
A simple and sensitive stability indicating HPLC-UV method was developed for the analysis of process impurities and degradation products in Sofosbuvir and Velpatasvir pharmaceutical formulations. Analysis was performed on C18 analytical column (250 mm × 4.6 mm, 5 μm), using a mobile phase of 0.05M ammonium acetate buffer (pH 6.5) with acetonitrile at 45 : 55 (v/v) ratio and a flow rate of 1.0 mL/min. The detection wavelength for simultaneous determination of both ingredients using UV detector was 268 nm. The method was validated for accuracy, precision, linearity, and specificity using reference standards of process impurities and degradation products. The calibration curve was linear and the limits of detection and quantification were determined using 3δ/slope and 10δ/slope expressions, respectively. Forced degradation studies for Sofosbuvir and Velpatasvir drug substances and products were designed under different environmental stress conditions. The effect of alkaline, acidic, oxidative, and thermal stress conditions on Sofosbuvir and Velpatasvir substances and dosage forms was studied. The influence of heat and humidity was studied at 80 ± 5°C and 75 ± 5% RH. Similarly, the photolytic studies were performed on visible light at 200 Wh/m2 UV and 1.2 million lux h/m2 in a climatic chamber. The proposed method is stability-indicating and has been successfully applied to the analysis of process impurities and degradation products in Sofosbuvir and Velpatasvir pharmaceutical formulations. The degradation products were characterized by MS, NMR and IR spectroscopy analysis for identification of degradation products and determination of mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Direct Acting Antiviral (DAA) drugs are new molecules for inhibition of Hepatitis-C Virus (HCV) polymerase and protease enzymes. DAA drugs have been recently developed for the treatment of HCV to prevent viral replication process by the selective binding mechanism [1]. Some of the new, highly potent DAA drugs like Sofosbuvir [2], Ledipasvir [3], Daclatasvir [4] and Velpatasvir [5, 6] are recently approved by the US Food and Drug Administration (FDA). Sofosbuvir (SOF) is a potent nucleotide analog formally known as GS-7977. It is the first selective non-structural protein (NS-5B) HCV polymerase enzyme inhibitor. SOF developed by Gilead Sciences USA was first approved by FDA in December 2013 [7]. SOF is white to off-white crystalline powder having molecular formula C22H29FN3O9P and structural formula shown in Fig. 1a. Velpatasvir (VEL) is a new antiviral drug, NS-5A inhibitor active against all major genotypes of HCV. VEL is also white to off-white, non-hygroscopic, crystalline solid having molecular formula C49H54N8O8 and structural formula shown in Fig. 1b. The pharmaceutical dosage forms of VEL are available in fixed dose combination (VEL-SOF) and approved by FDA on June 28, 2016 [8].

Chemical structures of (a) Sofosbuvir and (b ) Velpatasvir.
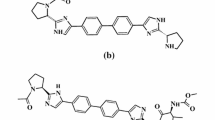
Analysis of the process of SOF and VEL synthesis shows that there are two major process impurities for both SOF and VEL. The process impurities of SOF are due to the two reaction sites of starting material B (Fig. 2a ). Similarly, VEL also has two major byproducts in reactions of synthesis (Fig. 2b ).

Process impurities of (a) Sofosbuvir and (b ) Velpatasvir.
Apart from the process impurities, degradation products (Figs. 3a and 3b ) have also been reported for SOF and VEL [9 – 11].

Degradation products of (a) Sofosbuvir and (b ) Velpatasvir.
The control of process impurities and degradation products in new lifesaving drugs is crucial. There are strict guidelines to limit the level of process impurities and degradation products in pharmaceutical formulations [12, 13]. Forced degradation studies are performed to evaluate the chemical stability of drug products under the influence of various stressed environmental conditions. Results provide information about potential degradation products that can pose risks to stability and efficacy of the product. Efficient and sensitive stability indicating methods are used to analyze the impurities and degradation products.
It is important to note that no pharmacopoeial monographs are available for analysis of the above mentioned new drug substances and pharmaceutical formulations. In the last few years, a number of methods such as LCMS/MS [14 – 17], UPLC-ESCI MS/MS and UPLC-MS/MS [18, 19] and HPTLC [20] have been reported for SOF-VEL. However, these methods can only be applied to the analysis of biological fluids, pharmacokinetics and bioequivalence studies. Various analytical methods including spectrofluorimetry [21], UPLC [22], spectrophotometry/UPLC [23] and spectrophotometry [24] can also be used for the simultaneous analysis of SOF and VEL in pharmaceutical formulations. Stability indicating methods based on UPLC [25], HPLC-PDA [26], RP-UFLC [27] and HPLC-UV [28, 29] were also reported very recently. However, these methods have some disadvantages due to low sensitivity and cannot be applied to analysis of the process impurities and degradation products. The forced degradation studies were conducted in different conditions but methods were not properly validated using impurity reference standards. To the best of our knowledge, no stability indicating HPLC-UV method has been reported so far for the determination of process impurities and degradation products in SOF-VEL dosage form.
For this purpose, we have developed a simple and sensitive stability indicating HPLC-UV method for simultaneous analysis of process impurities and degradation products in SOF-VEL combined dosage form. The method was properly validated using reference standards of process and degradation impurities according to the guidelines of United States Pharmacopeia (USP) and International Conference on Harmonization (ICH) [30, 31]. In addition, forced degradation studies of SOF-VEL formulations were performed under stressed environmental conditions and the degradation products were quantified and characterized.
EXPERIMENTAL
Materials and Chemicals
Pharmaceutical product SOF-VEL (Sofvel 400/100 mg tablets), drug substance of SOF having purity 99.8%, VEL purity 99.4%, and excipients, i.e., copovidone, microcrystalline cellulose, croscarmellose sodium, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol and purified talc were provided by Genome Pharmaceuticals (Pakistan). Reference standards of SOF purity 99.3%, and impurity standards SOF-IMP1 purity 94.6%, SOF-IMP2 purity 89.7%, SOF-DP1 purity 91.4%, SOF-DP2 purity 97.6% were provided by Ruyuan HEC Pharm Co. Ltd. (China). Reference standards of VEL purity 99.5%, and impurity standards VEL-IMP1 purity 98.5%, VEL-IMP2 purity 92.0% and VEL-DP1 purity 88.4% were provided by Nantong Chanyoo Pharmatech Co. Ltd (China). Methanol HPLC grade, acetonitrile HPLC grade, ammonium acetate, sodium hydroxide, hydrochloric acid (37% w/v) and hydrogen peroxide (30% w/v) were purchased from Sigma-Aldrich (Germany).
Instrumentation
Gradient HPLC system comprised a Cecil low pressure quaternary gradient pump Adept CE-4104, UV detector Adept CE 4200 (Cecil Instruments Limited UK). The system was controlled by power stream chromatography manager version 4.2. Symmetry analytical column 250 mm × 4.6 mm, 5 μm, packing C18 (Waters USA) was used for analysis. Other equipment used in this work included Nicolet iS5N FT-NIR spectrometer (Thermo Fisher Scientific USA), Agilent 6320 Iron Trap MS (USA), Bruker AVANCE-III HD 600 NMR (USA), Climatic chamber for thermal and photo stability studies with fluorescent light of 1.2 million lux h/m2 and UV light of 200 watt h/m2 (China).
Preparation of Solutions
Mobile phase and dilution medium. Ammonium acetate buffer pH 6.5 (0.05M) was prepared by dissolving 3.86 g ammonium acetate reagent grade in 400 mL purified water using a 1000 mL volumetric flask. The pH was adjusted to pH 6.5 with acetic acid solution and diluted to 1 L with purified water. Mobile phase was prepared by mixing ammonium acetate buffer pH 6.5 with acetonitrile at 45 : 55 (v/v) ratio and filtered through 0.2 μm nylon membrane filter. A homogeneous mixture of acetonitrile and water (1 : 1) was also prepared to be used as dilution medium.
Reference and sample stock solutions. Stock solution of SOF 0.4mg mL-1 was prepared by dissolving equivalent to 20.0 mg SBF reference standard in 50 mL dilution medium. Stock solution VEL 0.1 mg mL-1 was prepared by dissolving 5.0 mg reference standards of VEL in 50 mL dilution medium. Impurity reference solution 0.04 mg mL-1 was prepared by dissolving 2.0 mg reference standards of each impurity separately in 50 mL dilution medium.
Sample solution of tablet with known amount of SOF 0.4 mg mL-1 and VEL 0.1 mg mL-1 was prepared by dissolving ten tablets in 1 L dilution medium. This solution was filtered through 0.45 μm nylon filter (Millipore).
All the stock solutions were protected from daylight due to possible degradation of SOF, VEL or any impurity under high intensity light conditions. The above solutions were stored at 2 – 8°C and further diluted to SOF 40 μg mL-1and VEL 10 μg mL-1 for assay determination and validation study.
METHOD VALIDATION
Accuracy and Recovery
Reference and sample solutions of six concentration levels (20 – 120% of assay concentration) with known amount of SOF (8 – 48 μg mL-1) and VEL (2 – 12 μg mL-1) were prepared from stock solutions. To check the recovery of impurities based on ICH guidelines Q3B (R2), the reported threshold of impurities should be as low as 0.05%. The sample solutions were spiked with 0.1 to 0.5% of SOF-IMP1, SOF-IMP2, SOF-DP1 and SOF-DP2 (0.04 – 0.24 μg mL-1) and VEL-IMP1, VEL-IMP2 and VEL-DP1 (0.01 – 0.06 μg mL-1). The solution replicates (n=3) were analyzed for recovery. The recovery of SOF and VEL from the dosage form was determined by spiking of excipients with SOF and VEL drug substance. Excipient solutions were spiked with known amount of drug substance and analyzed for recovery. The recovery of each sample was calculated by comparing the peak response with individual reference solution. The values of RSD were evaluated against acceptance limits ± 2% for SOF, VEL and ±10% for impurities and relative error (RE) against ±20%.
Precision and Intermediate Precision
The precision was assured by analyzing replicates (n = 6) of three samples (high, medium and low concentration levels, 20, 60 and 120% of assay concentration). The combined samples containing known amounts of SOF (8, 24, and 48 μg mL-1) and VEL (2, 6, and 12 μg mL-1) were spiked with 0.1 to 0.5% of SOF impurity (0.04 – 0.24 μg mL-1) and VEL impurity (0.01 – 0.06 μg mL-1). For precision and intermediate precision, the values of overall relative standard deviation (RSD %) of peak response at different days were checked against acceptable limits (±2% for SOF, VEL and ±10% for impurities).
Robustness
Robustness of the method was checked by small changes (±2%) in the given values of dilution medium, column temperature, detection wavelength and mobile phase ratio. Replicates (n = 6) of reference solutions SOF (8, 24, and 48 μg mL-1) and VEL (2, 6, and 12 μg mL-1) spiked with 0.1 to 0.5% of SOF impurity (0.04 – 0.24 μg mL-1) and VEL impurity (0.01 – 0.06 μg mL-1) were analyzed. The influence of the changes on results was determined by evaluating the value of (RSD %) against acceptable limits ± 2 % of peak response and retention times.
Linearity and Range
For linearity assessment, replicates (n = 3) of seven concentrations were analyzed ranging from 10 to 120% having known amount of SOF (4 – 48 μg mL-1) and VEL(1 – 12 μg mL-1) spiked with 0.05 to 0.5% SOF impurity 0.04 – 0.2 μg mL-1) and VEL impurity (0.01 – 0.05 μg mL-1) respectively. The values of peak response were plotted on X axis against concentration on Y axis and based on least-square linear regression equation (A = slope C + Y intercept) the relation of peak response with concentration was evaluated. The limits of detection (LOD) and quantification (LOQ) were calculated from slop using expression (3.3δ/slope) and (10δ/slope) respectively.
Specificity and Placebo Interference
Composite solution of placebo was prepared in dilution medium using all excipients of tablet except active ingredients. This solution was analyzed on the same chromatographic condition and the baseline was evaluated for peak response. Placebo interference was also assured by spiking of reference solution with appropriate level of excipients and the baseline was evaluated for any interference or additional peak other than known peaks of SOF and VEL.
Studies of Solution Stability
Stress stability studies were performed to evaluate the degradation behavior of drug substance under various stressed environmental conditions. The forced degradation studies were performed in alkaline, acidic hydrolytic, photolytic, thermal and oxidative conditions following ICH guidelines [32, 33].
Solutions of SOF 400 μg mL-1 and VEL 100 μg mL-1 were used in forced degradation studies. The solutions were stored at room temperature in acidic (0.1 M HCl), alkaline (0.1 M NaOH) and neutral (in diluents) for 4 and 8h. The oxidative stress testing was performed using 3% H2O2 for 4 and 8 h. A composite solution of drug substance and all excipients was refluxed in neutral medium (diluents) for 4 and 8 h to check the compatibility of excipients. The effect of heat on drug product was studied by exposing the drug substance and product in solid to 105°C ± 5°C. Similarly the effect of heat and relative humidity (RH) on product was also studied at 80°C and 75 ± 5 %RH. For photo stability the drug substance and product were exposed to 200 W h/m2 UV light and 1.2 million lux h/m2 visible light at 40°C using photo stability chamber. The drug product in solid and solutions was also exposed to direct sunlight for 6 h to evaluate the maximum effect of light.
Stability of solutions exposed to room temperature (15 – 25°C) for 7 days, and cool temperature (2 – 08°C) for 14 days were also analyzed. The results for all solutions were compared to the results for freshly prepared solutions of reference standards.
RESULTS AND DISCUSSION
Method Development and Optimization
Due to conjugated group attached at Belt-K and benzene ring at Belt-B, SOF showed maximum absorbance at wavelength 261 ± 1 and VEL at 268 ± 1 nm and 303 ± 1 nm. To optimize the wavelength for simultaneous detection, the reference solutions SOF, VEL and all impurities each 20 μg mL-1 were scanned from 200 nm to 400 nm (Fig. 4). The optimum wavelength 268 nm, showing better absorbance for SOF, VEL and all impurities was selected for HPLC analysis.

UV absorbance spectra of SOF and VEL reference standards (1 and 5), impurity standards (2, 3, 6 and 7) and degradation products (4, 8 and 9).
For better resolution of all analytes, systematic elution of different mobile phases over different stationary phases was performed at different flow rates. The gradient and isocratic flow of mobile phase with different organic solvents and buffers was studied for analysis. Different results were obtained for analysis of composite reference solution using different mobile phases. Using mobile phase containing methanol with 0.1% phosphoric acid (35 : 65 v/v) at flow rate 1.2 mL min-1, separation of all the ingredients was achieved. The total run time was 35 minutes and resolution of SOF-IMP1 and SOF was not satisfactory. Mobile phase containing acetonitrile with potassium phosphate buffer solution, pH 2.0 (40 : 60 v/v) was also studied but the results of acidic mobile phase was not favorable for separation SOF and its impurities. Ammonium acetate (0.05 M) buffer (pH 6.0) in combination with acetonitrile was selected to reduce the sensitivity of retention with pH of mobile phase. For this purpose, analysis was performed using ammonium acetate buffer (pH 6.0) with acetonitrile having different ratio over different stationary phase C8 and C18. Mobile phase of ammonium acetate buffer (pH 6.0) with acetonitrile having ratio 45 : 55 v/v over C18 column showed better resolution and symmetrical peaks. Brand new symmetry analytical column (250 mm × 4.6 mm, 5 μm) packing C18 was selected for analysis. The retention time of each analyte was determined using individual and composite reference standard (Fig. 5). Better resolution of SOF, VEL and each impurity was achieved analyzing composite reference solution (Fig. 6).

Chromatograms of reference standards (a) SOF and VEL composite standard (SOF 8 μg mL-1, VEL 2 μg mL-1), (b ) SOF-DP1 0.1 μg mL-1, (c) SOF-IMP1 0.1 μg mL-1, (d ) SOF-IMP2 0.1 μg mL-1, (e) SOF-DP2 0.1 μg mL-1, ( f ) VEL-DP1 0.05 μg mL-1, (h ) VEL-IMP1 0.05 μg mL-1, (i ) VEL-IMP2 0.05 μg mL-1.

Chromatograms: (a) composite reference standards SOF 8.0 μg mL-1 and VEL 2 μg mL-1, SOF-IMP1, SOF-IMP2, SOF-DP1, SOF-DP2 0.1 μg mL-1 and VEL-DP1, VEL-IMP1 and VEL-IMP2 0.05 μg mL-1; (b ) composite impurities reference standards SOF-IMP2, SOF-DP1, SOF-DP2 0.1 μg mL-1 and VEL-DP1, VEL-IMP1 and VEL-IMP2 0.05 μg mL-1.
The selected chromatographic conditions were further validated for accuracy, precision and linearity. Specificity, limit of detection (LOD) and limit of quantification (LOQ) were determined as per USP and ICH guidelines.
System suitability was checked by replicate analysis (n = 5) of composite reference solution containing SOF 40 μg mL-1, VEL 10 μg mL-1, SOF impurity 0.04 μg mL-1 and VEL impurity 0.01 μg mL-1. Statistical data (Table 1) for theoretical plates (N), symmetry factor (As) and retention factor (Ko) were calculated from individual peak response of reference standards. The resolution and relative retention time of SOF, VEL and each impurity were calculated. The relative standard deviations (% RSD) of peak area for each analyte were calculated and the results were evaluated against acceptable limits ±2.0% for SOF and VEL and ±10.0% for each impurity. The values of retention factor and symmetry factor were also calculated from system suitability studies.
Method Validation
The results of recovery studies presented in Table 2 showed that overall recovery of SOF and VEL was 100 ± 1% and each impurity was 100 ± 20%. The value of RSD% for SOF and VEL was less than 2.0% and each impurity was less than 10%.
The results of precision and intermediate precision evaluation (Table 3) indicate that the given method is precise and reproducible within the acceptable limits and criteria. The value of RSD% was less than 2.0% for the peak response of SOF, VEL and it was less than 10% for each Impurity. Similarly in robustness studies, the minor change (±2%) in chromatographic condition i.e. mobile phase ratio and flow rate does not affect the results of SOF and VEL. The values of RSD% shown in Table 3 indicate that there is no influence of minor changes on results. Statistical data derived from linearity studies indicates a good correlation between concentration and peak response. The value of correlation coefficient (r) and intercept is shown in Table 3. The value of LOD for SOF and VEL was 0.015 and 0.021 μg mL-1 respectively. Similarly the value of LOD for each impurity was less than 0.015 μg mL-1. The value of LOQ for SOF was 0.046 and for VEL was 0.064 μg mL-1 and it was less than 0.04 μg mL-1 for each impurity. Results of placebo intemperance studies showed that there was no significant peak of any excipient at given retention time of any analyte. The influence of excipients on peaks of SOF, VEL and all impurities at given chromatographic condition is negligible. The validation results show that the proposed method is accurate, precise, sensitive and specific for quantification of process impurities and degradation products in SOF-VEL bulk and tablet dosage form.
Stability of Solutions
The results of stability studies of SOF and VEL drug substance and product in solid and solution are illustrated in Fig. 7 and summarized in Table 4. The results of normal studies showed that solutions of both drugs in dilution medium were stable for 7 days at room temperature (15 – 25°C) and for 14 days at cool temperature (2 – 8°C). Drug substance of SOF and VEL, and tablet dosage forms, stored at room temperature and at accelerated temperature 80 ± 5°C with relative humidity of 75 ± 5% were also stable. Similarly the drug substance stored at 105°C, protected from moisture was also stable for 8h and no degradation was noted.

Chromatograms of SOF-VEL degradation under (a) acidic conditions (0.1N HCL, 8 h), (b) alkaline conditions in basic solution (0.1M NaOH, 24 h), and (c) oxidative conditions (3% H2O2, 8 h).
The results of forced degradation showed that SOF is unstable in acidic and basic solution. In 0.1M HCl solution, SOF was degraded up to 6 ± 3% in 8 h. The degradation impurity SOF-DP1 was detected and quantified on HPLC analysis. Studies in 0.1N NaOH solutions showed that SBF degraded up to 85 ± 6% in 8 h. Interestingly same degradation product SOF-DP1 was detected during basic hydrolysis analysis. It was estimated that SOF degraded in both acidic and basic medium, to a hydrolysate of propyl ester (Fig. 2b ).
The results of VEL in acidic and alkaline solution showed that VEL is stable in both 0.1N HCl and 0.1M NaOH and no degradation was detected. The oxidative stress condition studies of SOF and VEL showed that both SOF and VEL are labile to oxidative condition. In 3% H2O2 solution SOF oxidizes to SOF-DP2 (Fig. 2b ) and VEL oxidizes to VEL-DP1 (Fig. 3b ).
The analysis of solutions in oxidative stress studies showed that SOF was degraded up to 3.5 ± 1% and VEL solution was degraded up to 18.4 ± 2% in 12 h. However the degradation rate of SOF and VEL was slow as compared to acid and base hydrolysis. The results of photo stability showed that SOF and VEL in solid and solution exposed to light, were stable and no degradation was noted in 8 h. The drug substance of VEL and SOF exposed to UV and fluorescence light for 7 days was also stable.
In overall stress studies, two degradation products (i.e. SOF-DP1 and SOF-DP2) were quantified for SOF and one degradation product (i.e. VEL-DP1) for VEL. All possible degradation impurities were resolved using the HPLC method and it was proved that the method is stability indicating and capable to isolate and quantify the possible degradation products.
Characterization of Degradation Products
The degradation products were further analyzed and confirmed by NMR, MS, and IR spectroscopy data. The IR spectrum ranging from 600 to 4000 cm-1 was obtained on Nicolet iS5 N FT-NIR spectrometer. The NMR analysis was performed on Bruker AVANCE-III HD 600 NMR using DMSO-d6 as solvent. Mass spectrum was obtained using both ESI and Auto MS mode using Agilent 6320 Iron Trap Mass spectrometer. The drying gas flow rate was 10 L min-1 at 350 ± 10°C, the nebulizer gas pressure was 45 psi and the capillary voltage was 3.5 kV.
During mass analysis of SOF-DP1 the parent ion peak at m/z of 488 correspond to the theoretical value of molecular mass (487.37 g mole-1) of SOF-DP1. Similarly the parent ion at m/z 488 produced 8 significant ions at m/z 399, 366, 366, 399, 287, 243, 200 and 167 corresponding to the fragmentation pattern of SOF-DP1. Similarly the parent ion peak at m/z 392.1 for SOF-DP2 and significant ions at m/z 302, 257, 180, 150 and 112 support the molecular mass (302.25 g mole-1) and fragmentation pattern of SOF-DP2. The VEL-DP1 spectrum full scan also exhibits a parent ion peak at m/z 735.9 that support the molecular mass of 735.8 g mole-1. The significant ions at m/z 622, 500, 443, 404, 296 and 142 also corresponded to the fragmentation pattern of VEL-DP1. The IR spectrum of all degradation products also showed characteristic peaks corresponding to the functional groups and aromatic ring respectively. Similarly, the data of NMR for number of protons and carbons in 1H NMR, C13 NMR also completely supported the structures of SOF-DP1, SBP-DP2 and VEL-DP1 as shown in Fig. 2b and Fig 3b.
Conclusion
It was concluded that the developed method is accurate, precise and stability indicating. Thus, the proposed method can be applied to the determination and analysis of process impurities and degradation products in SOF-VEL pharmaceutical formulations.
References
A. Aghemo and R. De Francesco, J. Hepatol., 58(1), 428 – 438 (2013).
E. Cholongitas and G. V. Papatheodoridis, Ann. Gastroenterol., 27(4), 331 – 337 (2014).
E. Tam, A. F. Luetkemeyer, P. S. Mantry, et al., Liver Int., 38(6), 1010 – 1021 (2018).
M. A. Smith, R. E. Regal, and R. A. Mohammad, Ann. Pharmacother., 50(1), 39 – 46 (2016).
S. Naggie, K. Patel and J. McHutchison, J. Antimicrob. Chemother., 65(10), 2063 – 2069 (2010).
L. Wei, S. G. Lim, Q. Xie, et al., Lancet Gastroenterol., 4(2), 127 – 134 (2019).
US Food and Drug Administration: Approval of Sovaldi (Sofosbuvir) Tablets for the Treatment of Chronic Hepatitis C (2014). Available from: https: // www.drugs.com / history/sovaldi.html
US Food and Drug Administration (2017). Available from: https: // www.fda.gov / news-events / press-announcements/fda-approves-epclusa-treatment-chronic-hepatitis-c-virus-infection
V. Pottabathini, V. Gugulothu, M. Kaliyaperumal, et al., Am. J. Chem., 7(11), 797 – 815 (2016).
M. Nebsen and E. S. Elzanfaly, J. Chromatogr. Sci., 54(9), 1631 – 1640 (2016).
D. Swain, G. Samanthula, S. Bhagat, et al., J. Pharm. Biomed. Anal., 120, 352 – 363 (2016).
International Conference on Harmonization: Impurities in New Drug Products, ICH, IFDMA, Geneva (2006).
S. B. Bari, B. R. Kadam, Y. S. Jaiswal, et al., Eurasian J. Anal. Chem., 2(1), 32 – 53 (2007).
E. F. Elkady and A. A. Aboelwafa, J. Chromatogr. B, 1102, 116 – 124 (2018).
O. M. Abdallah, A. M. Abdel-Megied and A. S. Gouda, Biomed. Chromatogr., 32(6), e4186 (2018).
K. Konam and S. Reddy, J. Young Pharm., 11(3) (2019).
M. van Seyen, M. J. de Graaff Teulen, N. P. van Erp, et al., J. Chromatogr. B, 1110, 15 – 24 (2019).
M. H. Semreen, H. Y. Alniss, M. K. Mousa, et al., Molecules, 24(7), 1302 (2019).
M. R. Rezk, E. B. Basalious, and K. A. Badr, Biomed. Chromatogr., 32(11), e4347 (2018).
M. R. Rezk, H. H. Monir, and H. M. Marzouk, Microchem. J., 146, 828 – 834 (2019).
R. M. El-Gamal, S. A. Abdel-Gawad, F. F. Belal, et al., RSC Adv., 8(57), 32909 – 32915 (2018).
M. E. Moustapha, R. M. El-Gamal, and F. F. Belal, BMC Chem., 13(1), 118 (2019).
A. H. Kamal, M. M. Mabrouk, L. I. Bebawy, and M. A. Mekky, Microchem. J., 103996 (2019).
M. R. Rezk, H. H. Monir and H. M. Marzouk, Spectrochim. Acta A, 213, 159 – 166 (2019).
A. G. Susmita and G. Rajitha, Int. J. Pharm. Sci. Res., 9(11), 4764 – 4769 (2018).
B. Balaswami, P. V. Ramana, B. S. Rao, and P. Sanjeeva, Res. J. Pharm. Technol., 11(9), 4147 – 4156 (2018).
S. Hemchand, R. R. C. Babu, and M. M. Annapurna, Res. J. Pharm. Technol., 11(12), 5637 – 5642 (2018).
D. Chinababu, Int. J. Pharm. Sci. Res., 1 – 10 (2019).
K. Swathi, P. V. Rao and N. S. Rao, Int. J. Pharm. Anal., 1(3), 59 – 67 (2019).
The United States Pharmacopeia, 37th Ed., United States Pharmacopeial Convention, Rockville Inc. (2008).
International Conference on Harmonization: Validation of Analytical Procedures. Text and Methodology, ICH, IFDMA, Geneva (1996).
A. Rignall, ICH Quality Guidelines: An Implementation Guide, 3 (2017).
International Conference on Harmonization: Photostability Testing of New Drugs and Products, ICH, IFDMA, Geneva (1996).
Acknowledgments
The authors are thankful to Genome Pharmaceuticals (Pakistan). for providing generic products, reference standards, and for support in this work.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zaman, B., Hassan, W. Development of Stability Indicating HPLC-UV Method for Determination of Process Impurities and Degradation Products in Sofosbuvir and Velpatasvir Tablets. Pharm Chem J 54, 1295–1305 (2021). https://doi.org/10.1007/s11094-021-02359-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-021-02359-3