
Benzylsulfamic acid has been prepared and introduced as a new heterogeneous acid catalyst. This reagent was used for the synthesis of new bis(indolyl)methanes 3a – 3f via reaction of bis-aldehydes 1 with indoles 2 at 100°C. All reactions were performed under solvent-free conditions with high to excellent yields. The synthesized bis(indolyl)methanes were evaluated for their antibacterial and antifungal activities.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1. INTRODUCTION
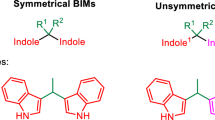
The study of bis(indolyl)methanes (BIMs) are one of the most active research areas for medicinal chemists because of the wide occurrence of BIMs in various natural products possessing biological activity [1,2,3]. This biological activity includes anti-inflammatory [4], anti-oxidant [5], antimicrobial [6, 7], anticancer [8], antibacterial properties [9], nontoxicity, DNA-damaging ability [10], and anti-tumorigenic action [11] reported for the indoles, BIMs and their derivatives. They are also effective in promoting estrogen metabolism and inducing apoptosis of cancer cells in humans [12]. In addition, a group of indole derivatives used as triplet energy materials [13], dyes [14], and colorimetric sensors [15, 16]. Because BIMs are important heterocycles in pharmaceutical chemistry, it was of interest to study the synthesis and properties of this group of compounds.
Ageneral approach to the synthesis of bis(indol-3-yl)methanes is based on the condensation of indoles with aldehydes or ketones in the presence of various catalysts. These catalysts can include ionic liquids [17,18,19,20,21], heteropolyacids [22], acid catalysts such as PEG-SO3H [23], Ph-PMO-SO3H [24], H6P2W18O62 [25], Fe(DS)3 [26], ZrOCl2 · 8H2O/SiO2 [27], trichloro-1,3,5-triazine [28], silica sulfuric acid (SSA) [29], sulfated zirconia [30], metal hydrogen sulfates [31]. It is also possible to use alternative energy sources including microwaves and ultrasound [32,33,34]. However, most of the reported methods involve toxic metal ions, corrosive and expensive reagents, and are characterized by low yield of products, long reaction times, tedious methods of separation of products, and use of environmentally harmful organic solvents. Performing organic reactions under solvent-free conditions has attracted much attention because of being safe, non-toxic, environmentally friendly, and cheap compared to organic solvents [35]. One of the logical solutions for the aforementioned problems is to use water-soluble catalyst under solvent-free conditions. When a water soluble catalyst is used, the insoluble crude products can be separated by simple filtration and the catalyst system can be recycled.
2. RESULTS AND DISCUSSION
2.1. Chemistry
The development of drug resistance by some germs and the basic need in new antimicrobial compounds on one hand, and various activities of BIMs such as anticancer, antibacterial and anti-fungal on the other hand encouraged us to study of the antimicrobial activity of some new BIM derivatives. In continuation of our research program on the development of new methods for the synthesis of heterocyclic derivatives, here, we describe the preparation of benzylsulfamic acid (Scheme 1) and its use as an inexpensive catalyst for the synthesis of new BIMs via the condensation reaction of one equivalent bis-aldehyde with four equivalent indoles under solvent-free conditions at 100oC (Scheme 2). The antimicrobial bacterial activity of these compounds was investigated against Gram-positive and Gram-negative species.

Preparation of benzylsulfamic acid.

Preparation of new bis(indolyl)methane derivatives.
Catalyst. To initiate the study, benzylsulfamic acid was prepared by straightforward addition of one equivalent of chlorosulfonic acid to benzylamine at 0oC. The structure of benzylsulfamic acid was identified by FT-IR, 1H and 13C NMR, TGA, and mass spectroscopy techniques.
The presence of an additional sulfonic acid group on benzylamine nitrogen in the catalyst increases the number of vibrational modes and causes completely different FT-IR spectrum. In the FT-IR spectrum, the broad band between 2422 and 3358 cm-1 that centered at 2959 cm-1 can be attributed to -OH stretching of the -SO3H group. Other characteristic absorption bands at 1267, 1156 and 887 cm-1 were assigned to the asymmetric and symmetric SO2 stretching, S-OH bending, and symmetric S-N stretching vibrations, respectively [37, 38]. Alternatively, in the IR spectra of catalyst, the bands 3412 at 3334 cm-1 disappeared and a band at 3396 cm-1 (related to the –NH stretching of–NHSO3H group) emerged. The 1H and 13C NMR spectra were taken in DMSO-d6 solvent. Important peak in 1H NMR spectrum of catalyst was attributed to acidic hydrogen (SO3H) and observed at δ = 5.64 ppm. The acidic hydrogen of ClSO3H at 13.45 ppm in the 1H NMR spectra of benzylamine in CDCl3 was reported in literature [39]. It is noteworthy that benzylamine and ClSO3H are readily soluble in CH2Cl2 while benzylsulfamic acid is insoluble in CH2Cl2 and readily soluble in water. Important peaks in the mass spectrum of the catalyst were observes at 188 (M++1), 187 (M+), 186 (M+-H), 107 (M+-SO3H), 92 (toluene) and 78 (benzene). The TGA curves of the catalyst were measured under nitrogen. The TGA curve indicated a weight loss below 100°C which corresponds to the loss of physically adsorbed water. Benzylsulfamic acid is degraded mainly via a two-stage process. The first weight loss observed in the vicinity of 160 – 170°C (Tmax = 163°C) is attributed to the loss of sulfonic acid. The second weight loss corresponding to the complete destruction of catalyst is observed in the region of 350 – 360°C. The TGA information affirms that the catalyst is stable at 160°C.
Synthesis of new bis(indolyl)methane derivatives. For this purpose, a model reaction of the condensation of 4,4′-(butane-1,4-diylbis(oxy))dibenzaldehyde (1 mmol) and indole (1 mmol) was carried out under solvent-free conditions in the presence of various quantities of benzylsulfamic acid at various temperatures. The results are summarized in Table 1. Excellent results were obtained in the presence of benzylsulfamic acid (Table 1, Entry 4), while in the absence of catalyst a minor product was detected (Table 1, Entry 1). Raising the amount of catalyst increased the yield of compound 3a. The optimum amount of the catalyst was 15 mol%, while a higher amount of the catalyst beyond this value did not increase the yield of 3a noticeably. Based on these results, this procedure was then extended to obtaining other bis-aldehydes and indoles in the optimized system. Thus, new BIM derivatives 3a – 3f were prepared via the one-pot reaction of one equivalents bis-aldehydes with four equivalents indoles under solvent-free conditions at 100oC. The general route for the synthesis of BIM derivatives is given in Scheme 2. The review of literature showed that BIM derivatives 3a – 3f have not been reported previously. Thestructures of new products were confirmed by FT-IR, 1H and 13C NMR, and elemental analyses.
Reusability of the catalyst was checked via the same model reaction under optimized conditions. After completion of the reaction, the catalyst was filtered after washing the reaction mixture with hot and dry ethanol, and reused in the same reaction process. As can be seen from Fig. 1, the catalyst could be reused at least four times with only small reduction in the catalytic activity.

Reusability of catalyst for the model reaction.
2.2. Antibacterial Activity
All synthesized compounds 3a – 3f were screened for their antibacterial activity against Pseudomonas aeruginosa (MTCC 1688), Escherichia coli (MTCC 443) and Pseudomonas oleovorans (MTCC 617) as Gram-negative bacteria, Staphylococcus aureus (MTCC 96) and Bacillus subtilis (MTCC 441) as Gram-positive bacteria. The inhibitory zones (in mm) were determined using the agar diffusion method (cup plate method). Streptomycin and DMSO were used as positive control and negative control against bacteria, respectively. All measurements were performed in duplicate and the results are reported as mean of at least three measurements.
As shown in Table 2, there is a significant difference between the antibacterial strength of synthesized compounds (3a – 3f) and streptomycin antibiotics (P = 0.001). The obtained data show that the antimicrobial activity of streptomycin is higher than that of other substances. However, among synthesized compounds, compounds 3f and 3a have the highest and lowest antibacterial activity, respectively. All compounds exhibited good antibacterial activity. The significant inhibition exhibited by compounds 3c and 3f might be due to the presence of nitrile group in the fifth position of the corresponding heteroaryl rings. The results presented in Table 2 show that, among these BIMs the activity is ordered as 3f > 3c > 3b > 3e. The replacement of various groups and changes in the alkyl chain of BMIs 3a – 3f also enhanced the inhibitory activity.
2.3. Antifungal Activity
As shown in Table 3, there is a significant difference between the antifungal strength of the synthesized compounds (3a – 3f) with amphotericin B antibiotic (P = 0.001). The obtained data show that, although the antifungal strength of amphotericin B is higher than that of other samples, compound 3c had the highest and compounds 3a and 3d had the lowest antifungal activity. The 5-nitrile heteroaryl derivatives 3c and 3f exhibited better inhibitory activity against all the three fungal strains than the other derivatives, whereas compound 3d was less active only against C. albicans and 3a was active only against R. oryzae. Compound 3f was found to be the most active against R. oryzae and A. niger, while 3c was the most active against C. albicans.
3. EXPERIMENTAL PART
3.1. General
Melting points were measured on Electrothermal 9100 apparatus. Chemicals were obtained from Merck and Fluka and used as purchased. The 1H NMR and 13C NMR spectra were obtained on Bruker DRX 500 Avance spectrometer using DMSO-d6 as solvent and TMS as internal standard. FT-IR spectra were recorded on Shimadzu FT-IR-8400S spectrophotometer. All bis-aldehydes (4) were prepared according to a reported method [36]. The mass spectra were obtained at 70 eV on Varian Mat CH-7 (Varian Medical Systems, Palo Alto, CA, USA). TGA analyses were performed on TGA-PL (Polymer Laboratories) thermal analysis system at a heating rate of 10 K/min. The scanning electron microscopy (SEM) microphotographs were obtained on LEO 1430VP (Germany) instrument.
3.2. Synthesis
General procedure for the preparation of benzylsulfamic acid catalyst. A 25-mL suction flask charged with benzylamine (1.092 mL, 10 mmol) solution in dry CH2Cl2 (5 mL) was equipped with a constant-pressure dropping funnel containing 1.165 g (10 mmol) chlorosulfonic acid and a gas inlet tube for HCl gas flow over an adsorbing aqueous solution. Chlorosulfonic acid was added dropwise over a period of 20 min while the reaction mixture was stirred slowly on an ice-cold water bath. Then, the mixture was heated to room temperature and stirred for additional 2 h. Finally, the mixture was filtered and the solid residue was washed with dry ether (3 × 5 mL) and dried under vacuum. The catalyst was obtained as a withe-colored solid in 1.794 g (98%) yields.
General procedure for the synthesis of bis(4-(bis-(4-substituent-1H-indol-3-yl)-methyl)-phenoxy)alkanes (3a – 3f). Amixture of bis-aldehyde (1 mmol), indole derivative (4 mmol), and benzylsulfamic acid catalyst (15 mol%) was stirred at 100°C for 40 – 45 min. The progress of reaction was monitored by TLC (n-hexane-EtOAc (3:1)). After completion of the reaction, the product was dissolved in hot dry ethanol (2 × 10 mL) and insoluble catalyst was removed by filtration with a filter paper. The organic phase including the product and EtOH was evaporated under vacuum. The resulting crude material was purified by recrystallization from EtOH to afford pure products. All synthesized compounds were characterized by their physical constants, FT-IR, 1H and 13C NMR spectroscopy, and elemental analysis.
3.3. Characteristics of Synthesized Compounds
Benzylsulfamic acid catalyst: white-colored solid; mp, 250 – 251°C; FT-IR (KBr, ν, cm(1): 3396 (N-H), 2422 – 3358 (O-Hacidic), 1592, 1559 (C=C), 1267, 1156 (SO2); 1H NMR (400 MHz, DMSO; δ, ppm): 4.02 (d, 2H, J = 5.6 Hz), 5.64 (s, 1H), 7.35 – 4.43 (m, 3H), 7.45 – 7.48 (m, 2H), 8.23 (s, 1H); 13C NMR (100 MHz, DMSO; δ, ppm): 42.8, 134.3, 128.9, 129.0, 129.3, 130.1; MS (70 eV; m/z) 188 (M+ + 1), 187 (M+), 186 (M+-H), 107 (M+-SO3H), 92 (toluene), 78 (benzene).
1,4-Bis(4-(di(1H-indol-3-yl)methyl)phenoxy)butane (3a): yield, 89%; mp, 218 – 220°C; FT-IR (KBr, ν, cm(1): 3409 (N-H), 3041 (C-Harom), 2937, 2867 (C-Haliph), 1604, 1459 (C=C), 1235 (C-O); 1H NMR (400 MHz, DMSO; δ, ppm): 1.83 (br, 4H), 3.97 (br, 4H), 5.77 (s, 2H), 6.80 – 6.88 (m, 16H), 7.03 (t, 4H, J = 7.2 Hz), 7.24 (d, 4H, J = 8.4 Hz), 7.35 (d, 4H, J = 8.4 Hz,), 10.80 (s, 4H); 13C NMR (100 MHz, DMSO; δ, ppm): 26.0, 39.3, 67.4, 111.9, 114.3, 118.6, 118.9, 119.6, 121.3, 123.9, 127.1, 129.6, 137.0, 137.3, 157.1; Calcd. for C50H42N4O2 (%): C 82.16; H 5.79; N 7.67; Found (%): C 85.19; H 5.77; N 7.64.
1,4-Bis(4-(bis(5-bromo-1H-indol-3-yl)methyl)phenoxy)butane (3b): yield, 88%; mp, 208 – 210°C; FT-IR (KBr, ν, cm(1): 3422 (N-H), 3047 (C-Harom), 2924, 2870 (C-Haliph), 1601, 1453 (C=C), 1238 (C-O); 1H NMR (400 MHz, DMSO; δ, ppm): 1.84 (s, 4H), 3.96 (br, 4H), 5.79 (s, 2H), 6.82 – 6.93 (m, 8H), 7.15 (d, 4H, J = 8.8 Hz), 7.25 (d, 4H, J = 8.8 Hz), 7.34 (d, 4H, J = 8.4 Hz), 7.42 (s, 4H), 11.06 (s, 4H); 13C NMR (100 MHz, DMSO; δ, ppm): 26.0, 38.5, 67.4, 111.3, 114.0, 114.5, 118.4, 121.7, 123.8, 125.6, 128.8, 129.5, 135.5, 136.6, 157.3; Calcd. for C50H38Br4N4O2 (%): C 57.39; H 3.66; N 5.35; Found (%): C 57.35; H 3.69; N 5.38.
3,3′,3″,3‴-(((Butane-1,4-diylbis(oxy))bis(4,1-phenylene)) bis(methanetriyl))tetrakis(1H-indole-5-carbonitrile) (3c): yield, 90%; mp, 214 – 216°C; FT-IR (KBr, ν, cm(1): 3332 (N-H), 3042 (C-Harom), 2923, 2870 (C-Haliph), 2217 (C≡N), 1506, 1465 (C=C), 1238 (C-O); 1H NMR (400 MHz, DMSO; δ, ppm): 1.87 (s, 4H), 3.98 (br, 4H), 5.97 (s, 2H), 6.87 (m, 4H), 7.11 (s, 4H), 7.26 (d, 4H, J = 8.4 Hz), 7.43 (d, 4H, J = 8.8 Hz), 7.54 (d, 4H, J = 8.4 Hz), 7.80 (s, 4H), 11.49 (s, 4H); 13C NMR (100 MHz, DMSO; δ, ppm): 22.9, 38.1, 67.4, 100.7, 113.3, 114.6, 119.7, 121.3, 124.2, 125.0, 126.6, 126.7, 129.5, 136.2, 138.7, 157.5; Calcd. for C54H38N8O2 (%): C 78.05; H 4.61; N 13.49; Found (%): C 78.08; H 4.58; N 13.51.
1,5-bis(4-(di(1H-indol-3-yl)methyl)phenoxy)pentane (3d): yield, 88%; mp, 222 – 224°C; FT-IR (KBr, ν, cm(1): 3409 (N-H), 3050 (C-Harom), 2933, 2865 (C-Haliph), 1503, 1458 (C=C), 1235 (C-O); 1H NMR (400 MHz, DMSO; δ, ppm): 1.54 (m, 2H), 1.74 (m, 4H), 3.92 (t, 4H, J = 8.4 Hz), 5.75 (s, 2H), 6.79 – 6.80 (m, 12H), 7.02 (t, 4H, J = 8.4 Hz), 7.22 (t, 4H, J = 7.6 Hz), 7.26 (d, 4H, J = 6.4 Hz), 7.33 (d, 4H, J = 6.4 Hz), 10.77 (s, 4H); 13C NMR (100 MHz, DMSO; δ, ppm): 22.8, 29.0, 39.3, 67.6, 111.8, 111.8, 114.3, 118.5, 118.8, 119.6, 121.2, 123.7, 127.0, 129.6, 137.0, 157.2; Calcd. for C51H44N4O2 (%): C 82.23; H 5.95; N 7.52; Found (%): C 82.26; H 5.91; N 7.55.
1,5-Bis(4-(bis(5-bromo-1H-indol-3-yl)methyl)phenoxy)pentane (3e): yield, 90%; mp, 193 – 195 °C; FT-IR (KBr, ν, cm(1): 3419 (N-H), 3045 (C-Harom), 2930, 2863 (C-Haliph), 1505, 1456 (C=C), 1239 (C-O); 1H NMR (400 MHz, DMSO; δ, ppm): 1.58 (m, 2H), 1.76 (br, 4H), 3.94 (t, 4H, J = 6.4 Hz), 5.79 (s, 2H), 6.84 – 6.88 (m, 8H), 7.15 (d, 4H, J = 8.8 Hz), 7.21 (d, 4H, J = 8.4 Hz), 7.34 (d, 4H, J = 8.4 Hz), 7.42 (s, 4H), 11.06 (s, 4H); 13C NMR (100 MHz, DMSO; δ, ppm): 22.8, 29.0, 38.4, 67.6, 111.3, 114.0, 114.5, 118.4, 121.6, 123.8, 125.4, 125.6, 128.8, 129.5, 135.7, 157.3; Calcd. for C51H40Br4N4O2 (%): C 57.76; H 3.80; N 5.28; Found (%): C 57.79; H 3.76; N 5.31.
3,3′,3″,3‴-(((Pentane-1,5-diylbis(oxy))bis(4,1-phenylene)) bis(methanetriyl))tetrakis(1H-indole-5-carbonitrile) (3): yield: 92%; mp, 223 – 225°C; FT-IR (KBr, ν, cm(1): 3422 (N-H), 3047 (C-Harom), 2936, 2867 (C-Haliph), 2218 (C≡N), 1506, 1437 (C=C), 1237 (C-O); 1H NMR (400 MHz, DMSO; δ, ppm): 1.55 (m, 2H), 1.75 (br, 4H), 3.94 (t, 4H, J = 6.4 Hz), 5.96 (s, 2H), 6.86 (d, 4H, J = 8.4 Hz), 7.10 (s, 4H), 7.26 (d, 4H, J = 8.0 Hz), 7.41 (d, 4H, J = 7.6 Hz), 7.54 (d, 4H, J = 8.4 Hz), 7.80 (s, 4H), 11.49 (s, 4H); 13C NMR (100 MHz, DMSO; δ, ppm): 22.8, 29.0, 38.1, 67.7, 100.7, 113.3, 114.6, 119.7, 121.3, 124.2, 125.0, 126.6, 126.7, 129.5, 136.2, 138.7, 157.5; Calcd. for C55H40N8O2, (%): C 78.18; H4.77; N 13.26 Found (%): C 78.15; H 4.79; N 13.23.
3.4. Antibacterial Assay
The antibacterial activity of the new bis(indolyl)methanes was determined by the well diffusion method according to Lindsay [29]. Four identical colonies from each agar plate were taken with a sterile wire loop and transferred into a tube containing 5 mL of nutrient agar. The turbidity of each suspension was corrected to attain an optical comparison to that of 0.5 turbidity McFarland standard, resulting in a suspension containing approximately 1 – 2 × 108 CFU/ml. Nutrient agar plates were inoculated using streaking the swab over the entire sterile agar surface. This method was repeated via streaking two more times, rotating the plate nearly 60° each time to ensure uniform diffusion of the inoculum. As the last step, the edge of the agar was swabbed. After allowing the inoculum to dry at 25°C, 6 mm diameter wells were ready in the agar by using a sterilized cork borer. The samples (100 μg/mL) were prepared by dissolving in dimethyl sulfoxide (DMSO) and introduced into duplicate wells. The plates were incubated at 37°C for 24 h. Then, the plates were studied for bacterial growth inhibition and the inhibition zone diameter measured to within nearest millimeter. DMSO and streptomycin were used as a negative control and positive control, respectively.
Antifungal Assay
The procedure followed for antifungal bioassay was analogous to that followed for antibacterial assay, where the medium was PDA 39 g/L. All compounds were studied for their antifungal activity at a of concentration 100 μg/mL by using DMSO as a solvent. DMSO did not exhibit any activity at the concentration used. The control and treated samples were kept in an incubator at 28 ± 2oC for 48 h and the inhibition zones were measured to within nearest millimeter. Three replicates were maintained for each treatment, and amphotericin B (50 μg/mL) was used as a positive control.
References
M. C. Bell, P. Crowley-Nowick, H. L. Bradlow, et al., Gynecol. Oncol., 78, 123 – 129 (2000).
J. J. Michnovicz and H. L. Bradlow, Nutr. Cancer,16, 59 – 66 (1991).
G. R. Humphrey and J. T. Kuethe, Chem. Rev.,106, 2875 – 2911 (2006).
K. Sujatha, P. T. Perumal, D. Muralidharan and M. Rajendran, Indian J. Chem.,48, 267 – 269 (2009).
S. H. Benabadji, R. Wen, J. Zheng, et al., Acta. Pharmacol. Sinica.,25, 666 – 671 (2004).
G. Sivaprasad, P. T. Perumal, V. R. Prabavathy and N. Mathivanan, Bioorg. Med. Chem. Lett.,16, 6302 – 6305 (2006).
A. Kamal, M. N. A. Khan, K. S. Reddy, et al., J. Enzyme. Inhib. Med. Chem.,24, 559 – 565 (2009).
K. R. M. Naidu, S. I. Khalivulla, S. Rasheed, et al., Int. J. Mol. Sci.,14, 1843 – 1853 (2013).
C. Hong, G. L. Firestone and L. F. Bjelanes, Biochem, Pharmacol., 63, 1085 – 1097 (2002).
T. Osawa and M. Namiki, Tetrahedron Lett.,24, 4719 – 4722 (1983).
W. Kassouf, S. Chintharlapalli, M. Abdelrahim, et al, Cancer Res.,66, 412 – 418 (2006).
J. S. Glasby, Encyclopedia of Aalkaloids, New York: Plenum Press (1975).
M. Kirkus, M. H. Tsai, J. V. Grazulevicius, et al., Synth. Met.,159, 729 – 734 (2009).
X. He, S. Hu, K. Liu, et al., Org. Lett.,8, 333 – 336 (2006).
R. Martinez, A. Espinosa, A. Tarraga and P. Molina, Tetrahedron,64, 2184 – 21891 (2008).
B. S. Liao, J. T. Chen and S. T. Liu, Synthesis,20, 3125 – 3128 (2007).
X. F. Zeng, S. J. Ji and S. Y. Wang, Tetrahedron,61, 10235–10241 (2005).
J. S. Yadav, B. V. S. Reddy, V. Sunitha and K. S. Reddy, Adv. Synth. Catal.,345, 1203 – 1206 (2003).
A. K. Chakraborti, S. R. Roy, D. Kumar and P. Chopra, Green Chem.,10, 1111 – 1118 (2008).
K. Rad-Moghadam and M. Sharifi-Kiasaraie, Tetrahedron,69, 8816 – 8820 (2009).
P. J. Das and J. Das, Tetrahedron Lett.,53, 4718 – 4720 (2012).
H. Firouzabadi, N. Iranpoor and A. A. Jafari, J. Mol. Catal.,244, 168–172 (2006).
V. Jhansi Rani, K. Veena Vani and C. Venkata Rao, Synth. Commun.,42, 2048–2057 (2012).
A. Karam, J. C. Alonso, T. I. Gerganova, et al., Chem. Commun., 45, 7000 – 7002 (2009).
M. M. Heravi, K. Bakhtiari, A. Fatehi and F. F. Bamoharram, Catal. Commun., 9, 289 – 292 (2008).
H. Veisi, B. Maleki, F. H. Eshbala, et al., RSC Adv.,4, 30683 – 30688 (2014).
H. Firouzabadi, N. Iranpoor, M. Jafarpour and A. Ghaderi, J. Mol. Catal. A: Chem.,253, 249 – 251 (2006).
G. V. M. Sharma, J. J. Reddy, P. S. Lakshim and P. R. Krishna, Tetrahedron Lett.,45, 7729 – 7732 (2004).
M. A. Zolfigol, P. Salehi, M. Shiri, et al., Mol. Divers., 2, 203 – 207 (2008).
B. M. Reddy, P. M. Sreekanth and P. Lakshmanan, J. Mol. Catal. A. Chem.,237, 93 – 100 (2005).
K. Niknam, M. A. Zolfigol, T. Sadabadi and A. Nejati, J. Iran. Chem. Soc.,3, 318–322 (2006).
M. Zahran, Y. Abdin, and H. Salama, ARKIVOC, 11, 256–265 (2008).
B. Das, R. Pal, J. Banerjee, et al., Indian J. Chem. B., 44, 327–730 (2005).
S. S. Sonar, S. A. Sadaphal, A. H. Kategaonkar, et al., Bull. Korean Chem. Soc.,30, 825–828 (2009).
F. Shirini, M. Safarpoor Langroodi and M. Abedini, Chin. Chem. Lett.,21, 1342 – 1345 (2010).
N. O. Mahmoodi, M. Mohammadi Zeydi, and E. Biazar, J. Sulfur. Chem.,37, 613 – 621 (2016).
S. Handy and N. M. Westbrook, Tetrahedron Lett.,55, 4969 – 4971 (2014).
H. Veisi, R. Gholbedaghi, J. Malakootikhah, et al., J. Heterocyclic. Chem., 47, 1398 – 1305 (2010).
M. H. Sarvari, Acta. Chim. Slov.,54, 354 – 359 (2007).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Soltani, S., Montazeri, N., Zeydi, M.M. et al. Synthesis of New Bis(Indolyl)Methanes Catalyzed by Benzylsulfamic Acid and Evaluation of Their Antimicrobial Activities. Pharm Chem J 53, 947–952 (2020). https://doi.org/10.1007/s11094-020-02103-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-020-02103-3