60.
Kaukonen AM, Boyd BJ, Porter CJ, Charman WN. Drug solubilization behavior during in vitro digestion of simple triglyceride lipid solution formulations. Pharm Res. 2004;21(2):245–53.
Kossena GA, Charman WN, Boyd BJ, Porter CJ. A novel cubic phase of medium chain lipid origin for the delivery of poorly water soluble drugs. J Control Release. 2004;99(2):217–29.
Larsen AT, Ohlsson AG, Polentarutti B, Barker RA, Phillips AR, Abu-Rmaileh R, et al. Oral bioavailability of cinnarizine in dogs: relation to SNEDDS droplet size, drug solubility and in vitro precipitation. Eur J Pharm Sci. 2013;48(1–2):339–50.
Hsieh YL, Ilevbare GA, Van Eerdenbrugh B, Box KJ, Sanchez-Felix MV, Taylor LS. pH-Induced precipitation behavior of weakly basic compounds: determination of extent and duration of supersaturation using potentiometric titration and correlation to solid state properties. Pharm Res. 2012;29(10):2738–53.
Hsieh YL, Box K, Taylor LS. Assessing the impact of polymers on the pH-induced precipitation behavior of poorly water soluble compounds using synchrotron wide angle x-ray scattering. J Pharm Sci. 2014;103(9):2724–35.
Lobmann K, Laitinen R, Grohganz H, Gordon KC, Strachan C, Rades T. Coamorphous drug systems: enhanced physical stability and dissolution rate of indomethacin and naproxen. Mol Pharm. 2011;8(5):1919–28.
Willart JF, Descamps M. Solid state amorphization of pharmaceuticals. Mol Pharm. 2008;5(6):905–20.
Miroshnyk I, Mirza S, Sandlert N. Pharmaceutical co-crystals-an opportunity for drug product enhancement. Expert Opin Drug Deliv. 2009;6(4):333–41.
Stillhart C, Imanidis G, Kuentz M. Insights into drug precipitation kinetics during in vitro digestion of a lipid-based drug delivery system using in-line raman spectroscopy and mathematical modeling. Pharm Res. 2013;30(12):3114–30.
Warren DB, Anby M, Hawley U, Boyd A, B. J. Real time evolution of liquid crystalline nanostructure during the digestion of formulation lipids using synchrotron small-angle X-ray scattering. Langmuir. 2011;27(15): 9528–34.
Salentinig S, Phan S, Khan J, Hawley A, Boyd BJ. Formation of highly organized nanostructures during the digestion of milk. ACS Nano. 2013;7(12):10904–11.
Phan S, Salentinig S, Prestidge CA, Boyd BJ. Self-assembled structures formed during lipid digestion: characterization and implications for oral lipid-based drug delivery systems. Drug Deliv Transl Res. 2014;4(3):275–94.
Khan J, Hawley A. Rades T, Boyd BJ. In situ lipolysis and synchrotron small-angle x-ray scattering for the direct determination of the precipitation and solid-state form of a poorly water-soluble drug during digestion of a lipid-based formulation. J Pharm Sci. 2015.
Ueda H, Ida Y, Kadota K, Tozuka Y. Raman mapping for kinetic analysis of crystallization of amorphous drug based on distributional images. Int J Pharm. 2014;462(1–2):115–22.
ACKNOWLEDGMENTS AND DISCLOSURES
This manuscript was supported by the Australian Research Council through the Discovery Projects scheme (DP120104032). B.J.B. holds an ARC Future Fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khan, J., Rades, T. & Boyd, B. The Precipitation Behavior of Poorly Water-Soluble Drugs with an Emphasis on the Digestion of Lipid Based Formulations. Pharm Res 33, 548–562 (2016). https://doi.org/10.1007/s11095-015-1829-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-015-1829-5