
Abstract
The microstructure and hot corrosion behavior of a Si–Y–Co-modified aluminide coating prepared on a nickel base superalloy DZ125 by the pack cementation process was studied. The Co–Al–Si–Y coated specimen had a mass gain of only 0.25 mg/cm2 after being exposed to NaCl + Na2SO4 salt for 100 h at 1173 K. Thus the addition of Si to a Y–Co-modified aluminide coating increased its hot corrosion resistance by 40 %. The improved hot corrosion resistance of the Co–Al–Si–Y coating was mainly attributed to the formation of an Al2O3 scale with SiO2 on the surface of the coating during the hot corrosion test. SiO2 can prohibit the high fluxing rate of dissolution of the protective Al2O3 scale and prevent rapid corrosion attack owing to its low solubility over a wide range of salt acidity and its weak oxygen permeability.
Graphical Abstract
The Co–Al–Si–Y coated specimen had a mass gain of only 0.25 mg/cm2 after being exposed to NaCl + Na2SO4 salt for 100 h at 1173 K. The addition of Si to a Y–Co-modified aluminide coating improved its hot corrosion resistance.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The durability of a gas turbine is mainly limited by the components which operate at high temperature in the turbine sections. In the last few decades, many efforts have been made to develop new heat resisting materials. Nevertheless, Ni-base superalloys are still the most promising structural materials in advanced gas turbines [1]. The turbine components are under severe oxidation and hot corrosion environments in the hot sections. Furthermore, when engines operate in locations where salt can be ingested, i.e., coastal areas and regions with high levels of airborne pollutants, salt will deposit on the surface of gas turbine hardware leading to severe hot corrosion of the turbine components [2, 3]. Thus, the protective coatings on the surface of the superalloys are the essential requirements which can shield the substrate materials from attack.
Owning to their low cost and simple technology, aluminide coatings have been widely used to protect superalloys from oxidation and hot corrosion environments by forming Al2O3 scales as protective barriers [4–7]. However, with the increasing thrust-weight ratio of turbine engines, conventional aluminide coatings can hardly meet the demand of service conditions. To improve the oxidation and corrosion resistance of aluminide coatings and increase the service life of coated superalloys at high temperatures, two approaches have been employed [8]: adding Si, Co, Hf, Pt and Cr [9–14] or adding reactive elements, such as Y, Ce, La, Dy etc. [15–20]. Recent research by Zhao et al. [21] showed that the addition of Y to Co-modified aluminide coatings on nickel base superalloys can improve its hot corrosion resistance by accelerating the diffusion of Co.
In addition to aluminide coatings, the silicide coatings on nickel-based superalloys have been used in gas turbines since 1960 [22]. The silicide coatings can lower the growth rate at high temperature by forming a protective SiO2 scale, but the brittleness of silicide coatings always leads to cracking and spalling [23]. However, when silicon was added as an alloying element as well as a coating element, it can improve the hot corrosion resistance of coatings [24, 25]. Early studies [26] had demonstrated that adding silicon to aluminide coatings could improve both oxidation and hot corrosion resistance. The study by Grünling [25] confirmed the excellence of silicon in protecting against high temperature hot corrosion for its inertness against sulphidation. Shirvani [27] discovered that the addition of silicon could improve hot corrosion resistance by forming the protective oxide SiO2 within the oxide scale. Recent research by He [28] demonstrated that the Si/Cr co-doped NiAlDy alloys exhibited lower oxidation rates and fewer spinels than Cr doped NiAlDy alloys, mainly due to the formation of a thin inert SiO2 film on the scale. Si has been one of the most favourable protective scale-forming or scale-modifying elements for gas turbine applications.
In this study, Si was doped into a Y–Co-modified aluminide coating on nickel base superalloys to improve hot corrosion resistance. The microstructure, composition and hot corrosion behavior of Si–Y–Co-modified aluminide coating were investigated. The effect of the Si addition on the hot corrosion behavior of the coating was discussed.
Experimental Procedures
Materials and Coating Preparation
The substrate material used in this study was nickel base superalloy DZ125 (Beijing Institute of Aeronautical Materials, China) prepared by arc melting and casting in an argon atmosphere. The composition of the nickel base superalloy DZ125 was 63.5Ni–6.2Co–8.9Cr–7.0W–6.1Al–3.8Ta–2.0Mo–1.5Hf–1.0Ti (wt%). The ingot was inverted and remelted more than five times to ensure compositional homogeneity and element parting. The alloy rod was sliced into buttons with a dimension of approximately 14 mm in diameter and 3 mm in thickness by wire-electrode cutting. Surfaces of the buttons were ground with 800 grit SiC paper. Then the buttons were cleaned by acetone solution ultrasonically to remove oxides and other contaminants from the surfaces.
The Co–Al–Si–Y coating was prepared by the pack cementation process with the pack composition of 20Co–7.6Al–4Si–2Y2O3–4NH4Cl–4NH4I–58.4Al2O3 (wt%). The Co, Al, Si and Y2O3 powders were used as donor sources and the Al2O3 powder was used as the filler. The halide salts NH4Cl and NH4I were used as activator which would react with the depositing elements to form a series of halide vapors. Each of the powders used in this study were commercial pure powders (≥99.8 % grade, Beijing Chemical Works, China) with average particle sizes of all less than 100 mesh. The powders of the pack mixture were weighed according to the ratio and mixed appropriately. Subsequently, the substrates were buried in the well-mixed pack powders in a sealed alumina retort with a dimension of approximately 20 mm in diameter and 35 mm in thickness,which was placed in an electric tube furnace. The deposition temperature was chosen as 1323 K and the holding time was chosen as 10 h. The furnace, which was filled with argon gas, was heated to the desired deposition temperature at a rate of 5 K/min. After holding at 1323 K for 10 h, the furnace was turned off while keeping the argon gas flowing. The retort together with specimens cooled down to room temperature naturally.
Hot Corrosion Tests
The corrosion medium in the hot corrosion test was the supersaturated salt solution with a mixture of 25 wt% NaCl + 75 wt% Na2SO4 in distilled water [29]. The specimen was heated to about 373 K in an oven in advance to ensure proper adhesion of the coated salt. Then the saturated salt solution was sprayed uniformly on the surface of the specimen by a sprayer. The water in solution soon evaporated at 373 K and then a layer of the mixed salt deposited on the surface of the specimen. The specimen was weighed before and after salting to ensure a salt supply of 3 mg/cm2 on the surface. In order to dry the alumina crucible, the alumina crucible used as the specimen container needed to be preheated to 1173 K. The specimen was placed into the square alumina crucible individually which was placed in an open-ended tube furnace at 1173 K for 100 h in the air atmosphere. The specimen was taken out after 1, 3, 5, 10, 20, 40, 60, 80, and 100 h, and weighed together with the square alumina crucible. The mass gains of the specimen were measured by the electronic balance (Model BS 224S Sartorious, Germany) with an accuracy of 0.1 mg. In order to improve the accuracy, the hot corrosion test was repeated three times. The mass gain in the corrosion kinetics curve is the average value of three tests. The mass gain of Co–Al–Si–Y coated specimen will be compared with that of Co–Al and Co–Al–Y coated specimens. Data for the Co–Al and Co–Al–Y coated specimens are obtained from previous experiments [20, 21].
Analyzing Methods
The surface phase composition of the coating was investigated by X-ray diffraction (XRD, Model D/Max 2500PC Rigaku, Japan) at 40 kV and 40 mA using Cu Kα radiation (λ = 0.154056 nm). The microstructure and elemental composition of the coating were analyzed by electron probe micro-analyzer (EPMA, Model JEOL JXA-8230) with wavelength dispersive spectrometer (WDS) and scanning electron microscopy (SEM, Model FEI Quanta600, USA).
Results and Discussion
Microstructure of Co–Al–Si–Y Coating
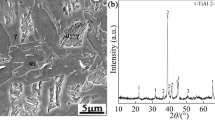
Figure 1 presents the surface morphology of as-coated Co–Al–Si–Y co-deposition coating on DZ125 superalloy prepared by the pack cementation process. It can be observed that the coating surface is flat and consists of polygonal network of grain boundary ridges and a small quantity of Al2O3. The Al2O3 particles from the filler powder are wrapped into the coating, which is typical characterization of outwardly growing aluminide coating. The cross-sectional SEM image of the Co–Al–Si–Y co-deposition coating on DZ125 superalloy is shown in Fig. 2. It is clearly observed that the coating consists of an outer layer and a diffusion zone beneath. The depths of the outer layer and the diffusion zone are about 26 and 25 μm, respectively.

Secondary electron image (SEI) of surface microstructure of the Co–Al–Si–Y coating on DZ125 superalloy prepared by pack cementation process

Cross-sectional microstructure (SEI) of the Co–Al–Si–Y coating on DZ125 superalloy prepared by pack cementation process
The quantitative elemental concentration of the Co–Al–Si–Y coating was detected by linear scanning with EPMA-WDS to investigate the elemental distribution and concentration. The linear scanning started from the coating surface going towards the substrate for approximately 55 μm. The elemental concentration profiles in Fig. 3 show the variation as a function of distance from the coating surface. The content of Ni, Co, Al, Si and Y remains almost unchanged in the outer layer but is variable in the diffusion zone. The elemental composition of the outer layer is about 62Ni–9.5Co–10Al–0.2Si–0.1Y (wt%).

Quantitative elemental concentration profiles of the Co–Al–Si–Y coating on DZ125 superalloy detected by linear scanning with EPMA-WDS
The Co–Al–Si–Y coating was also characterized with EPMA to investigate the elemental distribution. Figure 4 presents the backscattered electron image of the cross-sectional microstructure and the corresponding elemental mappings of Ni, Co, Al, Si and Y. The marked areas A and B in the backscattered electron image are the outer layer and the diffusion zone, respectively. The outer layer of Co–Al–Si–Y coating mainly consists of five elements namely Ni, Co, Al, Si and Y. These elements also exist in the diffusion zone. It can be concluded that the codeposition of Co, Al, Si and Y has been achieved. As clearly shown in Fig. 5, only characteristic peaks of Al9Ni11 and a small quantity of Al2O3 are observed. Because the depth of the outer layer is about 26 μm, the diffusion zone just beneath the scale can’t be detected by XRD. There are no other crystalline forms of Co-containing phases, Si-containing phases or Y-containing phases present in the XRD pattern of the outer layer (Fig. 5). It indicates that Co, Si and Y exist in the form of a solid solution in the coating, where part of the Ni is replaced by Co, Si and Y in the Al9Ni11 phase.

EPMA backscattered electron image and corresponding elemental mappings of the Co–Al–Si–Y coating on DZ125 superalloy prepared by pack cementation process

XRD pattern of surface of the Co–Al–Si–Y coating formed on DZ125 superalloy by pack cementation process
Hot Corrosion Behavior
Figure 6 presents the mass gain of Co–Al–Si–Y coated specimen during the hot corrosion test at 1173 K for 100 h. To recognize the effect of silicon additions on the hot corrosion kinetic of the Co–Al–Si–Y coated specimen, the corrosion kinetics curves of Co–Al and Co–Al–Y coated specimens with a salt deposit of 3 mg/cm2 at 1173 K for 100 h are also displayed in Fig. 6. Data for the Co–Al and Co–Al–Y coated specimens were obtained from previous studies by Liu et al. [20] and Zhao et al. [21]. As can be seen from the corrosion kinetic curves, the weight change for all three specimens is composed of three stages: rapid weight gain (0–20 h), slower weight gain (20–60 h), and stable weight (60–100 h).

At the rapid weight gain stage, the mass gain of Co–Al and Co–Al–Y coated specimens is 0.39 and 0.26 mg/cm2 while that of Co–Al–Si–Y is just 0.2 mg/cm2. After the steep rise, Co–Al, Co–Al–Y and Co–Al–Si–Y coated specimens have a mass gain of 0.08, 016 and 0.04 mg/cm2 at the slow weight gain stage (20–60 h), respectively. Until 100 h, the weight gain of Co–Al and Co–Al–Y coated specimens is up to 0.48 and 0.42 mg/cm2, respectively. However, the Co–Al–Si–Y coated specimen has a mass gain of only 0.25 mg/cm2 after 100 h hot corrosion at 1173 K. The addition of Si to Y–Co-modified aluminide coating decreases its mass gain by 40 % after 100 h hot corrosion.
Corrosion Product Characterization
The phase composition of the surface of Co–Al–Si–Y coating after hot corrosion testing was investigated by XRD. As clearly shown in Fig. 7, characteristic peaks of α-Al2O3, Al9Ni11 and minor SiO2 are observed.

XRD pattern of the surface of the Co–Al–Si–Y coating after being exposed to NaCl + Na2SO4 salt at 1173 K for 100 h
Figure 8 shows the surface morphology of the Co–Al–Si–Y coating after being exposed to NaCl + Na2SO4 salt at 1173 K for 100 h. The surface of Co–Al–Si–Y coating is dense and compact without obvious spallation and cracks, implying slight destruction and good corrosion resistance. Besides, a lot of round particles can be observed on the surface. The particles mainly consist of Al and O elements by WDS detection.

Surface morphology (SEI) of the Co–Al–Si–Y coating after hot corrosion at 1173 K for 100 h
Figure 9 displays the cross-sectional microstructure of the Co–Al–Si–Y coating with corrosion products after hot corrosion for 100 h. The oxide scale on the surface of Co–Al–Si–Y coating is thin, continuous and compact, which is the typical characteristic of good hot corrosion resistance. There are small amount of holes or spallation present in the interface of coating and corrosion products, indicating the strong adhesion of the oxide scale.

Cross-sectional microstructure (SEI) of the Co–Al–Si–Y coating after hot corrosion at 1173 K for 100 h
Figure 10 presents the cross-sectional backscattered electron image (BEI) and the corresponding elemental mappings of the Co–Al–Si–Y coating after hot corrosion for 100 h. The main elements of the oxide scale are Al, O and Si elements. Considering the surface XRD pattern of the Co–Al–Si–Y coating after hot corrosion (Fig. 7), the oxide scale on the coating surface is composed of α-Al2O3 and a small quantity of SiO2. Besides, since the oxide scale is very thin, the coating just beneath the scale is also detected with peaks of high intensity Al9Ni11. Besides, minor S can be observed around the oxide scale, which is likely to be present in the form of sulphides. The sulfide is formed by the reaction of elements in coatings with the film of molten sodium sulfate salt during the hot corrosion.

EPMA backscattered electron image of the cross-sectional microstructure for the Co–Al–Y–Ce coating after hot corrosion for 100 h and the corresponding elemental mappings of Al, O, Co, Ni, Si, S and Y
Discussion
As indicated above, the Co–Al–Si–Y coating exhibits superior hot corrosion resistance to Co–Al–Y and Co–Al coatings. The addition of Si improves its hot corrosion resistance as compared with Co–Al–Y. The less weight gain of Co–Al–Si–Y coating is mainly attributed to the formation of the Al2O3 oxide with SiO2 on the surface of coating during the hot corrosion test. SiO2 appears to have better corrosion resistance than Al2O3 when exposed to mixture of NaCl and Na2SO4 molten salt [25]. This can be explained as follows.
Hot corrosion could be seen as an accelerated oxidation in the presence of mixture of NaCl and Na2SO4 molten salt deposited on the surface of the coating [30, 31]. At the initial stage of hot corrosion, oxidation was the dominated feature and the protective Al2O3 scale was formed on the molten salt deposit/coating interface. When the molten salt Na2SO4 deposited on the surface of the coating, the protective Al2O3 scale was fluxed [32] via reaction (1). The protective Al2O3 scale was also dissolved by molten chlorine through the oxychlorination and chlorination/oxidation cyclic reaction (2) [33]. Due to the concentration gradient, the AlO2 − ion migrated from the molten salt deposit/coating interface to the molten salt deposit/air interface, where alkalinity of molten salt was low. Then Al2O3 can be re-precipitated by decomposition of AlO2 − and release of O2− in reaction (3). The alkalinity of the molten salt deposit increases during the re-precipitation of Al2O3, which promoted the fluxing of metal oxide. A basic fluxing-re-precipitation of metal oxides was formed in the presence of the molten salt deposition, which made the previously protective Al2O3 scale porous and easily spall.
The combined attack of NaCl and Na2SO4 molten salt results in high fluxing rate of dissolution for protective Al2O3 scale. However, SiO2 can prohibit the high fluxing rate and prevent rapid corrosion attack owning to its low solubility over a wide range of salt acidity and its weak oxygen permeability [34]. According to previous study by Rapp, the negative solubility gradient was regarded as a general criterion for continuing hot corrosion attack [35]. SiO2 does not form an ionic solute in a range of melt basicity. Therefore its low solubility was independent of basicity, which is favorable for hot corrosion resistance in acid Na2SO4. Besides, the corresponding thermodynamic treatment indicates that the reactions of SiO2 with NaCl and Na2SO4 will not take place under the standard state [25]. Consequently, the addition of Si can enhance the hot corrosion resistance of coating.
Conclusions
The codeposition of Co, Al, Si and Y has been achieved on the surface of the nickel base superalloys by pack cementation process. The Co–Al–Si–Y coated specimen had a mass gain of only 0.25 mg/cm2 after being exposed to mixture of NaCl and Na2SO4 molten salt after 100 h at 1173 K. The addition of Si to Y–Co-modified aluminide coating decreased its mass gain by 40 % after 100 h hot corrosion. The lower weight gain of Co–Al–Si–Y coating is mainly attributed to the formation of Al2O3 scale with SiO2 on the surface of the coating during the hot corrosion test. SiO2 can prohibit the high fluxing rate of dissolution for protective Al2O3 scale and prevent rapid corrosion attack owing to its low solubility over a wide range of salt acidity and its weak oxygen permeability.
References
K. A. Marino and E. A. Carter, Acta Materialia 58, 2726 (2010).
A. B. Smith, A. Kempster and J. Smith, Surface and Coatings Technology 112, 120 (1999).
C. H. Koo, C. Y. Bai and Y. J. Luo, Materials Chemistry and Physics 86, 259 (2004).
Z. H. Xu, J. W. Dai, J. Niu, L. M. He, R. Mu and Z. K. Wang, Journal of Alloys and Compounds 637, 343 (2015).
A. V. Put, D. Oquab, E. Pere, A. Raffaitin and D. Monceau, Oxidation of Metals 75, 247 (2011).
R. Burgel, Materials Science and Technology 2, 302 (1986).
A. H. Yaghtin, S. Javadpour and M. H. Shariat, Journal of Alloys and Compounds 584, 304 (2014).
F. Wang, H. Y. Lou, L. X. Bai and W. T. Wu, Materials Science and Engineering A 121, 388 (1989).
M. Qiao and C. G. Zhou, Corrosion Science 63, 241 (2012).
Z. H. Xu, Z. K. Wang, G. H. Huang, R. Mu and L. He, Journal of Alloys and Compounds 637, 226 (2015).
A. Rahman, V. Chawla, R. Jayaganthan, R. Chandra and R. Ambardar, Oxidation of Metals 74, 341 (2010).
C. Fu, S. Q. Wang, W. K. Kong and G. H. Cao, Oxidation of Metals 84, 151 (2015).
H. Asteman, W. Hartnagel and D. Jakobi, Oxidation of Metals 80, 3 (2013).
M. Qiao and C. G. Zhou, Surface and Coatings Technology 206, 2899 (2012).
J. Stringer, Materials Science and Engineering A 120, 129 (1989).
J. Lu, S. Zhu and F. Wang, Oxidation of Metals 76, 67 (2011).
Z. J. Liu, X. S. Zhao, H. M. Guo and C. G. Zhou, Corrosion Science 94, 135 (2015).
R. Thanneeru, S. Patil, S. Deshpande and S. Seal, Acta Materialia 55, 3457 (2007).
R. W. Jackson, J. P. Leonard, L. Niewolak, W. J. Quadakkers, R. Murray, S. Romani, G. J. Tatlock, F. S. Pettit and G. H. Meier, Oxidation of Metals 78, 197 (2012).
Z. J. Liu, X. S. Zhao and C. G. Zhou, Corrosion Science 92, 148 (2015).
X. S. Zhao and C. G. Zhou, Corrosion Science 86, 223 (2014).
G. Becker and W. Spyra, Metalloberfliiche 17, 271 (1963).
J. P. Riviere, L. Pichon and M. Drouet, Surface and Coatings Technology 201, 8343 (2007).
A. S. Dorcheh and M. C. Galetz, Oxidation of Metals 84, 89 (2015).
H. W. Grünling and R. Bauer, Thin Solid Films 95, 3 (1982).
G. S. Young and D. L. Deadmore, Thin Solid Films 73, 373 (1980).
K. Shirvani, M. Saremi, A. Nishikata and T. Tsuru, Materials Science Forum 461, 335 (2004).
J. He, H. B. Guo, Y. L. Zhang and S. K. Gong, Corrosion Science 85, 232 (2014).
K. Zhang, M. M. Liu, S. L. Liu, C. Sun and F. H. Wang, Corrosion Science 53, 1990 (2011).
P. Hancock, Material Science and Technology 3, 536 (1987).
N. Eliaz, G. Shemesh and R. Latanision, Engineering Failure Analysis 9, 31 (2002).
P. S. Liu, K. M. Liang, H. Y. Zhou, S. R. Gu, X. F. Sun, H. R. Guan, T. Jin and K. N. Yang, Surface and Coatings Technology 145, 75 (2001).
D. J. Young and B. Gleeson, Corrosion Science 44, 345 (2002).
D. Z. Shi and R. A. Rapp, Journal of Electrochemical Society 133, 84 (1986).
R. A. Rapp, Corrosion Science 44, 209 (2002).
Acknowledgments
This project is supported by the National Natural Science Foundation of China under Contract 51371021 and the Aviation Science Foundation under Contract 2014ZE51053.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liu, Z., Zhou, C. Hot Corrosion Behavior of Si–Y–Co-Modified Aluminide Coating Exposed to NaCl + Na2SO4 Salt at 1173 K. Oxid Met 85, 205–217 (2016). https://doi.org/10.1007/s11085-015-9599-3
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-015-9599-3