
Abstract
Si–Co–Y diffusion coatings were deposited on TiAl–Nb alloy using pack cementation process. The influence of activators and deposition temperatures on the coating structures was investigated, alongside the coating formation process and hot corrosion performance of the optimized coating in molten salt of 25%NaCl + 75%K2SO4 at 850 °C. The results show that a dense and compact Si–Co–Y diffusion coating can be prepared on TiAl–Nb alloy, with a multi-layered structure including an outmost layer of (Ti, X)Si2 (X represents Co, Al, Nb, Y), an outer layer composed of TiSi2 + Ti5Si4 + Ti5Si3 mixtures, a middle layer of Ti5Si3, and an inner layer of TiAl2. The coating prepared with AlCl3·6H2O and NH4Cl had many pores. Increase in deposition temperature led to a higher coating growth rate within the range of 1050–1100 °C, but temperature exceeding 1100 °C caused the formation of intensive holes in the coating. Hot corrosion tests at high temperatures proved that the Si–Co–Y diffusion coating prepared on TiAl–Nb alloy exhibited excellent hot corrosion resistance in 25%NaCl–75%K2SO4 molten at 850 °C. A protective hot corrosion product scale composed of a TiO2 + Na2SiO3 + Na2TiO3 outer layer and an Al2O3 middle layer, formed on coating after hot corrosion for 50 h. The scale can effectively inhibit the inward diffusion of corrosion medial of O, Cl and S elements.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
TiAl alloys possess low density, high strength and specific strength, excellent mechanical properties, making them potential lightweight alternatives to nickel-based alloys in high-temperature structural applications [1,2,3]. When serving as hot-end components in aero-engines and gas turbines, TiAl alloys need to withstand long-term exposure to high levels of air salinity, particularly in offshore or marine environments. Unfortunately, TiAl alloys exhibit relatively poor anti-hot corrosion resistance, as they are prone to react with corrosive substances such as oxygen, Cl− and SO2 at high temperatures. The corrosion products formed are loose, porous, and easily peel off, significantly compromising safety and service life [4, 5]. Therefore, enhancing the hot corrosion resistance of TiAl alloy is imperative to enable their use as hot-end components.
Preparing surface protective coatings is an effective way to improve the hot corrosion resistance of TiAl alloys. In recent decades, various methods have been utilized to prepare protective coatings for TiAl alloys, such as aluminide coatings (TiAl3, TiAl2, etc.) [5], M-CrAlY (M = Ni, Co or NiCo) coatings [6, 7], ceramic coatings [8, 9], silicide coatings [10,11,12] and composite coatings doped with noble metals [13]. Among the these coating systems, silicide coatings exhibit excellent thermal stability and can form a flowable and self-healing SiO2 protective scale at high temperatures, rendering them suitable for load-bearing applications in TiAl alloys [14]. At present, several studies have examined the high-temperature oxidation resistance of silicide coatings applied to TiAl alloys, while limited studies has focused on their hot corrosion behavior. Based on the available literature, it appears that the hot corrosion resistance of the silicide coatings is insufficient due to the continuous erosion of silicon dioxide formed by silicides through sulfates and chlorides. This hinders the formation of a protective product film, leading to ongoing internal sulfidation and eventual cracking failure. Studies have indicated that the addition of appropriate 'third-elements' such as Co, Ni, Hf and B, etc., can effectively promote the formation of protective scales of Al2O3, Cr2O3 or SiO2 [15]. Rare earth elements (REEs) such as Ce, La, and Y have been proven to be crucial in enhancing the high-temperature performance of alloy coatings by inhibiting high-temperature diffusion and delaying degradation. REEs also play a significant role in refining and purifying the coating/oxide film structure, promoting the formation of oxide films, and enhancing their adhesion. Among the REEs, Y is widely distributed and commonly used as a modification element for high-temperature coatings. Wang et al. [16] have found that Y atoms, characterized by their high chemical activity and low electronegativity, can effectively improve the activity of co-deposited element and facilitate their adsorption onto the substrate’s surface. Additionally, the large atomic radius of Y induces lattice distortion in the coating, increasing dislocation density and vacancies. Consequently accelerates the diffusion rate of Si into the substrate.
The pack cementation technique, characterized by its simplicity, versatility, and high repeatability, is essentially a gas-phase deposition technique. With using this method, compact silicon-based coatings or modified silicon-based coatings with strong bonding to the substrate can be conveniently prepared on TiAl–Nb alloys. In this study, a Si–Co–Y coating was prepared on a TiAl–Nb alloy by pack cementation method. The structural characteristics and hot corrosion resistance of the coatings prepared with different activators and temperatures were investigated. The primary goal was to develop a protective coating with good anti-hot corrosion performance for TiAl–Nb alloys, and provide support for promoting the application of TiAl–Nb alloys in high-temperature structural components.
Experimental Procedures
The TiAl–Nb substrate with a composition of Ti45Al–8Nb–0.3Y was prepared by vacuum consumable arc-melting method. Before smelting, the raw materials were sequentially subjected to degreasing → acid cleaning → alkali cleaning → alcohol cleaning to remove surface oxides and contaminants. The smelting equipment used was a self-made high-temperature, high-vacuum water-cooled crucible arc melting furnace, with a tungsten rod (arc gun) as the cathode and the copper crucible as the anode. During smelting, high-purity argon gas was filled into the furnace to prevent the oxidation of highly active elements such as Ti, Al, and Y, and to suppress the volatilization of low-melting-point element of Al. To minimize the component segregation, each alloy component was re-melted for three times.
The specimens to be coated were cut from the ingot into cuboid measuring 4 mm × 4 mm × 3 mm. Before packing, each surface of the specimens was polished using 1000 # SiC paper, and then ultrasonically cleaned in an acetone bath. According to the previous research of our team, the pack powders of 15Si–10Co–3Y–4M–68Al2O3 (wt%) were employed, in which, Si, Co and Y powders were used as the donor sources, M represented the activators of NaF, NaCl, NH4Cl and AlCl3·6H2O, and Al2O3 was used as the filler. All the powders were produced by Sinopharm Chemical Reagent Co., Ltd in Shanghai, China. The total weight of the pack powders utilized for each sample was 50 g. Before packing, the powders were ball milled in a planetary ball mill to be fully mixed and refined. Each sample to be coated was buried nearly in the center of the pack powders contained in an alumina crucible with a capacity of 50 ml, which was then sealed with Na2SiO3. The sealed crucible was then placed in an atmosphere furnace. After vacuuming, the furnace was heated to 1080 °C at a rate of 10 °C/min for 0–4 h. After coating, each face of the samples was slightly brushed, and then ultrasonically cleaned in an acetone bath to remove the residual pack powders.
Hot corrosion testing was conducted in an SX2-2.5-12A type muffle furnace at a temperature of 850 °C for 1 h, 10 h, 25 h and 50 h, using 25% NaCl + 75% K2SO4 (wt%) as the corrosion medium. During hot corrosion, the specimens were placed in a corundum crucible after heating without changing mass to obtain the spalling corrosion products. Every 12 h, the samples were pulled out of the furnace to supplement the corrosive solution on their surfaces using a dropper, resulting in the deposition of approximately 2.0 ± 0.2 mg/cm2 of solid salt each time. After hot corrosion, the specimens were placed in distilled water for 10 min before weighing, during which they were slightly stirred to dissolve the surface salt, and then dried before weighing. The detached insoluble corrosion products during the cleaning process will be collected and, after drying, weighed together with the samples after hot corrosion.
An electronic balance with an accuracy of 0.1 mg was used to measure the mass change of the specimens before and after oxidation, and the average value was determined by weighing the sample five times. X-ray diffraction (XRD, Panalytical X ‘Pert PRO, Cu Kα) was employed to identify the constituent phases of the coatings and their hot corrosion products. A scanning electron microscopy (SEM, JSM-6360LV) equipped with an energy dispersive spectroscopy (EDS) were employed to identify the microstructure and compositions of the coatings and their hot corrosion products.
Results
Coating Structure
Effects of the Activators
Figure 1 presents the cross-sectional BSE images and element distribution curves the Si–Co–Y diffusion coatings prepared with NaF, NH4Cl, AlCl3·6H2O and NaCl activators at 1080 °C for 4 h. Figure 2 shows the surface XRD patterns of the coatings prepared with different activators, as well as the outer layer of the coating prepared using NaF activator. Upon observing the microstructure of the coating as a whole, it is noted that the coatings prepared with NaF, AlCl3·6H2O and NH4Cl exhibit a greater coating thickness prepared with NaCl. Furthermore, the coatings prepared with NaF and NaCl display a denser microstructure than those prepared with AlCl3·6H2O and NH4Cl. In addition, some Al2O3 diffraction peaks can be observed in the surface XRD patterns of the coatings prepared with different activators, which should result from the adhesion of Al2O3 from the pack powders.

Cross-sectional BSE images (a–d) and elemental concentration profiles (a1–d1) of the Si–Co–Y coatings prepared at 1080 °C for 4 with using different activators. (a) (a1) NaF, (b) (b1) NH4Cl, (c) (c1) AlCl3·6H2O, (d) (d1) NaCl

XRD patterns conducted on (a) the surfaces of the Si–Co–Y coatings prepared with different activators and (b) the outer layer of the coating prepared with NaF as the activator. The XRD pattern in (b) was obtained by stripping the coating from its original surface by about 10 μm using 1500 #SiC paper, and then conducted XRD analysis
It can be observed from Fig. 1a that the coating prepared with NaF has a total thickness of about 63 μm, exhibiting a dense and multi-layered structure. The light-gray outermost layer of the coating has a thickness of about 5 μm, with small amounts of dispersed gray phases present inside. Based on the Ti–Si binary phase diagram [17], EDS analysis results (Fig. 1(a1)) and XRD pattern (Fig. 2), it can be determined that the outermost layer of the coating is mainly composed of TiSi2 phase.
The outer layer of the coating, with a thickness of around 31 μm, is mainly composed of dark gray and light gray phases. The XRD pattern in Fig. 2 and EDS analysis results in Fig. 1(a1) indicate that it is mainly composed a mixture of TiSi2, Ti5Si4 and Ti5Si3 phases. The middle layer of the coating has a thickness of about 15 µm, and mainly consists of three distinctive phases with different contrasts. EDS analysis results (Fig. 1(a1)) show that the atomic percentage of Ti and Si in each phase is approximately 5:3, while the Al content varies from top to bottom, with values of 4.26 at%, 2.90 at%, and 0.42 at%, respectively. This reveals that the middle layer is mainly consisted of the Ti5Si3, and the variation in phase contrast is attributed to the varying Al content. The dark gray inner layer of the coating has a thickness of about 12 µm. EDS analysis results (Fig. 1(a1)) indicate that the contents of Ti and Al in the layer are respectively about 27.52 at% and 58.15 at%, indicating the formation of TiAl2 inner layer [18].
Moving on to Fig. 1b, it can be observed that the coating prepared with NH4Cl has a thickness of approximately 108 µm. The microstructure of the coating is similar to the one prepared with NaF, but numerals pores have formed in the outer and middle layers. The EDS analysis results in Fig. 1(b1) indicate that both the surface and outer layers of the coating are rich in Al. However, the content of Al in the inner layer of the coating is only about 43.15 at%, indicating that the inner layer has not completely transformed into TiAl2. The coating prepared with NaCl has a three-layer structure with a total thickness of about 15μm. EDS analysis (Fig. 1(d1)) and XRD analysis results (Fig. 2) reveal the absence of TiSi2 outer layer and TiAl2 inner layer formation in the coating. Furthermore, the Al element is enriched in the outermost layer, and a certain amount of Al2O3 is present.
Effects of Deposition Temperature
The coating prepared with NaF exhibited a relatively dense coating structure, and possessed a proper coating growth rate. Consequently, NaF was chosen to examine the effects of packing temperatures on the structure of the coating. Figure 3 presents the cross-sectional BSE images and elemental concentration profiles of the coatings prepared at 1050 °C and 1100 °C using NaF as the activator. Figure 4 shows the surface XRD patterns of the corresponding coatings.

Cross-section BSE images (a) (b) and element distribution (a1) (b1) of the Si–Co–Y diffusion coatings prepared with NaF at 1050 °C and 1100 °C. (a) (a1) 1050 °C, (b) (b1) 1100 °C

Surface XRD patterns of the Si–Co–Y diffusion coatings prepared at 1050 °C and 1100 °C using NaF as the activator
Together with the coating prepared at 1080 °C, as shown in Fig. 1a and (a1), it is observable that the coatings prepared at different deposition temperatures possess similar structure. As the deposition temperature increased, there was a noticeable increase in the coating thickness. Moreover, intense holes formed in the middle and inner layers of the coating prepared at 1100 °C. According to the EDS analysis results of the coating cross-sections at different temperatures and the surface XRD patterns (Figs. 2 and 4), it can be concluded that there were no significant differences in the composition of the various coatings. They all consisted of an outmost layer of TiSi2, an outer layer of TiSi2 + Ti5Si3 + Ti5Si4, a middle layer of Ti5Si3, and an inner layer of TiAl2.
Coating Formation Process
The formation of diffusion coatings is generally considered to be mainly controlled by two processes. The first step mainly involves the formation, transmission, and adsorption of the active atoms from the deposited elements, which is mainly influenced by the vapor partial pressures of the halides of the deposited elements produced in the pack. During the packing process, the activator reacts with the deposited elements at high temperatures, leading to the formation of the respective vapor halides. Deposition of a single element is relatively straightforward when the partial pressures of the vapor halides of the deposited element are high. However, simultaneously depositing multiple elements becomes more challenging since it requires maintaining a reasonable range of partial pressures for each deposited element [15].
Figure 5 presents the calculated equilibrium partial pressures of the main vapor halides of the deposited elements using different activators. These calculations are based on the Gibbs free energy of each independent reaction and state equation. It is observed that the equilibrium partial pressures of the vapor halides of Si and Co in the pack using NaF, NH4Cl and AlCl3·6H2O as the activators are generally higher than those using NaCl as the activator. In other words, the equilibrium partial pressures of the vapor halides produced in the NaCl containing pack are generally low, resulting in thinner coating thickness as shown in Fig. 1d. However, numerous voids formed in coatings prepared by NH4Cl and AlCl3·6H2O. This should be mainly attributed to the formation of HCl and Cl2, which is released by decomposition of NH4Cl and AlCl3·6H2O, respectively, and can penetrate into the substrate at high temperatures and react to form volatile halides, as shown by Eqs of (1)–(4) [19].

Gas phase partial pressures of the main halides in different catalyst packs. a NaF, b NH4Cl, c AlCl3·6H2O, d NaCl
For activator AlCl3·6H2O, decomposition of AlCl3·6H2O would also occur in the pack, and release active [Al] atoms through reactions of (5) and (6):
It is noted that the reaction (6) is an endothermic process, and the content of AlCl3·6H2O in the pack is only 4 wt%, thereby limiting the availability of aluminum resources in the pack. The calculated partial pressures of the aluminum-containing species are also significantly lower than those of silicon and cobalt-containing species, as shown in Fig. 5c. Therefore, it is considered that the deposition of aluminum is limited during the co-deposition process. This can also explain the relatively low content of Al in the coating prepared with AlCl3·6H2O.
In NaF containing pack, the partial pressures of Si containing species are generally higher compared to those of Co and Y containing species. Among these species, the partial pressures of Y containing species are the lowest. These observations suggest that the transport and adsorption of Si atoms on the surface of the substrate should be easier compared to those of Co and Y atoms. Consequently, deposition of Si atoms is considered to be more favorable compared to Co and Y. Fortunately, the differences in partial pressures between Si, Co, and Y-containing halides fall within a reasonable range, providing the necessary thermodynamic conditions for co-deposition.
The second step of the coating formation process mainly involves the inward diffusion or reaction diffusion of the active atoms within the substrate. Once the active atoms are adsorbed on the surface of the substrate, the diffusion temperature, the reactivity between the deposited atoms and the substrate, and the diffusion activation energy of the deposited elements become the main factors influencing the growth of the coating. According to the empirical formula Q = 32Tm (Tm is the melting point of the material), the growth rate of the diffusion coating is lower at the lower temperature of 1050 °C, resulting in a thinner coating thickness. Increasing the deposition temperature to a higher temperature of 1100 °C resulted in a larger coating thickness. However, an excessively high diffusion rate can result in insufficient deposition atoms in the pack, leading to the formation of intensive pores in the coating. It is also important to note that the atomic radius and melting points of Co and Si are similar and much lower than those of Y. As a result, the diffusion rates of Si and Co within the TiAl–Nb alloy will be higher compared to that of the Y atoms.
From the perspective of the reactivity of deposited elements with the TiAl–Nb substrate at a fixed temperature, it is observed that the standard formation Gibbs free energy of Ti5Si3 is the lowest, followed by Ti5Si4 and TiSi2. On the other hand, the formation of Co–Si and Co–Al compounds exhibits relatively higher energies. Considering that the partial pressures of Si containing halide species are much higher compared to Co and Y containing halide species (Fig. 5), it is reasonable to deduce that Ti5Si3 will initially form in the early stage of co-deposition, followed by Ti5Si4 and TiSi2. Thus, Ti5Si3 acts as the growth frontier of the diffusion coating, leading to a multi-layer structure as presented in Figs. 1 and 3. It is worth noting that although direct reactions between the Si and Co are favorable in the pack, it is difficult to form a dense coating through these direct reactions. Thus, the formation of Co-silicides in the coating is considered to be primarily achieved through the reaction of Co atoms with diffusion-formed Ti-silicides. Unfortunately, the cobalt-silicide compounds such as Co2Si, CoSi and CoSi2 can hardly form during this period because the Gibbs formation energies of Co reacting with Ti5Si3 or Ti5Si4 to form these compounds are positive values universally higher than 200 kJ/mol at the employed deposition temperatures. However, once the TiSi2 outmost layer is formed, the formation of cobalt-silicide compounds through Eqs. (6) to (8) became possible. The above analysis revealed that the formation process of the Si–Co co-deposited coatings on TiAl–Nb alloy follows an orderly progression of depositing Si first, followed by Co and Y. In this process, the inward react-diffusion of Si into the substrate is the dominant factor for the growth of the co-deposition coating.
Hot Corrosion Properties
Hot Corrosion Kinetics
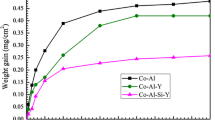
Figure 6 shows the hot corrosion kinetics curves and macroscopic morphology of the TiAl–Nb substrate and Si–Co–Y diffusion coating in 25% NaCl + 75% K2SO4 mixed molten salt at 850 °C. The weight gain of the TiAl–Nb substrate increases sharply to about 7.07 mg/cm2 within 1 h of hot corrosion, and then becomes a relatively slow increase to about 3.17 mg/cm2 with prolonging the hot corrosion time to 10 h. When the hot corrosion time exceeds 10 h, the weight gain of the substrate resumes a rapid growth to about 32.81 mg/cm2 at 25 h and 81.83 mg/cm2 at 50 h. In contrast, the weight gain of the Si–Co–Y diffusion coating remains relatively stable throughout the entire hot corrosion period, and it is significantly lower compared to the TiAl–Nb substrate. After 25 h of hot corrosion, the weight gain for the coating is around 7.91 mg/cm2, and slowly increases to about 11.09 mg/cm2 after 50 h of hot corrosion. This indicates that the hot corrosion resistance of the Si–Co–Y diffusion coating is obviously better than that of the TiAl–Nb substrate.

Hot corrosion kinetic curves of the TiAl–Nb substrate and Si–Co–Y diffusion coating in NaCl + K2SO4 molten salt at 850 °C. The macroscopic morphology of the specimens after hot corrosion for 50 h are also presented for comparison
Morphology of Hot Corrosion Products
Figure 7 shows the surface and cross-section BSE images, along with the EDS analysis maps, of the hot-corrosion products scale formed on TiAl substrate and Si–Co–Y diffusion coating after hot corrosion for 50 h. Figure 8 presents the surface XRD patterns of the hot corrosion product scales. It can be seen from Fig. 7a that the hot corrosion products formed on TiAl–Nb substrate are mainly composed of strip-like dark gray phase with a small amount of bright white phase embedded within it. The EDS analysis maps reveal that the dark gray phase has a typical composition of 22.51Ti–0.17Al–0.56Nb–76.70O–0.05Na–0.01K (at%), and the bright white phase has a typical composition of 15.02Ti–3.08Al–5.26Nb–72.88O–3.69Na–0.06K–0.01Cl (at%). Combined with the XRD pattern in Fig. 8, the dark gray phase should be TiO2 and the bright white phase is rich in NaNbO3. The cross-section BSE images of the corrosion products formed on the TiAl–Nb substrate, as shown in Fig. 7(a1), reveal that the hot-corrosion product scale with a thickness exceeding 200 μm, appears rather loose. Additionally, noticeable cracking also formed between the products scale and the substrate. The EDS analysis maps confirm that the hot-corrosion product scale of the TiAl–Nb substrate is mainly composed of TiO2 and Al2O3 mixtures, without the formation of a dense and continuous Al2O3 layer. Research by Guan et al. also reported that a single dense protective Al2O3 layer can hardly form on TiAl–Nb alloys when exposed to hot corrosion in a mixed molten salt of K2SO4 + NaCl.

Surface and cross-section BSE images, EDS analysis maps of the hot corrosion product scales formed on (a) (a1) TiAl–Nb substrate and (b) (b1) the Si–Co–Y diffusion coating after hot corrosion for 50 h

Surface XRD patterns of the hot corrosion product scales formed on TiAl–Nb substrate and Si–Co–Y diffusion coating, after hot corrosion for 50 h
From Fig. 7b, it is seen that the hot-corrosion product scale formed on the Si–Co–Y diffusion coating is mainly composed of small blocky gray phases. EDS analysis determined that these phases possess a typical composition of 26.40Ti–65.56O–0.61Nb–2.35Al–0.11Si–0.16Co–4.56Na–0.23Cl–0.02K (at%). Combined with the XRD pattern in Fig. 8, the superficial zone of the hot-corrosion product scale formed on the Si–Co–Y diffusion coating is mainly composed of TiO2, Na2SiO3 and Na2TiO3. The cross-sectional image in Fig. 7(b1) and its EDS analysis maps show that the hot-corrosion product scale exhibits a double layered structure that is rather dense and tightly adherent to the residual coating. The thin outer layer has a typical composition of 25.14Ti–66.01O–4.71Al–3.37Na–0.21Nb–0.17Si–0.08Y–0.13Co–0.03Cl–0.15S (at%), confirming the formation of TiO2, Na2SiO3 and Na2TiO3 once again. Beneath the outer layer, an dense inner layer with a typical composition of 33.89Al–60.81O–3.27Si–0.93Ti–0.62Nb–0.11Y–0.28Cl–0.09S (at%) is observed, indicating that the inner layer is composed of A2O3 with a small amount of SiO2. Such an inner layer may be a result of the outward diffusion of Al from the TiAl2 layer in the coating, as the TiAl2 layer has undergone a certain degree of degradation compared to the original coating. Moreover, It is considered that the protective TiO2 + Na2SiO3 + Na2TiO3 outer hindered the inward diffusion of oxygen, created the conditions for the selective oxidation of Al in the inner layer, thus promoted the formation of the dense, Al2O3 containing inner layer.
From the EDS analysis maps, it can be observed that the hot-corrosion product scale primarily contains Ti, Al, O, Nb and Si elements. The presence of S element, which is considered as a main invader, is slightly enriched at the interface of the scale and the remained coating. This indicates that only a few S ions have successfully permeated into the scale. Fortunately, there is very limited Cl element, which is considered as the main factor causing cracking, observed in both scale and the remained coating. This suggests that the inward diffusion of Cl ions has been effectively suppressed.
Discussion
According to the acid–base melting theory, the following equilibrium reactions would occur during hot corrosion in a NaCl + K2SO4 molten salt environment [20]:
NaCl can also react with the decomposition products of K2SO4 [21, 22]:
The hot corrosion behavior of TiAl–Nb alloys in the NaCl + K2SO4 molten salt environment has been reported in detail in References [19,20,21,22,23,24,25]. Briefly, in the early stage of hot corrosion, the higher aluminum content in γ-TiAl leads to the initial formation of Al2O3 on top of this phase. However, this tendency can change very quickly. The diffusion coefficients of aluminum in γ-TiAl and α2-Ti3Al (110–19 [m2/s] and 210–20 [m2/s], respectively) are approximately one order of magnitude lower than those of titanium (1.410–18 [m2/s] and 2.510–19 [m2/s], respectively) [23]. As a result, the α2-Ti3Al phase, with the higher titanium content, corroded faster and led to preferential consumption of α2-Ti3Al lamellas [19, 23]. The oxides produced during this process then reacted with NaCl to form Cl2 [24]. The as-generated Cl2 was considered to be a highly corrosive agent that reacted with the metal elements, leading to the formation of volatile species such as TiCl4, AlCl3. These volatile chlorides with low melting points and high vapor pressures (the boiling point of TiCl4 is 135.9 °C and the boiling point of AlCl3 is 178 °C) would volatilize outward to a region with an appropriate oxygen partial pressure, initiating the oxidation to form oxides and meanwhile, releasing Cl2, S2, SO3 or SO2. Consequently, a large number of voids, holes and even cracks, could be easily generated, creating more pathways for transport of corrosive agents and oxygen. Therefore, throughout the entire reaction process, chlorine acted as a catalyst, accelerating the failure of the corrosion product scale [25].
In the case of the Si–Co–Y diffusion coating under hot corrosion, the TiSi2 outer layer came into direct contact with corrosive medium of O2, Cl2 and S at the initial stage and underwent reactions at high temperatures [25, 26]:
Meanwhile, the sulfides and chlorides produced by reactions (16) and (17) are gaseous at high temperatures, and tended to volatilize outside of the diffusion coating. Upon exposure to O2, they underwent oxidation reactions [26]:
S2 and Cl2, generated by reactions (18)–(21), can infiltrate into the coating and react with Ti, Si again, resulting in a self-sustaining sulfurization/oxidation process. Thus, S2 and Cl2 can act as catalysts, accelerating the oxidation process of the coating. Fortunately, SiO2 possesses low solubility and good stability in the salts, and can hardly dissolve in the melt as either basic or acidic solutes [27, 28]. Additionally, the amorphous silicate can also play a beneficial role in sealing the loose TiO2, and enhanced the protective nature of the corrosion product scale. The formed oxides of TiO2, SiO2 (or silicate) etc., can also react with Na2O through the following reactions [29]:
Therefore, the hot corrosion product scales shown in Figs. 7(b1) and 8 reveal the formation of Na2SiO3 and Na2TiO3. The amorphous silicate generated from SiO2 possesses a certain degree of fluidity at high temperatures and can adhere to the surface, forming a continuous scale that acts as the first barrier to enhance the protective performance of the corrosion product scale.
As hot corrosion progresses, both Ti and Al elements undergo outward diffusion. Dudziak et al. [30] have found that diffusion of Ti is faster compared to Al within the temperature range of 750–950 °C. As a result, Ti reacted with the inward diffusing oxygen first, followed by the reaction of Al with oxygen. This diffusion mechanism effectively explains the dual-layer structure of the corrosion product scale, which consists of an outer layer of TiO2 + Na2SiO3 + Na2TiO3 and an inner layer of Al2O3. Once a dense and continuous Al2O3 layer formed in the corrosion product scale, the inward diffusion of corrosive media such as O, S, Cl, etc., will be effectively inhibited. Studies have shown that both Co and Y can promote the selective oxidation of Al to form a continuous Al2O3 protective layer [15, 31, 32]. During the hot corrosion process, there was external diffusion of Al element from the TiAl2 inner layer of the Si–Co–Y coating. Under the synergistic effect of Co and Y, a continuous and dense corrosion product scale mainly composed of Al2O3 layer was formed, Zhou et al. [33] found that the addition of Y can promote the diffusion of Co during the coating formation process, resulting in an even distribution of Co. Wei et al. [5] have also found that Co has the ability to delay the diffusion of S, which slows down the sulfidation-oxidation rate, thereby improving the hot corrosion resistance of the coating.
Conclusions
-
(1)
Si–Co–Y diffusion coating with a dense structure and close bonding with the substrate can be prepared on TiAl–Nb alloy by pack cementation process at 1080 °C, using NaF as the activator. The activator and deposition temperature imposed obvious influences on the microstructure of the coating: the coating prepared with NaF, AlCl3·6H2O and NH4Cl exhibited a higher growth rate compared to the one prepared with NaCl. However, there were many pores formed in the coating prepared with AlCl3·6H2O and NH4Cl. Increase in the deposition temperature within the range of 1050–1100 °C led to a higher coating growth rate; but a higher temperature of 1100 °C results in the formation of intensive holes in the coating.
-
(2)
The coating prepared with NaF at 1080 °C for 4 h is dense and compact, which has a multi-layer structure of (Ti, X) Si2 (X represents Co, Al, Nb, Y) outermost layer, an outer layer composed of TiSi2 + Ti5Si4 + Ti5Si3 mixtures, a Ti5Si3 middle layer and a TiAl2 inner layer.
-
(3)
Si–Co–Y diffusion coating prepared on TiAl–Nb alloy possesses excellent hot corrosion resistance in 25%NaCl + 75%K2SO4 molten environments at 850 °C. After 50 h of hot corrosion, a protective hot corrosion product scale composed of TiO2 + Na2SiO3 + Na2TiO3 outer layer, a dense Al2O3 middle layer with a small amount of SiO2 formed on the coating, which can effectively inhibit the inward diffusion of corrosion media of O, Cl and S elements.
References
H. Wakabayashi, L. J. Signori, A. Shaaban, et al., MRS Advances 4, 1465 (2019).
D. V. Pavlenko, Y. O. Belokon, and D. V. Tkach, Materials Science 55, 908 (2020).
S. Q. Wang, F. Q. Xie, X. Q. Wu, and L. Y. Chen, Journal of Alloys and Compounds 788, 632 (2019).
J. Dai, C. Sun, A. Wang, et al., Corrosion Science 184, 109336 (2021).
Z. K. Wei, X. Li, W. Lv, et al., Journal of Materials Engineering and Performance 32, 4796 (2023).
S. Tian, Y. Zhang, A. He, et al., Surface and Coatings Technology 444, 128687 (2022).
X. Gong, R. R. Chen, Y. Wang, et al., Frontiers in Materials 8, 710431 (2021).
J. J. Wu, H. J. Yan, F. H. Cao, et al., Surface and Coatings Technology 422, 127495 (2021).
S. Wang, F. Xie, X. Wu, et al., Journal of Alloys and Compounds 828, 154271 (2020).
P. Zhang and X. P. Guo, Corrosion Science 71, 10 (2013).
Y. Q. Qiao, Z. Shen, and X. P. Guo, Corrosion Science 93, 126 (2015).
J. Huang, F. Zhao, X. Cui, et al., Applied Surface Science 582, 152444 (2022).
X. Ma, Y. He, J. Lin, et al., Surface and Coatings Technology 206, 2690 (2012).
Y. Qiao, J. Kong, R. Zhou, et al., Vacuum 161, 314 (2019).
M. Qiao and C. Zhou, Corrosion science 75, 454 (2013).
K. L. Wang, Q. B. Zhang, M. L. Sun, et al., Journal of materials processing technology 139, 448 (2003).
A. S. Ramos, C. A. Nunes, and G. C. Coelho, Materials Characterization 56, 107 (2006).
H. Clemens and W. Smarsly, Advanced Materials Research 278, 551 (2011).
Z. Sun, W. Wu, Y. Chen, et al., Corrosion Science 185, 109399 (2021).
Y. Q. Li, J. L. Li, C. Qin, et al., Journal of Central South University 27, 381 (2020).
J. Xiang, F. Xie, X. Wu, and S. Wang, Intermetallics 132, 107151 (2021).
M. K. Marzena and G. Elzbieta, Corrosion Science 115, 18 (2017).
Y. Mishin and Chr Herzig, Acta Materialia 48, 589 (2000).
J. He, X. Guo, and Y. Qiao, Transactions of Nonferrous Metals Society of China 31, 207 (2021).
K. Rubacha, E. Godlewska, and K. Mars, Corrosion Science 118, 158 (2017).
J. Xiang, F. Xie, X. Wu, et al., Surface and Coatings Technology 419, 127282 (2021).
L. K. Wu, J. J. Wu, W. Y. Wu, H. J. Yan, et al., Corrosion Science 174, 108827 (2020).
T. Ishitsuka and K. Nose, Corrosion Science 44, 247 (2002).
Y. Qiao, X. Guo, and X. Li, Corrosion Science 91, 75 (2015).
T. Dudziak, P. Datta, H. Du, et al., Central European Journal of Engineering 3, 722 (2013).
V. Babic, C. Geers, and I. Panas, Oxidation of Metals 93, 229 (2019).
H. Li, M. Qiao, and C. Zhou, Materials Chemistry and Physics 143, 915 (2014).
X. Zhao and C. Zhou, Corrosion Science 86, 223 (2014).
Acknowledgements
This research was financially supported by The National Natural Science Foundation of China (No. 51961003), the Major Project of Science and Technology Department of Sichuan province (No. 22SYSX0141), and the Project of the Key Laboratory of Mechanical Structure Optimization & Material Application Technology of Luzhou (SCHYZSA-2023-01, SCHYZSB-2023-01).
Author information
Authors and Affiliations
Contributions
The authors declare that they have no known competing fnancial interests or personal relationships that could have appeared to influence the work reported in this paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing fnancial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, X., Lv, W., Xie, X. et al. Microstructure and Hot Corrosion Property of a Si–Co–Y Diffusion Coating Deposited on TiAl–Nb Alloy. High Temperature Corrosion of mater. 101, 169–185 (2024). https://doi.org/10.1007/s11085-023-10206-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-023-10206-x