
The purpose of this study was to investigate the effects of cilostazol on the bioavailability and pharmacokinetics of nifedipine and its main metabolite, dehydronifedipine, in rats. The pharmacokinetic parameters of nifedipine and dehydronifedipine were determined following oral and intravenous administration of nifedipine (1.5 and 6.0 mg ・ kg-1) in rats. Cilostazol inhibited CYP3A4 enzyme activity at a 50% inhibitory concentration (IC50) of 4.1 μM. The areas under the plasma concentration–time curve (AUC 0-∞) and the peak concentration (C max) of nifedipine were significantly increased, respectively, in the presence of cilostazol compared to that in the control. The total body clearance (CL/F) was significantly decreased by cilostazol. Consequently, the absolute bioavailability (AB) of nifedipine with cilostazol was significantly higher than that in the control. The metabolite to parent AUC ratio (MR) in the presence of cilostazol was significantly decreased compared to that in the control. The AUC 0-∞ of intravenous nifedipine was significantly increased with cilostazol compared to that in the control. The increased bioavailability of nifedipine in rats can be mainly due to the inhibition of CYP3A4-mediated metabolism in the small intestine and/or liver by cilostazol. In addition, the reduction of CL/F of nifedipine by cilostazol may also be a factor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nifedipine is predominantly metabolized by cytochrome p450 (CYP) 3A4 to its primary pyridine metabolite, dehydronifedipine [1, 2]. CYP enzymes are responsible for the oxidative metabolism of many xenobiotics and play a major role in the phase I metabolism of many drugs [3]. CYP3A4 is the most abundant CYP enzyme (30 – 40%) in the adult liver and it metabolizes more than 50% of the clinically administered drugs including nifedipine, cyclosporine, and midazolam [4, 5]. There are some reports that nifedipine is a substrate of CYP3A4 in humans [6,7,8].
Nifedipine is a calcium channel blocking agent that is widely used for the treatment of essential hypertension, coronary artery spasm, and angina pectoris [9]. Nifedipine inhibits the influx of extracellular calcium through myocardial and vascular membrane pores by physically plugging the channel, leading to decreased intracellular calcium levels, inhibition of the contractile processes of smooth muscle cells as well as dilation of the coronary and systemic arteries [10, 11]. In addition, it also causes increased oxygen delivery to the myocardial tissue as well as decreased total peripheral resistance, systemic blood pressure, and afterload [10, 11].
Cilostazol is an antiplatelet vasodilator agent that has been used for more than a decade in Japan for the treatment of chronic peripheral arterial occlusive disease [12]. Cilostazol was also approved by the US Food and Drug Administration (FDA) in 1998 for the treatment of symptoms related to intermittent claudication [13]. Cilostazol exerts its pharmacologic effects through selective inhibition of phosphodiesterase-3, which is abundant in platelets and vascular smooth muscle cells. These inhibitory effects mediate the antiplatelet properties of cilostazol and suppress vascular smooth muscle cell proliferation [14]. Cilostazol is extensively metabolized, primarily by the CYP3A4 enzymes, into the active metabolites 3,4-dehydrocilostazol and 4′-transhydroxycilostazol [15, 16].
It is known that the substrates and/or inhibitors of CYP3A4 and P-glycoprotein (P-gp) overlap with each other [17]. P-gp appears to play a key role in the absorption, distribution, and elimination of many drugs [18, 19]. It was previously reported [20] that nifedipine belongs to the group of P-gp substrates. Since P-gp is co-localized with CYP3A4 in the small intestine when co-administered, they may act synergistically to promote presystemic drug metabolism, resulting in the limited absorption of drugs. However, the effect of cilostazol on CYP3A4 and P-gp activity has not been reported. Therefore, we attempted to evaluate P-gp activity using the rhodamine-123 retention assay in P-gp-overexpressed adriamycin-resistant human breast cancer cell line (MCF-7/ADR). Furthermore, we evaluated the effect of cilostazol on CYP3A4 activity.
Drug–drug interaction between cilostazol and nifedipine is possible, since they could be concomitantly prescribed for the prevention or treatment of cardiovascular diseases as a combination therapy. However, the effect of cilostazol on the pharmacokinetics of nifedipine in vivo has not been reported. Therefore, the purpose of this study was to investigate the effects of cilostazol on the CYP3A4 and P-gp activity. In addition, we investigated the effects of cilostazol on the bioavailability and pharmacokinetics of orally and intravenously administered nifedipine and on its active metabolite, dehydronifedipine, in rats.
Experimental
Materials
Nifedipine, dehydronifedipine, cilostazol and amlodipine [internal standard for the high-performance liquid chromatography (HPLC) analysis of nifedipine] were purchased from the Sigma-Aldrich Co. (St. Louis, MO, United States). Methanol, isooctane, methyl-tert-butyl ether (MTBE), analytical grade acetic acid, and triethylamine (TEA) were obtained from Merck Co. (Darmstadt, Germany). Rhodamine was obtained from Calbiochem (USA), and the CYP inhibition assay kit was obtained from Gentest Corp. (Woburn, MA, USA). Other chemicals used were of reagent or HPLC grade. The apparatus used in this study included an HPLC system comprising a Waters 1515 isocratic HPLC pump, a Waters 717 plus autosampler, aWaters 2487 scanning UV detector (Waters Co., Milford, MA, USA), an HPLC column temperature controller (Phenomenex Inc., CA, USA), a BransonicR Ultrasonic Cleaner (Branson Ultrasonic Co., Danbury, CT, USA), a vortex-mixer (Scientific Industries Co., NY, USA), and a high-speed microcentrifuge (Hitachi Co., Tokyo, Japan).
Animal Studies
All animal study protocols were approved by the Animal Care Committee of Chosun University (Gwangju, Republic of Korea). Male Sprague-Dawley rats (270 – 300 g) were purchased from the Dae Han Laboratory Animal Research Co. (Eumsung, Republic of Korea). Animals were allowed free access to a normal standard chow diet (No. 322-7-1, Superfeed Co., Wonju, Republic of Korea) and tap water. Throughout the experiments, the animals were housed, four or five per cage, in laminar flow cages maintained at a temperature of 22 _ 2°C, 50 – 60% relative humidity, and a 12 h light/dark cycle. The rats were acclimated under these conditions for at least one week prior to experiments. Rats were fasted for at least 24 h prior to the experiments. The left femoral artery (for blood sampling) and left femoral vein (for drug administration in the intravenous study) were cannulated using a polyethylene tube (SP45; i.d., 0.58 mm; o.d., 0.96 mm; Natsume Seisakusho Company, Tokyo, Japan) while each rat was under light ether anesthesia.
Intravenous and Oral Administration of Drugs
The rats were divided into six groups (n = 6) as follows: oral groups administered 10 mg ・ kg-1 of nifedipine dissolved in distilled water (1.0 mL ・ kg-1) without (control) or with 1.5 and 6.0 mg ・ kg-1 of cilostazol (mixed in distilled water; total oral volume of 1.0 mL ・ kg-1), and intravenous groups administered 2.5 mg ・ kg-1 of nifedipine in 0.9% NaCl injectable solution (total injection volume of 1.0 mL ・ kg-1) without (control) or with 1.5 and 6.0 mg ・ kg-1 of cilostazol. A feeding tube was used to administer nifedipine and cilostazol intragastrically. Cilostazol was administered 30 min prior to oral administration of nifedipine. Blood samples were collected into heparinized tubes via the femoral artery at 0.017 (end of infusion), 0.1, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h for the intravenous study, and 0.25, 0.5, 0.75, 1, 2, 4, 8, 12, and 24 h for the oral study. Whole blood (approximately 1.2 mL) collected from untreated rats was infused via the femoral artery at 0.75, 4, and 8 h to replace blood loss from blood sampling. The blood samples were centrifuged (13000 rpm, 3 min), and 200 μL aliquots of plasma samples were stored in the deep freezer at -40°C prior to HPLC analysis.
HPLC Measurements
The plasma concentrations of nifedipine were determined using an HPLC assay with a modification of the method reported by Grundy, et al. [21]. Briefly, 50 μL of amlodipine (3 μg ・ mL-1) as the internal standard and 50 μL of 1.0 M sodium hydroxide were added to 0.2 mL plasma sample. This was mixed for 3 sec, and 1 mL of MTBE-isooctane (75 : 25, v/v) was added. The resultant mixture was vortex-mixed for 1 min and centrifuged at 3000 rpm for 5 min. The organic layer (0.8 mL) was transferred into a clean test tube and evaporated under a gentle stream of nitrogen gas (no heat applied). The dried extract was reconstituted with 200 μL of the mobile phase vortex-mixed for 1 min and aliquots of 160 μL were transferred to a clean autosampler vial. A 70 μL aliquot of the supernatant was injected into the HPLC system. The UV detector wavelength was set to 350 nm; and a Nova-pack C8 column (100mm × 8 mm I. D., 4 μm; Waters Co., Milford, MA, USA), was used at room temperature. A methanol – water mixture (62 : 38, v/v, pH 4.5 adjusted with acetic acid, and 320 μL TEA/1000 mL mixture added) was used as the mobile phase at a flow rate of 1.0 mL ・ min-1. The retention times were as follows: 16.8 min for internal standard, 8.2 min for nifedipine, and 6.5 min for dehydronifedipine. The detection limits of nifedipine and dehydronifedipine in rat plasma were 5 ng ・ mL-1. The coefficients of variation for nifedipine and dehydronifedipine were below 5.0%.
CYP3A4 Inhibition Assay
This experiment was performed to study the inhibitory effect of cilostazol on CYP3A4 activity, as nifedipine and cilostazol are both substrates of CYP3A4. The inhibition assay for the human CYP3A4 enzyme activity was performed in a multi-well plate using CYP inhibition assay kit (GENTEST, Woburn, MA) as described previously [22]. Briefly, human CYP enzyme was obtained from baculovirus-infected insect cells. The CYP substrate (7-BFC for CYP3A4) was incubated with or without the test compounds in the enzyme/substrate buffer with 1.0 pmol of P450 enzyme and an NADPH-generating system (1.3 mM NADP, 3.54mM glucose 6-phosphate, 0.4 U ・ mL-1 glucose 6-phosphate dehydrogenase and 3.3 mM MgCl2) in potassium phosphate buffer (pH 7.4). Reactions were terminated by adding the stop solution after incubation for 45 min. Metabolite concentrations were measured with spectrofluorometer (Molecular Device, Sunnyvale, CA) at an excitation wavelength of 409 nm and an emission wavelength of 530 nm. The positive control (1 μM ketoconazole for CYP3A4) was run on the same plate and showed 99% inhibition. All experiments were done in duplicate and the results were expressed as percentage inhibition.
Rhodamine-123 Retention Assay
This experiment was done to study the effect of cilostazol on P-gp, because it was not reported whether cilostazol is a substrate of P-gp. The procedures used for the rhodamine-123 retention assay were similar to previously reported methods [23]. The MCF-7/ADR cells were seeded in 24-well plates. At 80% confluence, the cells were incubated in fetal bovine serum (FBS)-free Dulbeccos modified eagles medium (DMEM) for 18 h. The culture medium was changed to Hanks′ balanced salt solution (HBSS) and the cells were incubated at 37°C for 30 min. After incubation of the cells with 20 μM rhodamine-123 in the presence or absence of cilostazol (10, 30, and 100 μM) and verapamil (positive control) for 90 min, the medium was completely removed. The cells were washed three times with ice-cold phosphate buffer (pH 7.0) and lysed in EBC buffer. Then, rhodamine-123 fluorescence in the cell lysates was measured at the excitation and emission wavelengths of 480 and 540 nm, respectively. Fluorescence values were normalized to the total protein content of each sample and presented as ratios to the control.
Pharmacokinetic Analysis
The plasma concentration data were analyzed by the non-compartment method using the Thermo Kinetica Software Version 5.0 (Thermo Fisher Scientific Inc., Miami, OK,
USA). The values of parameters were obtained by fitting the data to the pharmacokinetic model using the simplex algorithm. The area under the plasma concentration–time curve (AUC 0-∞) was calculated by the trapezoidal rule extrapolation method [24]. The peak concentration (C max) of nifedipine in plasma and the time to reach C max(T max) were obtained by the visual inspection of data from the concentration–time curve. The terminal half-life (t 1/2) was calculated as 0.693/K el. Total body clearance (CL/F) was calculated as dose/AUC. The extent of absolute bioavailability (F) was calculated as AUC oral/AUCi.v. × dosei.v./doseoral, and the relative bioavailability (RB) of nifedipine was calculated as AUC nifedipine with cilostazol/AUC control. The metabolite–parent AUC ratio (MR) was calculated as AUC dehydronifedipine/AUC nifedipine.
Statistical Analysis
The data are presented as mean ± standard deviation (SD). The pharmacokinetic parameters were compared using a one-way analysis of variance (ANOVA), followed by a posteriori test with the Dunnett correction. A confidence value of p < 0.05 was considered statistically significant.
Results and Discussion
Effect of Cilostazol on CYP3A4 Activity
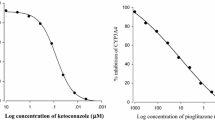
The inhibitory effect of cilostazol on CYP3A4 activity is shown in Fig. 1. Cilostazol inhibited CYP)A4 activity in a concentration-dependent manner with a 50% inhibitory concentration (IC50value) of 4.1 μM. Ketoconazole inhibited CYP3A4 activity with IC50 = 0.14 μM, which was the standard index of CYP3A4 activity inhibition.

Inhibitory effects of ketoconazole and cilostazol on CYP3A4 activity.
The importance of first-pass metabolism for limiting systemic drug availability is well established; however, intestinal drug metabolism can further diminish its systemic availability. Through functional enzyme activity studies and immunoblot analyses, CYP3A expression in mature enterocytes, located mainly in the villi tips, of jejunal mucosa was shown to be comparable to or even exceed the expression of CYP3A in hepatocytes [25]. Total CYP p450 content was reported to increase slightly on the passage from the duodenum to the jejunum and then decreased sharply to the ileum [26]. Using in situ hybridization with a probe specific for CYP3A4, McKinnon and McManus [27] confirmed CYP3A expression throughout the entire small intestine, with highest levels in the proximal regions. The most abundant CYP isoenzyme in the intestine is 3A4 [28]. In vitro metabolism studies in human liver microsomes revealed that hepatic cytochrome p450 enzymes, primarily CYP3A4, were involved in the metabolism of cilostazol [15, 16].
Considering that nifedipine is a substrate of both CYP3A4 and P-gp [9,10,11, 20], this experiment was performed to find the effect of cilostazol on the inhibition of CYP3A4, because nifedipine and cilostazol are both substrates of CYP3A4. In addition, this experiment was aimed to find the effect of cilostazol on P-gp, since it was never reported whether cilostazol was a substrate of P-gp.
Cilostazol significantly (p < 0.05) inhibited CYP3A4 activity. The inhibitory effect of cilostazol (IC50= 4.1 μM) on CYP3A4 was weaker than that of ketoconazole (IC50 = 0.14 μM). In general, when the inhibitory effect (IC50) of CYP3A4 was below 12 μM, the bioavailability (AUC) of CYP3A4 substrates was only increased by CYP3A4 inhibitors [29,30,31]. The inhibitory effect of cilostazol on CYP3A4-mediated metabolism was confirmed by using recombinant CYP enzymes. Therefore, cilostazol (an inhibitor of CYP3A4) may significantly influence the bioavailability of nifedipine, a substrate of CYP3A4. As CYP3A9 expressed in rat is corresponding to the ortholog of CYP3A4 in humans [32], CYP3A4 of rats are similar to human CYP3A4 [29,30,31, 33, 34]. The human CYP3A4 and rat 3A4 have 73% protein homology [33,34,35]. On the basis of above theory, this study evaluated the influence of cilostazol on the pharmacokinetics of nifedipine in rats in order to assess the potential drug interactions between cilostazol and nifedipine, a substrate of CYP enzymes.
Effect of Cilostazol on the P-gp Activity
The accumulation of rhodamine-123, a P-gp substrate, increased in MCF-7/ADR cells overexpressing P-gp compared to that observed in MCF-7 cells lacking P-gp, as shown in Fig. 2. The concurrent use of cilostazol did not enhance the cellular uptake of rhodamine-123. This result suggests that cilostazol does not inhibit the P-gp activity.

Effect of cilostazol on the cellular accumulation of rhodamine-123 in MCF-7 and MCF-7/ADR cells (mean ± SD (n = 6); ** p < 0.01 vs. control group).
Effects of Cilostazol on the Pharmacokinetics of Oral Nifedipine
The mean plasma concentration–time profiles of nifedipine in the presence and absence of cilostazol (1.5 and 6.0 mg ・ kg-1) are shown in Fig. 3. The pharmacokinetic parameters of nifedipine are summarized in Table 1. Cilostazol (6.0 mg ・ kg-1) significantly (p < 0.05) increased AUC 0–∞ of nifedipine (by 41.0%) and C max of nifedipine (by 35.3%). The total body clearance CL/F was significantly decreased (6.0 mg ・ kg-1, p < 0.05) by cilostazol (31.0%). Accordingly, the absolute bioavailability (F) of nifedipine in the presence of cilostazol (6.0 mg ・ kg-1) was significantly (p < 0.05) higher (20.2%) than that in the control group. Cilostazol increased the relative bioavailability (RB) of nifedipine by a factor pf 1.14 – 1.41.

Mean plasma concentration vs. time profiles of nifedipine in rats after oral (10 mg ・ kg-1) administration in the presence and absence of cilostazol (mean ± SD (n = 6)): (●) control (nifedipine 10 mg ・ kg-1 alone); (○) with cilostazol 1.5 mg ・ kg-1; (▼) with cilostazol 6.0 mg ・ kg-1 (* p < 0.05 vs. control group).
The obtained results are consistent with previous reports, according to which ketoconazole and coenzyme Q10 significantly increase the AUC 0–∞ and C max of nifedipine in rats [36, 37]. These results are also similar to our previous data [29,30,31] showing that fluvastatin, simvastatin, and lovastatin significantly enhance the AUC 0–∞ and C max of repaglinide, losartan, and diltiazem, substrates of CYP enzymes.
Effect of Cilostazol on the Pharmacokinetics of Dehydronifedipine
The plasma concentration–time profiles of dehydronifedipine are shown in Fig. 4. The pharmacokinetic parameters of dehydronifedipine are summarized in Table 2. The AUC 0–∞ of dehydronifedipine was not significantly increased (6.0 mg ・ kg-1, p < 0.05) by cilostazol. The MR ratios were significantly (p < 0.05, 6.0 mg ・ kg-1) decreased (20.0%) by cilostazol, this result suggesting that the formation of dehydronifedipine was considerably altered by cilostazol. The increase in the bioavailability of nifedipine may be mainly because of the inhibition of CYP3A4 activity by cilostazol in the small intestine or liver or both.

Mean plasma concentration vs. time profiles of dehydronifedipine in rats after oral administration of nifedipine (10 mg ・ kg-1) in the presence and absence of cilostazol (mean ± SD (n = 6)): (●) control (nifedipine 10 mg ・ kg-1 alone); (○) with cilostazol 1.5 mg ・ kg-1; (▼) with cilostazol 6.0 mg ・ kg-1.
Effect of Cilostazol on the Pharmacokinetics of Intravenous Nifedipine
The mean arterial plasma concentration–time profiles of nifedipine following an intravenous administration of nifedipine (2.5 mg ・ kg-1) in rats in the presence or absence of cilostazol (1.5 and 6.0 mg ・ kg-1) are shown in Fig. 5, and the corresponding pharmacokinetic parameters are shown in Table 3. The AUC 0–∞ and CLt of nifedipine were significantly (p < 0.05) changed by 19.0 and 22.2%, respectively, compared to those in the control group. This result (increasing AUC 0–∞) is not consistent with the previous studies showing that HMG-CoA reductase inhibitors (simvastatin) did not increase the AUC 0–∞ of intravenous losartan in rats [30].

Mean plasma concentration vs. time profiles of nifedipine in rats after intravenous (2.5 mg ・ kg-1) administration of nifedipine in the presence and absence of cilostazol (mean ± SD (n=6)): (●) control (nifedipine 2.5 mg ・ kg-1 alone); (○) with cilostazol 1.5 mg ・ kg-1; (▼) with cilostazol 6.0 mg ・ kg-1 (* p < 0.05 vs. control group).
Accordingly, the increased bioavailability of nifedipine in the presence of cilostazol can be mainly due to inhibition of the CYP3A4-mediated metabolism of nifedipine in the liver by cilostazol.
Conclusion
The increased bioavailability of nifedipine in rats can be mainly due to the inhibition of CYP3A4-mediated metabolism in the small intestine and/or liver. In addition, the reduction of MR and CL/F may also contribute to the increased bioavailability (AUC 0–∞ and C max) of nifedipine by cilostazol. Therefore, concomitant use of cilostazol and nifedipine may require close monitoring of potential drug interactions and also dosage regimen of nifedipine probably needs to be readjusted for safe combined therapy of cardiovascular diseases. The clinical importance of these findings should be further investigated in clinical trials.
References
E. M. Sorkin, S. P. Clissold, and R. N. Brogden, Drugs, 30, 182 – 274 (1985).
J. L. Blackshear, C. Orlandi, N. K. Hollenberg, et al., J. Cardiovasc. Pharmacol., 8, 37 – 43 (1986).
S. R. Hamann, M. T. Piascik, and R. G. McAllister, Biopharm. Drug Dispos., 7, 1 – 10 (1986).
T. Funaki, P. A. Soons, F. P. Guengerich, et al., Biochem. Pharmacol., 38, 4213 – 4216 (1989).
P. B. Watkins, Pharmacogenetics, 4, 171 – 184 (1994).
F. He, H. C. Bi, Z. Y. Xie, et al., Rapid Commun. Mass Spectrom., 21, 635 – 643 (2007).
T. Shimada, H. Yamazaki, M. Mimura, et al., J. Pharmacol. Exp. Ther., 270, 414 – 423 (1994).
S. Rendic and F. J. Di Carlo, Drug Metab. Rev., 29, 413 – 580 (1997).
J. H. Lin, M. Chiba, and T. A. Baillie, Pharmacol. Rev., 51, 135 – 158 (1999).
G. K. Dresser, J. D. Spence, and D. G. Bailey, Clin. Pharmacokinet., 38, 41 – 57 (2000).
M. M. Doherty and W. N. Charman, Clin. Pharmacokinet., 41, 235 – 253 (2002).
K. Schror, Diabetes Obes. Metab., 4, S14 – S19 (2002).
M. P. Reilly and E. R. Mohler, Ann. Pharmacother., 35, 48 – 56 (2001).
K. P. Kim, B. H. Kim, K. S. Lim, et al., Clin. Ther., 31, 2098 – 2106 (2009).
R. Abbas, C. P. Chow, N. J. Browder, et al., Hum. Exp. Toxicol., 19, 178 – 184 (2000).
S. L. Bramer, W. P. Forbes, and S. Mallikaarjun, Clin. Pharmacokinet., 37 (Suppl 2), 1 – 11 (1999).
V. J. Wacher, C. Y. Wu, and L. Z. Benet, Mol. Carcinog., 13, 129 – 134 (1995).
D. Leveque and F. Jehl, Anticancer Res., 15, 231 – 336 (1995).
M. V. Relling, Ther. Drug Monit., 18, 350 – 356 (1996).
M. Dorababu, A. Nishimura, and T. Prabha, Biomed. Pharmacother., 63, 697 – 702 (2009).
J. S. Grundy, R. Kherani, and R. T. Foste, J. Chromatogr. B: Biomed. Appl ., 654, 146 – 151 (1994).
C. L. Crespi, V. P. Miller, and B. W. Penman, Anal. Biochem., 248, 188 – 190 (1997).
C. Y. Han, K. B. Cho, H. S. Choi, et al., Carcinogenesis, 29, 1837 – 1844 (2008).
W. L. Chiou, J. Pharmacokinet. Biopharm., 6, 539 – 546 (1978).
P. B. Watkins, Gastroenterol Clin. North Am., 21, 511 – 526 (1992).
Q. Zhang, D. Dunbar, A. Ostrowska, et al., Drug Metab. Dispos., 27, 804 – 809 (1999).
R. McKinnon and and M. McManus, Pathology, 28, 148 – 155 (1996).
M. Tubic-Grozdanis, J. M. Hilfinger, G. L. Amidon, et al., Pharm. Res., 25, 1591 – 1600 (2008).
C. K. Lee, J. S. Choi, and J. S. Bang, Korean J. Physiol. Pharmacol., 17, 245 – 251 (2013).
S. H. Yang, J. S. Choi, and D. H. Choi, Pharmacology, 88, 1 – 9 (2011).
D. H. Choi, J. S. Choi, J. S. Choi, et al., J. Pharm. Pharmacol., 63, 129 – 135 (2011).
P. A. Kelly, H. Wang, K. L. Napoli, et al., Eur. J. Drug Metab. Pharmacokinet., 24, 321 – 328 (1999).
J. J. Bogaards, M. Bertrand, P. Jackson, et al., Xenobiotica, 30, 1131 – 1152 (2000).
F. P. Guengerich, M. V. Martin, P. H. Beaune, et al., J. Biol. Chem., 261, 5051 – 5060 (1986).
D. F. V. Lewis, Cytochrome P450 Structure, Function, and Mechanisms, Taylor & Francis, Bristol (1996), pp._122 – 123.
M. Kuroha, H. Kayaba, S. Kishimoto, et al., J. Pharm. Sci., 91, 868 – 873 (2002).
A. Nishimura, M. Fujimura, F. Hasegawa, et al., J. Health Sci., 56, 310 – 320 (2010).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lee, CK., Choi, JS. & Choi, DH. Effects of Cilostazol on the Pharmacokinetics of Nifedipine After Oral and Intravenous Administration in Rats. Pharm Chem J 51, 748–755 (2017). https://doi.org/10.1007/s11094-017-1686-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-017-1686-0